
A Biologically-Based Computational Approach to Drug Repurposing for Anthrax Infection

Abstract
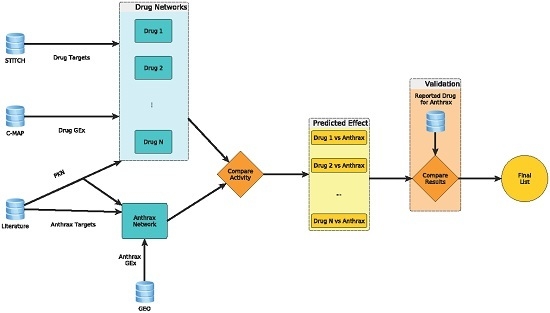
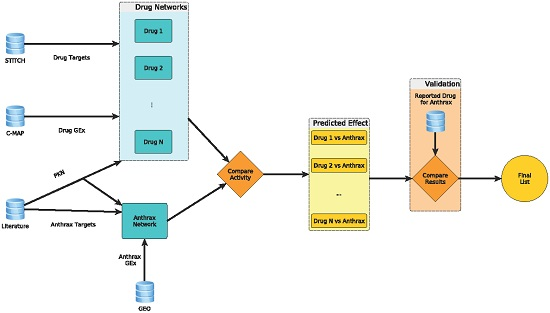
:
1. Introduction
2. Results
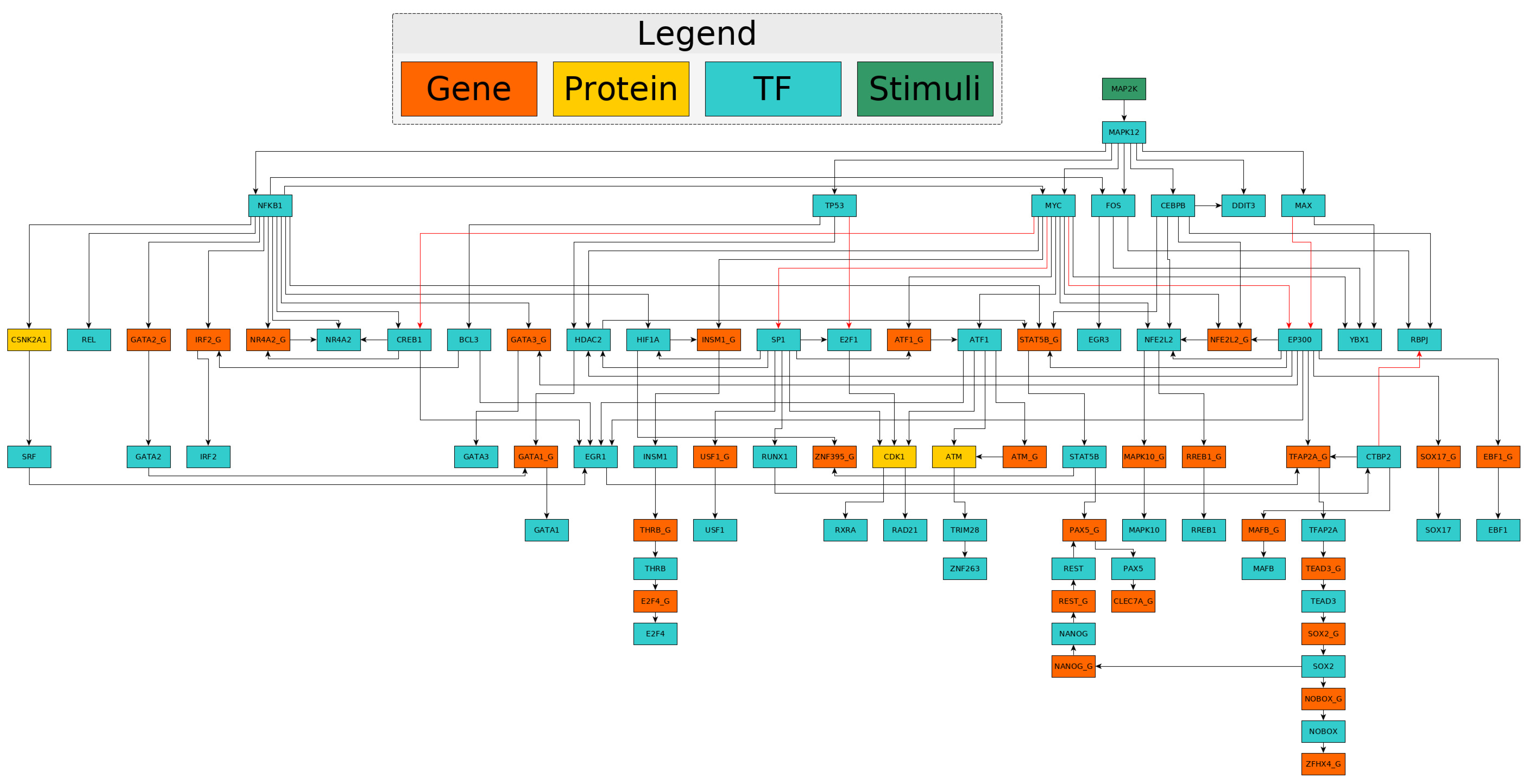
2.1. Anthrax Network for Anthrax Toxicity
2.2. Computation of Network Activities to Identify Candidates for Repurposing
2.3. Confirmation of Candidates with Reported In Vitro Studies in Literature
3. Discussion
4. Conclusions
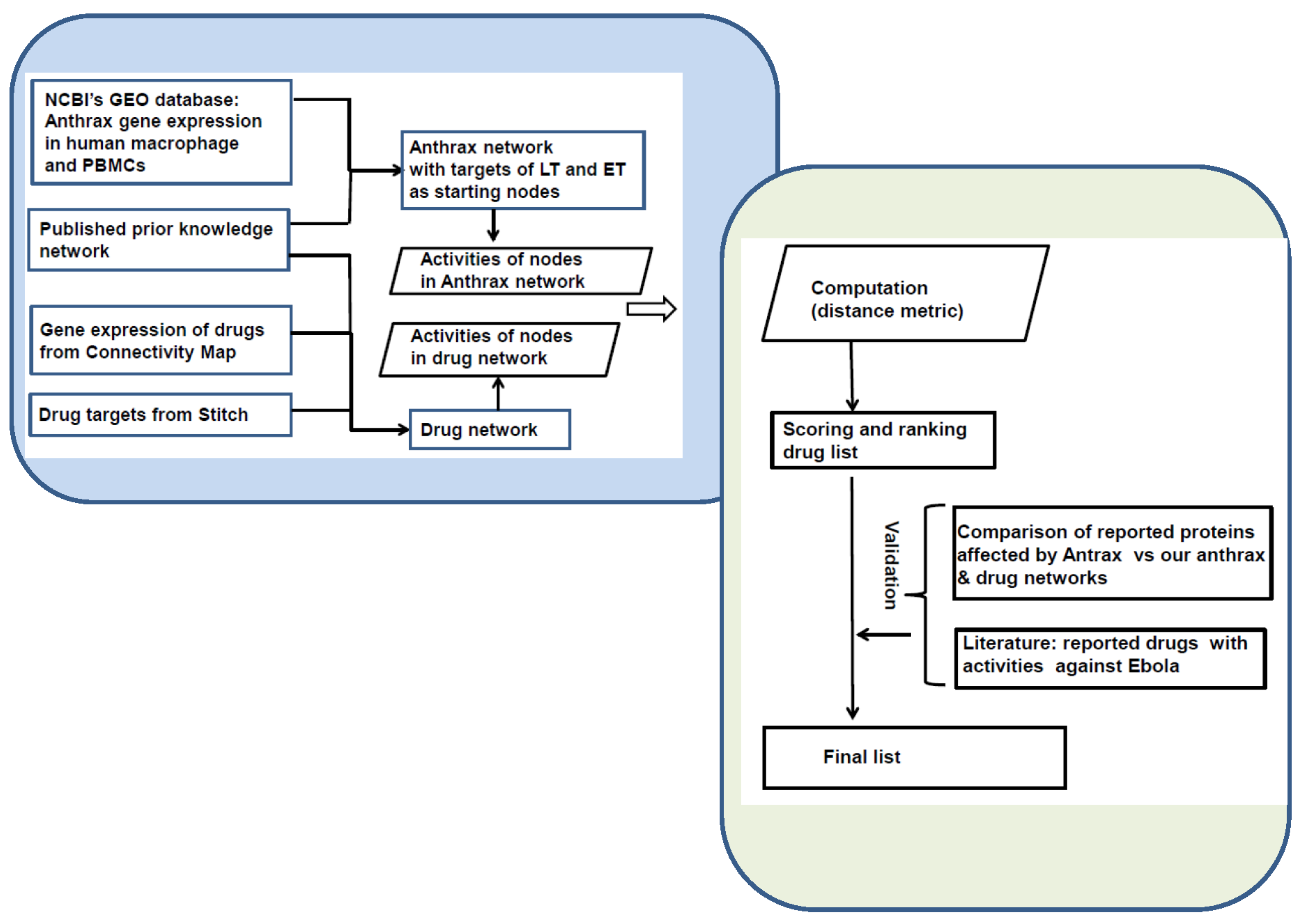
5. Experimental Section
5.1. Construction of Anthrax Network
- GSE14390: Alveolar Macrophages treated with Anthrax Spores
- GSE34407: Peripheral Monocytes treated with Lethal Toxin
- GSE12131: Umbilical Vein Endothelial Cells treated with Lethal Toxin
- GSE17777: Microvascular Endothelial Cells treated with Edema Toxin
- GSE4478: Peripheral Monocytes treated with Lethal Toxin
- GSE12533: Peripheral Monocytes treated with Protective Antigen
5.2. Construction of Drug Network
5.3. Computational Identification of Potential Drugs
5.4. Validation
Supplementary Materials
Acknowledgements
Author contributions
Conflicts of Interest
References
- Navdarashvili, A.; Doker, T.J.; Geleishvili, M.; Haberling, D.L.; Kharod, G.A.; Rush, T.H.; Maes, E.; Zakhashvili, K.; Imnadze, P.; Bower, W.A.; et al. Human anthrax outbreak associated with livestock exposure: Georgia, 2012. Epidemiol. Infect. 2016, 144, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Review of the Scientific Approaches Used during the FBI’s Investigation of the 2001 Anthrax Letters; The National Academies Press: Washington, DC, USA, 2011.
- Meselson, M.; Guillemin, J.; Hugh-Jones, M.; Langmuir, A.; Popova, I.; Shelokov, A.; Yampolskaya, O. The Sverdlovsk anthrax outbreak of 1979. Science 1994, 266, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Abramova, F.A.; Grinberg, L.M.; Yampolskaya, O.V.; Walker, D.H. Pathology of inhalational anthrax in 42 cases from the Sverdlovsk outbreak of 1979. Proc. Natl. Acad. Sci. USA 1993, 90, 2291–2294. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.M.; Abramova, F.A.; Yampolskaya, O.V.; Walker, D.H.; Smith, J.H. Quantitative pathology of inhalational anthrax I: Quantitative microscopic findings. Mod. Pathol. 2001, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Moayeri, M.; Leppla, S.H. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 2014, 22, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.W.; Cui, X.; Sweeney, D.A.; Li, Y.; Barochia, A.; Eichacker, P.Q. The potential contributions of lethal and edema toxins to the pathogenesis of anthrax associated shock. Toxins (Basel) 2011, 3, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Klimpel, K.R.; Arora, N.; Leppla, S.H. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol. Microbiol. 1994, 13, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352 Pt 3, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Duesbery, N.S.; Vande Woude, G.F. Anthrax lethal factor causes proteolytic inactivation of mitogen-activated protein kinase kinase. J. Appl. Microbiol. 1999, 87, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Levin, T.C.; Wickliffe, K.E.; Leppla, S.H.; Moayeri, M. Heat shock inhibits caspase-1 activity while also preventing its inflammasome-mediated activation by anthrax lethal toxin. Cell. Microbiol. 2008, 10, 2434–2446. [Google Scholar] [CrossRef] [PubMed]
- Wickliffe, K.E.; Leppla, S.H.; Moayeri, M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell. Microbiol. 2008, 10, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [PubMed]
- Ribot, W.J.; Panchal, R.G.; Brittingham, K.C.; Ruthel, G.; Kenny, T.A.; Lane, D.; Curry, B.; Hoover, T.A.; Friedlander, A.M.; Bavari, S. Anthrax lethal toxin impairs innate immune functions of alveolar macrophages and facilitates bacillus anthracis survival. Infect. Immun. 2006, 74, 5029–5034. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Lingappa, J.; Leppla, S.H.; Agrawal, S.; Jabbar, A.; Quinn, C.; Pulendran, B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 2003, 424, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Pulendran, B. Anthrax lethal toxin: A weapon of multisystem destruction. Cell. Mol. Life Sci. 2004, 61, 2859–2865. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.G.; Espina, V.; Zhou, W.; Mueller, C.; Liotta, L.; Popov, S.G. Whole proteome analysis of mouse lymph nodes in cutaneous anthrax. PLoS ONE 2014, 9, e110873. [Google Scholar] [CrossRef] [PubMed]
- Tournier, J.N.; Quesnel-Hellmann, A.; Mathieu, J.; Montecucco, C.; Tang, W.J.; Mock, M.; Vidal, D.R.; Goossens, P.L. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J. Immunol. 2005, 174, 4934–4941. [Google Scholar] [CrossRef] [PubMed]
- Comer, J.E.; Chopra, A.K.; Peterson, J.W.; Konig, R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect. Immun. 2005, 73, 8275–8281. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.L.; Friedlander, A.M.; Rogers, L.C.; Yoon, I.K.; Warren, R.L.; Cross, A.S. Anthrax edema toxin differentially regulates lipopolysaccharide-induced monocyte production of tumor necrosis factor alpha and interleukin-6 by increasing intracellular cyclic AMP. Infect. Immun. 1994, 62, 4432–4439. [Google Scholar] [PubMed]
- Yeager, L.A.; Chopra, A.K.; Peterson, J.W. Bacillus anthracis edema toxin suppresses human macrophage phagocytosis and cytoskeletal remodeling via the protein kinase A and exchange protein activated by cyclic amp pathways. Infect. Immun. 2009, 77, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Milia, E.; Warburton, R.R.; Hill, N.S.; Gaestel, M.; Kayyali, U.S. Anthrax lethal toxin disrupts the endothelial permeability barrier through blocking p38 signaling. J. Cell. Physiol. 2012, 227, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Warfel, J.M.; Steele, A.D.; D’Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar] [CrossRef]
- Maddugoda, M.P.; Stefani, C.; Gonzalez-Rodriguez, D.; Saarikangas, J.; Torrino, S.; Janel, S.; Munro, P.; Doye, A.; Prodon, F.; Aurrand-Lions, M.; et al. cAMP signaling by anthrax edema toxin induces transendothelial cell tunnels, which are resealed by MIM via Arp2/3-driven actin polymerization. Cell Host Microbe 2011, 10, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Doebele, R.C.; Lingen, M.W.; Quilliam, L.A.; Tang, W.J.; Rosner, M.R. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J. Biol. Chem. 2007, 282, 19781–19787. [Google Scholar] [CrossRef] [PubMed]
- Melas, I.N.; Sakellaropoulos, T.; Iorio, F.; Alexopoulos, L.G.; Loh, W.Y.; Lauffenburger, D.A.; Saez-Rodriguez, J.; Bai, J.P. Identification of drug-specific pathways based on gene expression data: Application to drug induced lung injury. Integr. Biol. (Camb.) 2015, 7, 904–920. [Google Scholar] [CrossRef] [PubMed]
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of NLRP1 is required for activation of the inflammasome. PLoS Pathog. 2012, 8, e1002638. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Sastalla, I.; Leppla, S.H. Anthrax and the inflammasome. Microbes Infect. 2012, 14, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Finger, J.N.; Lich, J.D.; Dare, L.C.; Cook, M.N.; Brown, K.K.; Duraiswami, C.; Bertin, J.; Gough, P.J. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 2012, 287, 25030–25037. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [PubMed]
- Sanchez, A.M.; Thomas, D.; Gillespie, E.J.; Damoiseaux, R.; Rogers, J.; Saxe, J.P.; Huang, J.; Manchester, M.; Bradley, K.A. Amiodarone and bepridil inhibit anthrax toxin entry into host cells. Antimicrob. Agents Chemother. 2007, 51, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Artenstein, A.W.; Opal, S.M.; Cristofaro, P.; Palardy, J.E.; Parejo, N.A.; Green, M.D.; Jhung, J.W. Chloroquine enhances survival in bacillus anthracis intoxication. J. Infect. Dis. 2004, 190, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.J.; Hobson, J.P.; Southall, N.; Qiu, C.; Thomas, C.J.; Lu, J.; Inglese, J.; Zheng, W.; Leppla, S.H.; Bugge, T.H.; et al. Quantitative high-throughput screening identifies inhibitors of anthrax-induced cell death. Bioorg. Med. Chem. 2009, 17, 5139–5145. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, R.; Singh, Y.; Leppla, S.H.; Friedlander, A.M. Calcium is required for the expression of anthrax lethal toxin activity in the macrophagelike cell line J774A.1. Infect. Immun. 1989, 57, 2107–2114. [Google Scholar] [PubMed]
- deCathelineau, A.M.; Bokoch, G.M. Inactivation of Rho gtpases by statins attenuates anthrax lethal toxin activity. Infect. Immun. 2009, 77, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Squires, R.C.; Muehlbauer, S.M.; Brojatsch, J. Proteasomes control caspase-1 activation in anthrax lethal toxin-mediated cell killing. J. Biol. Chem. 2007, 282, 34260–34267. [Google Scholar] [CrossRef] [PubMed]
- Brook, I. The prophylaxis and treatment of anthrax. Int. J. Antimicrob. Agents 2002, 20, 320–325. [Google Scholar] [CrossRef]
- Lee, L.V.; Bower, K.E.; Liang, F.S.; Shi, J.; Wu, D.; Sucheck, S.J.; Vogt, P.K.; Wong, C.H. Inhibition of the proteolytic activity of anthrax lethal factor by aminoglycosides. J. Am. Chem. Soc. 2004, 126, 4774–4775. [Google Scholar] [CrossRef] [PubMed]
- Agren, J.; Finn, M.; Bengtsson, B.; Segerman, B. Microevolution during an anthrax outbreak leading to clonal heterogeneity and penicillin resistance. PLoS ONE 2014, 9, e89112. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, V.; Zeitlin, R. Cotinine: A potential new therapeutic agent against Alzheimer’s disease. CNS Neurosci. Ther. 2012, 18, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Duez, H.; Blanquart, C.; Berezowski, V.; Poulain, P.; Fruchart, J.C.; Najib-Fruchart, J.; Glineur, C.; Staels, B. Statin-induced inhibition of the Rho-signaling pathway activates PPARα and induces HDL apoA-I. J. Clin. Investig. 2001, 107, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Tan, G.; Guo, Q.; Pang, R.; Zeng, F. Abnormal activation of the Akt-GSK3β signaling pathway in peripheral blood T cells from patients with systemic lupus erythematosus. Cell Cycle 2009, 8, 2789–2793. [Google Scholar] [CrossRef] [PubMed]
- Eckels, P.C.; Banerjee, A.; Moore, E.E.; McLaughlin, N.J.; Gries, L.M.; Kelher, M.R.; England, K.M.; Gamboni-Robertson, F.; Khan, S.Y.; Silliman, C.C. Amantadine inhibits platelet-activating factor induced clathrin-mediated endocytosis in human neutrophils. Am. J. Physiol. Cell Physiol. 2009, 297, C886–C897. [Google Scholar] [CrossRef] [PubMed]
- Von Borstel Smith, M.; Crofoot, K.; Rodriguez-Proteau, R.; Filtz, T.M. Effects of phenytoin and carbamazepine on calcium transport in Caco-2 cells. Toxicol. In Vitro 2007, 21, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Sitges, M.; Chiu, L.M.; Reed, R.C. Effects of levetiracetam, carbamazepine, phenytoin, valproate, lamotrigine, oxcarbazepine, topiramate, vinpocetine and sertraline on presynaptic hippocampal Na+ and Ca2+ channels permeability. Neurochem. Res. 2016, 41, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Lochner, M.; Lummis, S.C. The antimalarial drugs quinine, chloroquine and mefloquine are antagonists at 5-HT3 receptors. Br. J. Pharmacol. 2007, 151, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Kayser, V.; Aubel, B.; Hamon, M.; Bourgoin, S. The antimigraine 5-HT 1B/1D receptor agonists, sumatriptan, zolmitriptan and dihydroergotamine, attenuate pain-related behaviour in a rat model of trigeminal neuropathic pain. Br. J. Pharmacol. 2002, 137, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Hinton, J.M.; Hill, P.; Jeremy, J.; Garland, C. Signalling pathways activated by 5-HT(1B)/5-HT(1D) receptors in native smooth muscle and primary cultures of rabbit renal artery smooth muscle cells. J. Vasc. Res. 2000, 37, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.R.; Chong, I.W.; Chen, C.C.; Lin, S.R.; Sheu, C.C.; Hwang, J.J. Mitogen-activated protein kinase pathway was significantly activated in human bronchial epithelial cells by nicotine. DNA Cell Biol. 2006, 25, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.W.; Singh, A.K. Nicotine and lung cancer. J. Carcinog. 2013, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.T.; Zimmerman, K.; Wexler, L.H.; Blaney, S.; Jarosinski, P.; Weaver-McClure, L.; Izraeli, S.; Balis, F.M. A prospective evaluation of ifosfamide-related nephrotoxicity in children and young adults. Cancer 1995, 76, 2557–2564. [Google Scholar] [CrossRef]
- Wisler, J.W.; DeWire, S.M.; Whalen, E.J.; Violin, J.D.; Drake, M.T.; Ahn, S.; Shenoy, S.K.; Lefkowitz, R.J. A unique mechanism of beta-blocker action: Carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar] [CrossRef] [PubMed]
- Cochran, J.C.; Gatial, J.E., 3rd; Kapoor, T.M.; Gilbert, S.P. Monastrol inhibition of the mitotic kinesin Eg5. J. Biol. Chem. 2005, 280, 12658–12667. [Google Scholar] [CrossRef] [PubMed]
- Sarker, K.P.; Biswas, K.K.; Rosales, J.L.; Yamaji, K.; Hashiguchi, T.; Lee, K.Y.; Maruyama, I. Ebselen inhibits NO-induced apoptosis of differentiated PC12 cells via inhibition of ASK1-p38 MAPK-p53 and JNK signaling and activation of p44/42 MAPK and Bcl-2. J. Neurochem. 2003, 87, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ham, K.; Gao, X.; Castro, L.; Yan, Y.; Kissling, G.E.; Tucker, C.J.; Flagler, N.; Dong, R.; Archer, T.K.; et al. Epigenetic regulation of transcription factor promoter regions by low-dose genistein through mitogen-activated protein kinase and mitogen-and-stress activated kinase 1 nongenomic signaling. Cell Commun. Signal. 2016, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, F.S.; Thaa, B.; Amrun, S.N.; Simarmata, D.; Rausalu, K.; Nyman, T.A.; Merits, A.; McInerney, G.M.; Ng, L.F.; Ahola, T. The antiviral alkaloid berberine reduces chikungunya virus-induced mitogen-activated protein kinase (MAPK) signaling. J. Virol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.H.; Wu, X.J.; Liu, J.S.; Gao, Y.B. Withaferin a activates stress signalling proteins in high risk acute lymphoblastic leukemia. Int. J. Clin. Exp. Pathol. 2015, 8, 15652–15660. [Google Scholar] [PubMed]
- Kau, J.H.; Sun, D.S.; Tsai, W.J.; Shyu, H.F.; Huang, H.H.; Lin, H.C.; Chang, H.H. Antiplatelet activities of anthrax lethal toxin are associated with suppressed p42/44 and p38 mitogen-activated protein kinase pathways in the platelets. J. Infect. Dis. 2005, 192, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.T.; Chen, H.M.; Cheng, S.J.; Chiang, C.P.; Kuo, M.Y. Arecoline-stimulated connective tissue growth factor production in human buccal mucosal fibroblasts: Modulation by curcumin. Oral Oncol. 2009, 45, e99–e105. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, X.; Yang, G.; Min, X.; Deng, M. Apigenin inhibits cell migration through MAPK pathways in human bladder smooth muscle cells. Biocell 2011, 35, 71–79. [Google Scholar] [PubMed]
- Boncompagni, S.; Arthurton, L.; Akujuru, E.; Pearson, T.; Steverding, D.; Protasi, F.; Mutungi, G. Membrane glucocorticoid receptors are localised in the extracellular matrix and signal through the MAPK pathway in mammalian skeletal muscle fibres. J. Physiol. 2015, 593, 2679–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remy, K.E.; Qiu, P.; Li, Y.; Cui, X.; Eichacker, P.Q. B. anthracis associated cardiovascular dysfunction and shock: The potential contribution of both non-toxin and toxin components. BMC Med. 2013, 11, 217. [Google Scholar] [CrossRef] [PubMed]
- Brojatsch, J.; Casadevall, A.; Goldman, D.L. Molecular determinants for a cardiovascular collapse in anthrax. Front. Biosci. 2014, 6, 139–147. [Google Scholar]
- Silswal, N.; Parelkar, N.; Andresen, J.; Wacker, M.J. Restoration of endothelial function in Pparα−/− mice by tempol. PPAR Res. 2015, 2015, 728494. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, A.; Schoonjans, K.; Jesel, L.; Carpusca, I.; Auwerx, J.; Andriantsitohaina, R. Activation of the peroxisome proliferator-activated receptor alpha protects against myocardial ischaemic injury and improves endothelial vasodilatation. BMC Pharmacol. 2002, 2, 10. [Google Scholar] [CrossRef]
- Gardner, O.S.; Dewar, B.J.; Earp, H.S.; Samet, J.M.; Graves, L.M. Dependence of peroxisome proliferator-activated receptor ligand-induced mitogen-activated protein kinase signaling on epidermal growth factor receptor transactivation. J. Biol. Chem. 2003, 278, 46261–46269. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.; Linde, C.; Putsep, K.; Pohanka, A.; Normark, S.; Henriques-Normark, B.; Andersson, J.; Bjorkhem-Bergman, L. Studies on the antibacterial effects of statins—In vitro and in vivo. PLoS ONE 2011, 6, e24394. [Google Scholar] [CrossRef] [PubMed]
- Vainio, P.J.; Tornquist, K.; Tuominen, R.K. Cotinine and nicotine inhibit each other’s calcium responses in bovine chromaffin cells. Toxicol. Appl. Pharmacol. 2000, 163, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Beitzinger, C.; Bronnhuber, A.; Duscha, K.; Riedl, Z.; Huber-Lang, M.; Benz, R.; Hajos, G.; Barth, H. Designed azolopyridinium salts block protective antigen pores in vitro and protect cells from anthrax toxin. PLoS ONE 2013, 8, e66099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi Paccani, S.; Tonello, F.; Patrussi, L.; Capitani, N.; Simonato, M.; Montecucco, C.; Baldari, C.T. Anthrax toxins inhibit immune cell chemotaxis by perturbing chemokine receptor signalling. Cell. Microbiol. 2007, 9, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D’Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Rossi Paccani, S.; Benagiano, M.; Capitani, N.; Zornetta, I.; Ladant, D.; Montecucco, C.; D’Elios, M.M.; Baldari, C.T. The adenylate cyclase toxins of Bacillus anthracis and bordetella pertussis promote TH2 cell development by shaping T cell antigen receptor signaling. PLoS Pathog. 2009, 5, e1000325. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Benagiano, M.; Savino, M.T.; Finetti, F.; Tonello, F.; D’Elios, M.M.; Baldari, C.T. The adenylate cyclase toxin of Bacillus anthracis is a potent promoter of TH17 cell development. J. Allergy Clin. Immunol. 2011, 127, 1635–1637. [Google Scholar] [CrossRef] [PubMed]
- Tancevski, I.; Nairz, M.; Duwensee, K.; Auer, K.; Schroll, A.; Heim, C.; Feistritzer, C.; Hoefer, J.; Gerner, R.R.; Moschen, A.R.; et al. Fibrates ameliorate the course of bacterial sepsis by promoting neutrophil recruitment via CXCR2. EMBO Mol. Med. 2014, 6, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.H.; Tsai, W.C.; Lee, T.J.; Huang, C.C.; Chang, P.H.; Su Pang, J.H. Simvastatin inhibits IL-5-induced chemotaxis and CCR3 expression of HL-60-derived and human primary eosinophils. PLoS ONE 2016, 11, e0157186. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Burger, F.; Pelli, G.; Poku, N.K.; Berlier, C.; Steffens, S.; Mach, F. Statins inhibit C-reactive protein-induced chemokine secretion, ICAM-1 upregulation and chemotaxis in adherent human monocytes. Rheumatology (Oxford) 2009, 48, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Dozmorov, M.; Wu, W.; Chakrabarty, K.; Booth, J.L.; Hurst, R.E.; Coggeshall, K.M.; Metcalf, J.P. Gene expression profiling of human alveolar macrophages infected by B. Anthracis spores demonstrates TNF-alpha and NF-kappab are key components of the innate immune response to the pathogen. BMC Infect. Dis. 2009, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Chauncey, K.M.; Lopez, M.C.; Sidhu, G.; Szarowicz, S.E.; Baker, H.V.; Quinn, C.; Southwick, F.S. Bacillus anthracis’ lethal toxin induces broad transcriptional responses in human peripheral monocytes. BMC Immunol. 2012, 13, 33. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Published Results | References |
|---|---|
| Studied in dendritic cells, LT (10 μg/mL, 6-h incubation) was shown to impaired adaptive immunity. | Agrawal et al. [16] |
| Inhibition of MAPK signaling pathways by LT impaired innate and adaptive immunity, as well as vascular barrier integrity (review). | Agrawal et al. [17] |
| LT (1 μg/mL, 12–72 h of incubation) induced a concentration- and time-dependent increase in vascular permeability (primary human lung microvascular endothelial cells). | Warfel et al. [24] |
| Studied in nonhuman primate alveolar macrophages, LT (1 μg/mL, 4-h incubation) impaired host’s innate immune responses. | Ribot et al. [15] |
| LT (2 μg/mL, 45-min incubation) decreased activation of p38 substrate kinase, MK2, and reduced phosphorylation of HSP27, leading to damaging endothelial barrier and vascular integrity (rat pulmonary microvascular endothelial cells). | Liu et al. [23] |
| Studied in PBMCs, LT (0.5 μg/mL, 15-h incubation) caused apoptosis and reduced production of pro-inflammatory cytokines. | Popova et al. [18] |
| Published Results | References |
|---|---|
| In human monocytes, edema toxin (20 ng/mL, 1-h incubation) induced cAMP accumulation and damaged cellular antimicrobial activity. | Hoover et al. [21] |
| Cooperating with lethal toxin, edema toxin impaired innate immune responses (maurine bone marrow-derived dendritic cells, 40ng/mL for both ER and LT, 1-h incubation). | Tournier et al. [19] |
| Studied in mice, edema toxin and lethal toxin (PA:10 μg/mL, LF and EF: 7.5 μg/mL, injection) inhibited T cell activation, implying impairment of adaptive immune response. | Comer et al. [20] |
| Edema toxin inhibited endothelial cell chemotaxis via Epac, the effector of RAP1 (endothelial cell line:HMVECs; PA:5 μg/mL, EF: 1 μg/mL, 1-h incubation). | Hong et al. [26] |
| Edema toxin (PA:5 μg/mL, EF: 1.15 μg/mL, 24-h incubation) suppresses human macrophage phagocytosis by deregulating cAMP-dependent protein kinase pathway. | Yeager et al. [22] |
| Edema toxin (PA:70 mg/mouse, EF: 70 mg/mouse, tail end injection) induced transendothelial cell macroaperture (TEM) tunnels (intestine) via affecting c-AMP signaling (human umbilical vein endothelial cells, 1 μg/mL ET, 1-h incubation). | Maddugoda et al. [25] |
| Drug | Biological Evidences | References |
|---|---|---|
| Fenofibrate (PPARα) activator | Cross talks between mevalonate pathway and PPARα; inhibition of LT cytotoxicity by statins mediated via inhibiting Rho GTPase and activating PPARα. | deCathelineau et al. [36]; Martin et al. [42] |
| Dihydroergotamine | 5-HT 1B/1D agonist Stimulation of 5-HT 1B/1D receptors activated MAPK and reduced cAMP level. | Kayser et al. [48]; Hinton et al. [49] |
| Cotinine | Activated mitogen-activated protein kinases. | Warren et al. [51]; Tsai et al. [50] |
| Simvastatin | Statins inhibited LT cytotoxicity by inactivating Rho GTPase. | deCathelineau et al. [36] |
| Amantadine | Cancelled activation of p38/MAP. p38/MAP kinase inhibitors (SB-203580 and SB-202190) protected cells from LT-mediated cytotoxicity. | Eckels et al. [44]; Sanchez et al. [32] |
| Mephenytoin | A derivative of phenytoin. Phenytoin inhibited active transport of Ca+2 via enterocytes, and Ca+2 channel in the brain. | von Borstel Smith et al. [45]; Sitges et al. [46] |
| Mefloquine | Mefloquine is an analog of chloroquine that had in vitro activity protecting cells from LT toxicity. | Thompson et al. [47] |
| Bepridil | Calcium channel blocker; Ca+2 is required for LT toxicity. | Sanchez et al. [32]; Bhatnagar et al. [35] |
| Sotalol | Decreased intracellular accumulation of cAMP, an action that is opposite to that of ET. | Wisler et al. [53] |
| Ifosfamide | Increased renal recreation of Ca+2 that could lead to disturbance of Ca+2 homeostasis and depletion of Ca+2. | Ho et al. [52] |
| Drug | Biological Evidences | References |
|---|---|---|
| Monastrol | Arresting cells in mitosis. | Cochran et al. [54] |
| Colforsin | An agonist of adenyl cyclase that converts ATP to cAMP. Such action would increase intracellular cAMP and synergistically increase ET toxicity. | Johannessen et al. [57] |
| Berberine | Reduced activation of MAPK signaling by chikungunya virus. | Varghese et al. [58] |
| Withaferin a | Activated p38 MAPK, a downstream kinase of MAPK signaling. | Shi et al. [59] |
| Arecoline | Its action is opposite to that of P38 MAPK inhibitors (Its induction of CTGF expression was inhibited by P38 MAPK inhibitors). | Deng et al. [61] |
| Ebselen | Inhibited ASK1-p38 MAPK-p35 and JUK signaling and activated MPAK p44/42. | Sarker et al. [55] |
| Genistein | Activated MAPK p44/42. | Yu et al. [56] |
| Apigenin | Inhibited MAPK (an action similar to LT). | Liu et al. [62] |
| Beclometasone | Activated p38 MAPK (an action opposite to that of p38 MAPK inhibitors in protecting cells from LT). | Boncompagni et al. [63]; Sanchez et al. [32] |
| Enilconazole | Antifungal drug for animals. | Merck veterinary manual |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, J.P.F.; Sakellaropoulos, T.; Alexopoulos, L.G. A Biologically-Based Computational Approach to Drug Repurposing for Anthrax Infection. Toxins 2017, 9, 99. https://doi.org/10.3390/toxins9030099
Bai JPF, Sakellaropoulos T, Alexopoulos LG. A Biologically-Based Computational Approach to Drug Repurposing for Anthrax Infection. Toxins. 2017; 9(3):99. https://doi.org/10.3390/toxins9030099
Chicago/Turabian StyleBai, Jane P. F., Theodore Sakellaropoulos, and Leonidas G. Alexopoulos. 2017. "A Biologically-Based Computational Approach to Drug Repurposing for Anthrax Infection" Toxins 9, no. 3: 99. https://doi.org/10.3390/toxins9030099
APA StyleBai, J. P. F., Sakellaropoulos, T., & Alexopoulos, L. G. (2017). A Biologically-Based Computational Approach to Drug Repurposing for Anthrax Infection. Toxins, 9(3), 99. https://doi.org/10.3390/toxins9030099