Molecular Modeling of the Catalytic Domain of CyaA Deepened the Knowledge of Its Functional Dynamics


Abstract
:
{kind=link}
{kind=link}
{kind=link}
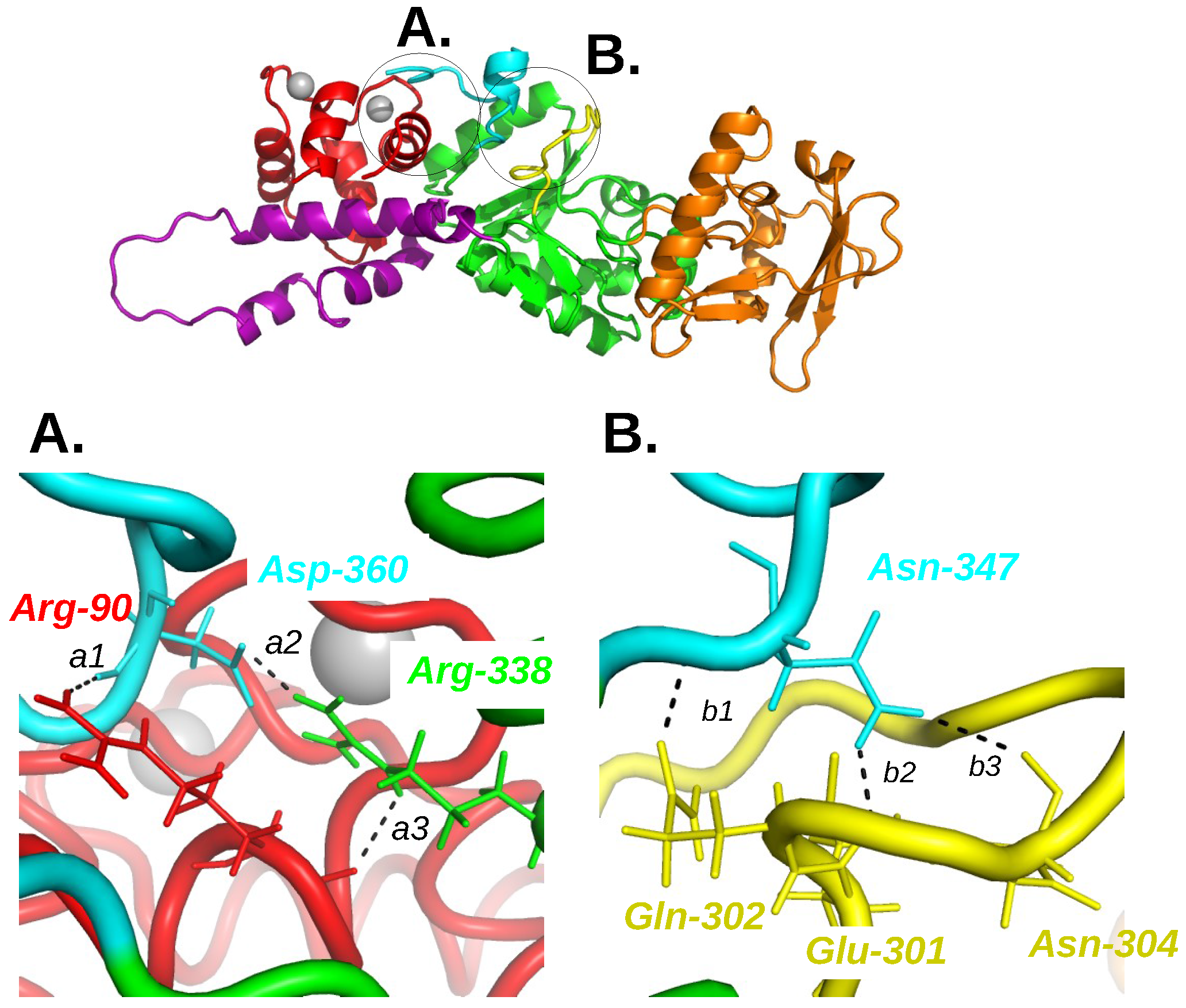{kind=link}
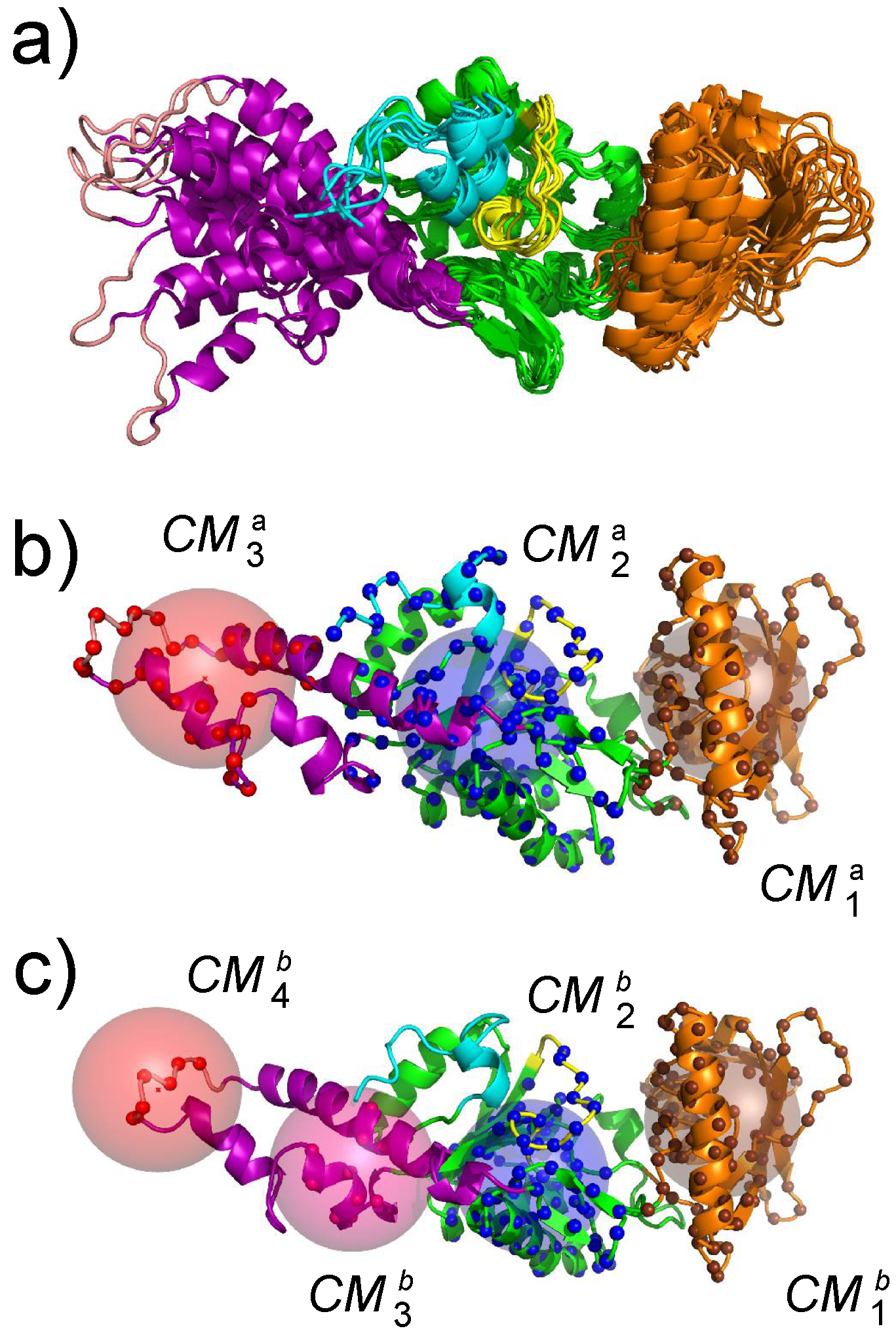{kind=link}
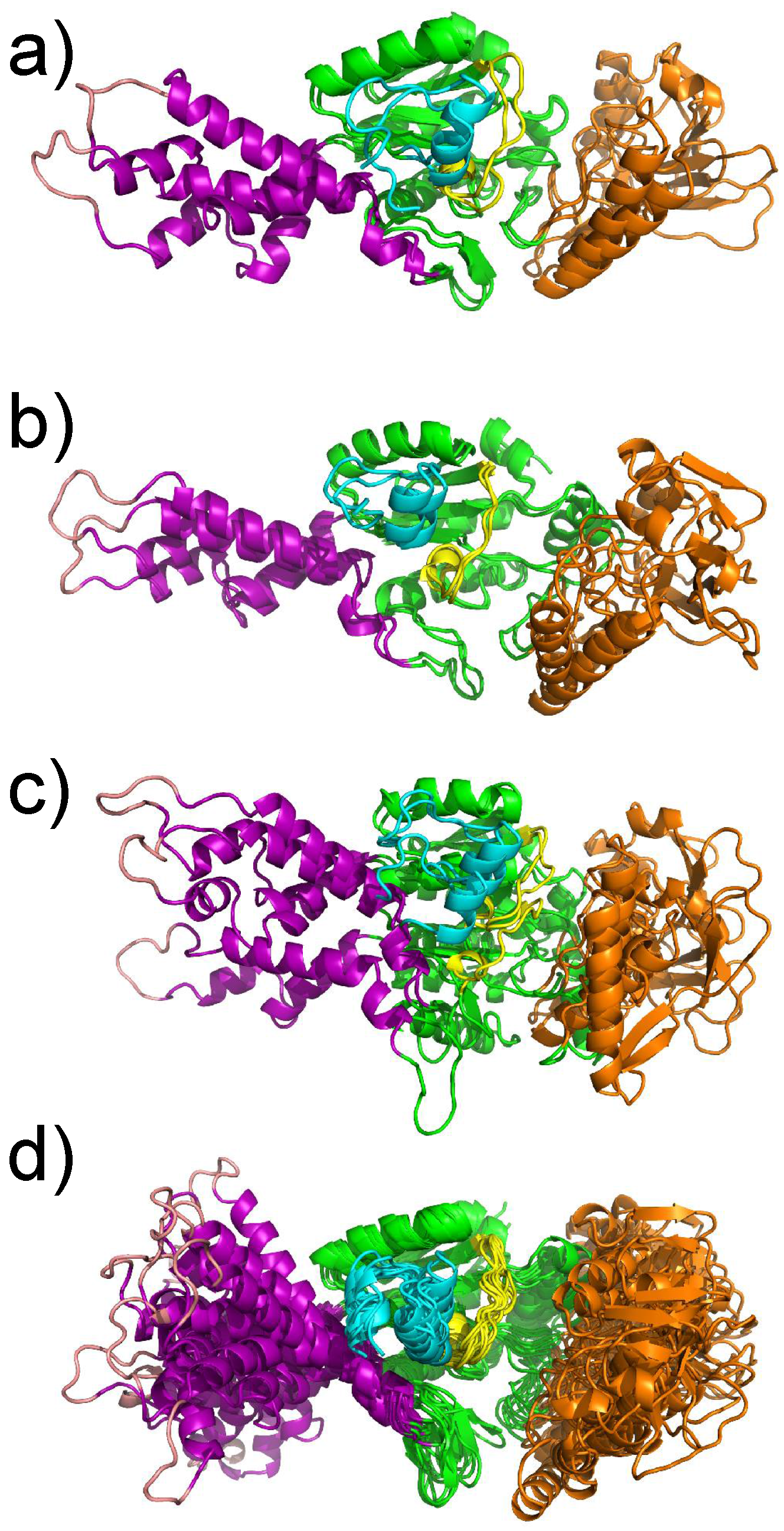{kind=link}
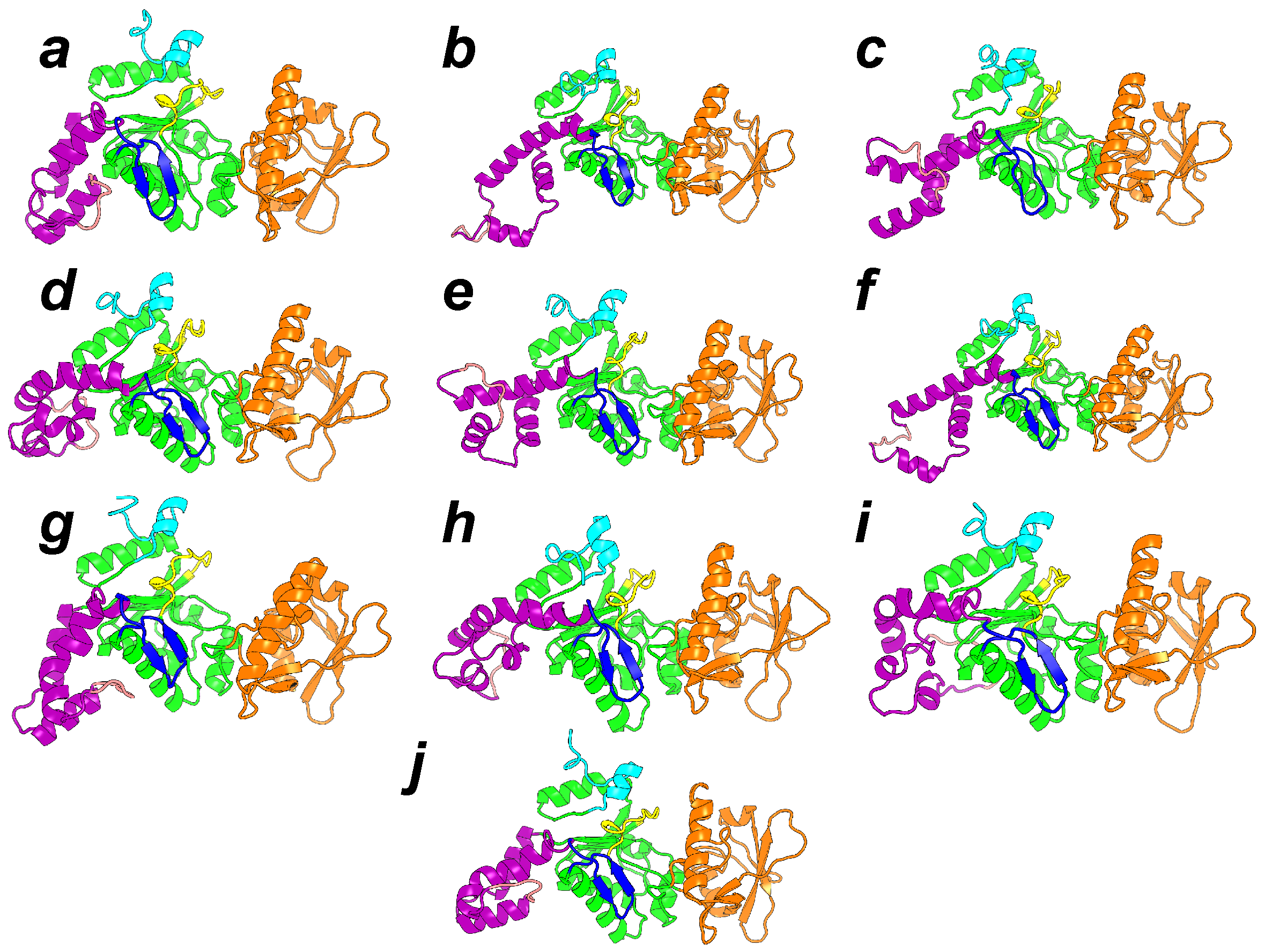{kind=link}
{kind=link}
{kind=link}
1. Introduction
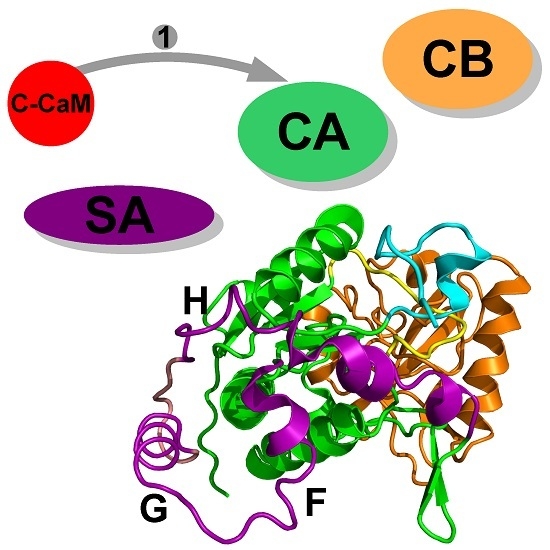
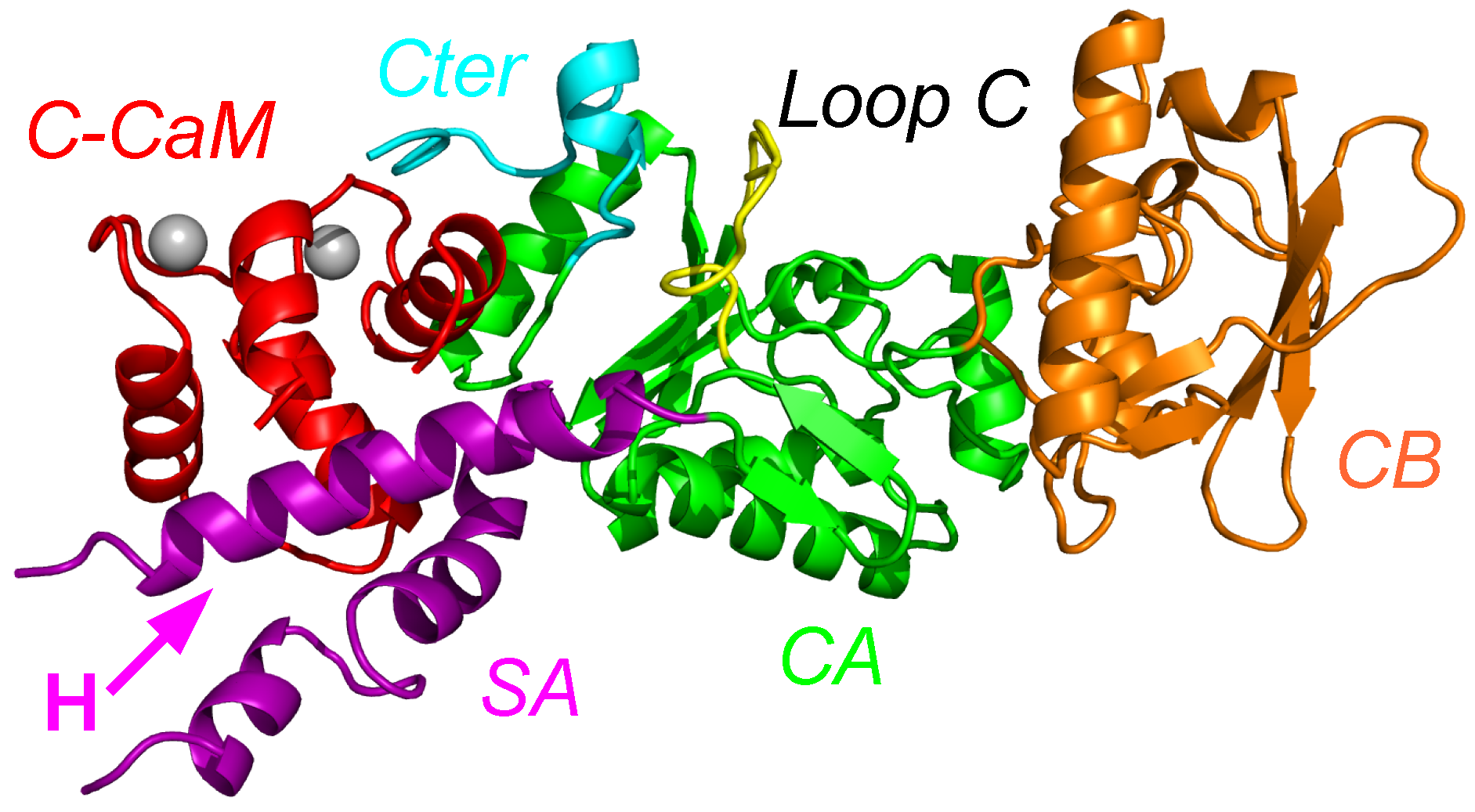
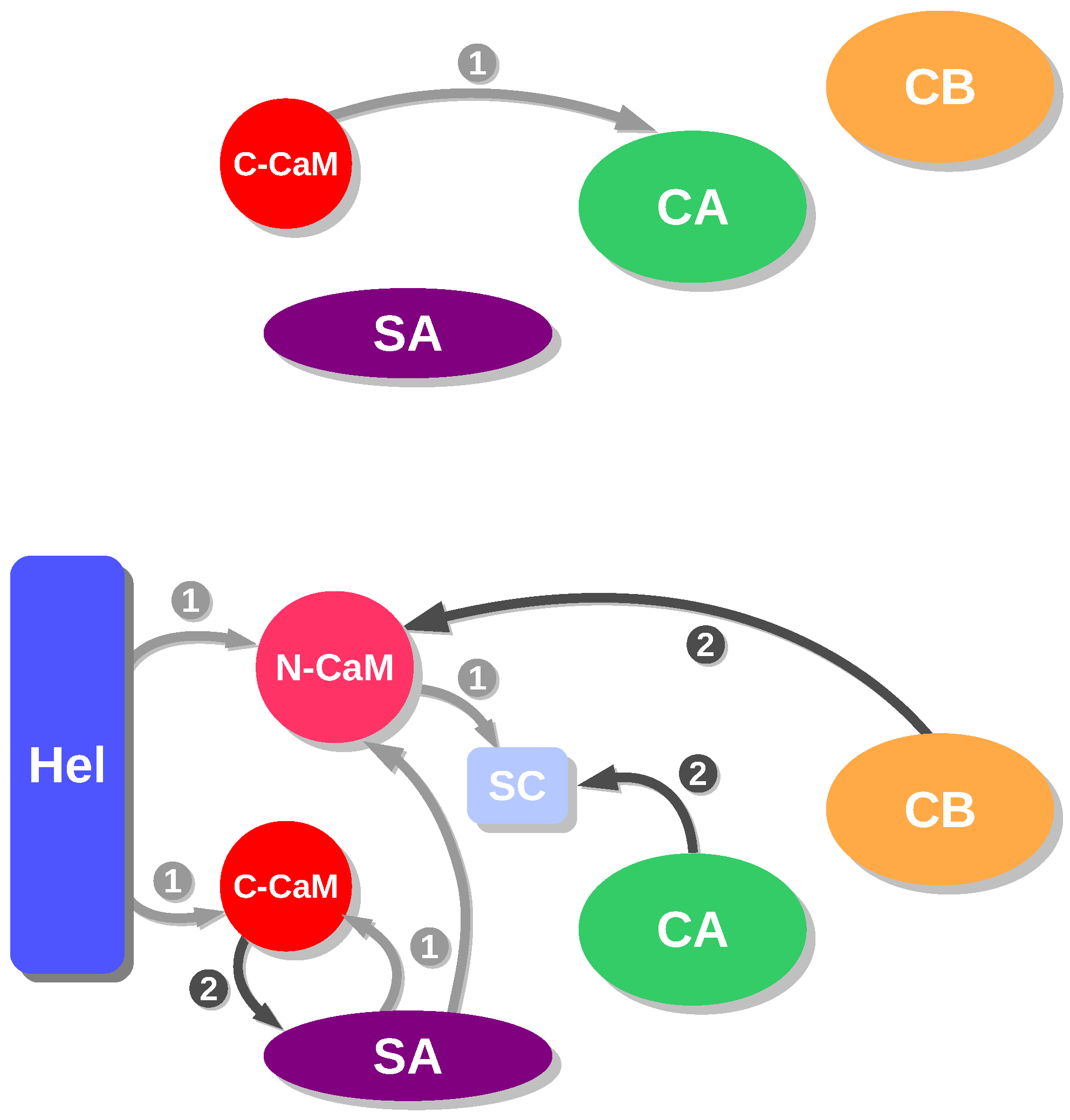
2. Interaction between Calmodulin and AC
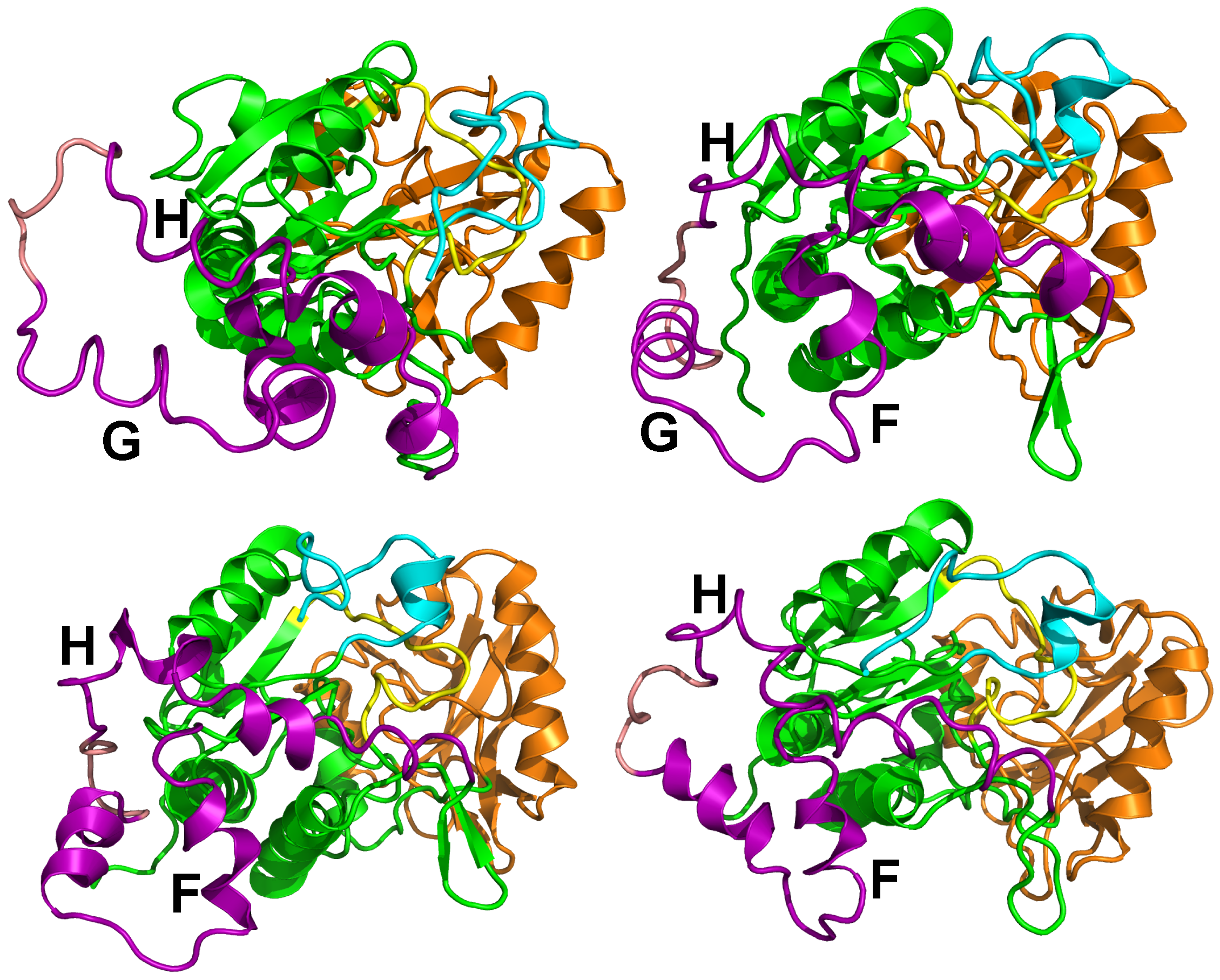
3. Conformational Landscape of Free AC
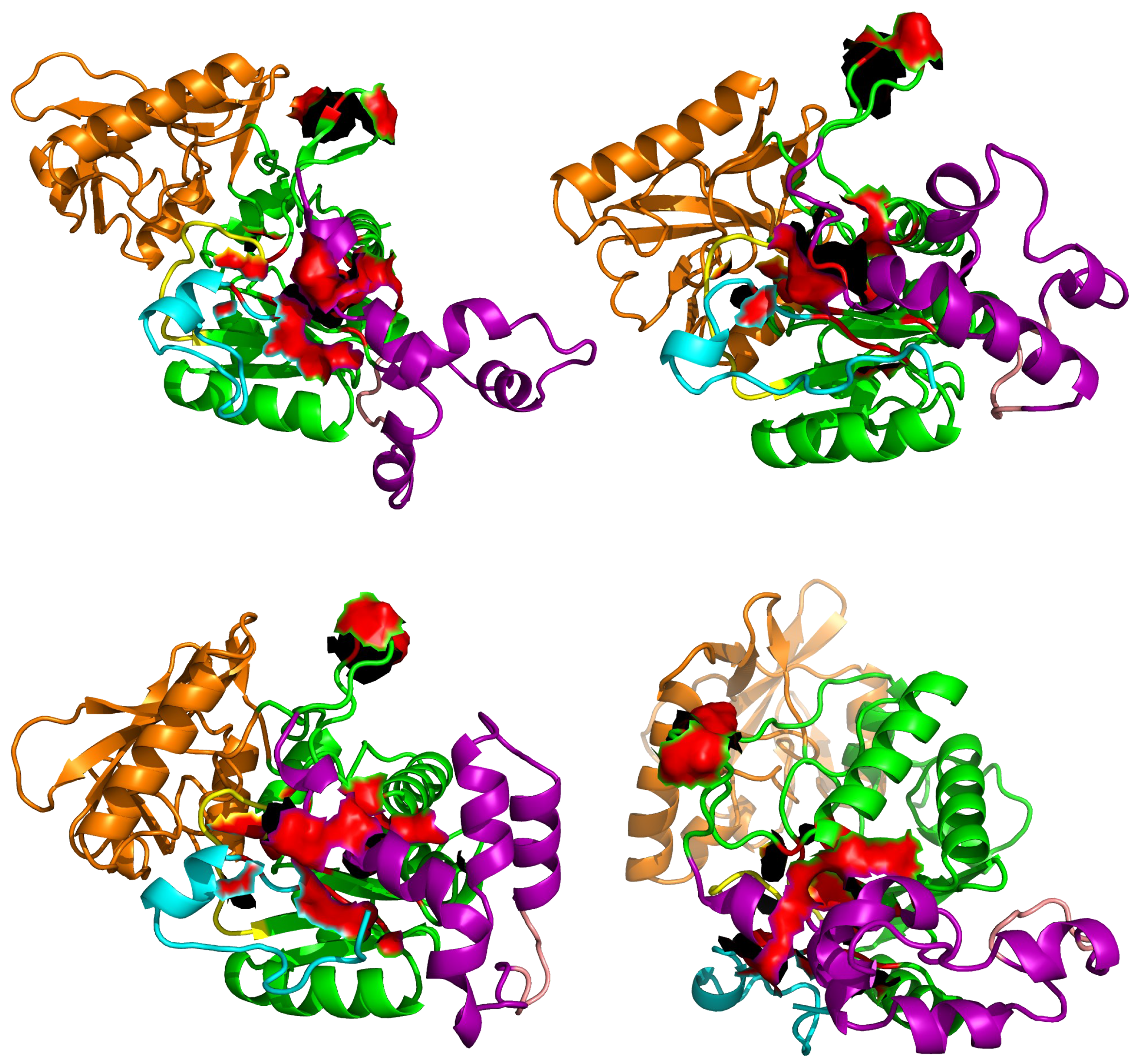
4. Searching Inhibitors of AC Activation
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Drum, C.; Yan, S.; Bard, J.; Shen, Y.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Shen, Y.; Lee, Y.; Gibbs, C.; Mrksich, M.; Tang, W. Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 2005, 24, 3190–3201. [Google Scholar] [CrossRef] [PubMed]
- Chenal, A.; Karst, J.; Sotomayor-Pérez, A.; Wozniak, A.; Baron, B.; England, P.; Ladant, D. Calcium-induced folding and stabilization of the intrinsically disordered RTX domain of the CyaA toxin. Biophys. J. 2010, 99, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor-Pérez, A.; Subrini, O.; Hessel, A.; Ladant, D.; Chenal, A. Molecular Crowding Stabilizes Both the Intrinsically Disordered Calcium-Free State and the Folded Calcium-Bound State of a Repeat in Toxin (RTX) Protein. J. Am. Chem. Soc. 2013, 135, 11929–11934. [Google Scholar] [CrossRef] [PubMed]
- Bumba, L.; Masin, J.; Macek, P.; Wald, T.; Motlova, L.; Bibova, I.; Klimova, N.; Bednarova, L.; Veverka, V.; Kachala, M.; et al. Calcium-Driven Folding of RTX Domain β-Rolls Ratchets Translocation of RTX Proteins through Type I Secretion Ducts. Mol. Cell 2016, 62, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor-Pérez, A.; Ladant, D.; Chenal, A. Disorder-to-order transition in the CyaA toxin RTX domain: Implications for toxin secretion. Toxins 2014, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, D.; Hernandez, B.; Durand, D.; Hourdel, V.; Sotomayor-Pérez, A.; Vachette, P.; Ghomi, M.; Chamot-Rooke, J.; Ladant, D.; Brier, S.; et al. Structural models of intrinsically disordered and calcium-bound folded states of a protein adapted for secretion. Sci. Rep. 2015, 5, 14223. [Google Scholar] [CrossRef] [PubMed]
- Ladant, D.; Ullmann, A. Bordetella pertussis adenylate cyclase: A toxin with multiple talents. Trends Microbiol. 1999, 7, 172–176. [Google Scholar] [CrossRef]
- Karimova, G.; Pidoux, J.; Ullmann, A.; Ladant, D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5752–5756. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Dove, S. Towards selective inhibitors of adenylyl cyclase toxin from Bordetella pertussis. Trends Microbiol. 2012, 20, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yao, K.; Ma, X.; Shi, W.; Yuan, L.; Yang, Y. Susceptibility to Erythromycin and Virulence-Related Genotype Changes in China (1970–2014). PLoS ONE 2015, 10, e0138941. [Google Scholar]
- Bernardi, R.C.; Melo, M.C.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, H.; Moritsugu, K.; Matsunaga, Y.; Morishita, T.; Maragliano, L. Extended Phase-Space Methods for Enhanced Sampling in Molecular Simulations: A Review. Front. Bioeng. Biotechnol. 2015, 3, 125. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; McCammon, J.A. Unconstrained Enhanced Sampling for Free Energy Calculations of Biomolecules: A Review. Mol. Simul. 2016, 42, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lee, Y.; Soelaiman, S.; Bergson, P.; Lu, D.; Chen, A.; Beckingham, K.; Grabarek, Z.; Mrksich, M.; Tang, W. Physiological calcium concentrations regulate calmodulin binding and catalysis of adenylyl cyclase exotoxins. EMBO J. 2002, 21, 6721–6732. [Google Scholar] [CrossRef] [PubMed]
- Glaser, P.; Elmaoglou-Lazaridou, A.; Krin, E.; Ladant, D.; Bárzu, O.; Danchin, A. Identification of residues essential for catalysis and binding of calmodulin in Bordetella pertussis adenylate cyclase by site-directed mutagenesis. EMBO J. 1989, 8, 967–972. [Google Scholar] [PubMed]
- Vougier, S.; Mary, J.; Dautin, N.; Vinh, J.; Friguet, B.; Ladant, D. Essential role of methionine residues in calmodulin binding to Bordetella pertussis adenylate cyclase, as probed by selective oxidation and repair by the peptide methionine sulfoxide reductases. J. Biol. Chem. 2004, 279, 30210–30218. [Google Scholar] [CrossRef] [PubMed]
- Prêcheur, B.; Siffert, O.; Bârzu, O.; Craescu, C.T. NMR and circular dichroic studies on the solution conformation of a synthetic peptide derived from the calmodulin-binding domain of Bordetella pertussis adenylate cyclase. Eur. J. Biochem. 1991, 196, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Craescu, C.; Bouhss, A.; Mispelter, J.; Diesis, E.; Popescu, A.; Chiriac, M.; Bârzu, O. Calmodulin binding of a peptide derived from the regulatory domain of Bordetella pertussis adenylate cyclase. J. Biol. Chem. 1995, 270, 7088–7096. [Google Scholar] [CrossRef] [PubMed]
- Karst, J.; Sotomayor-Pérez, A.; Guijarro, J.; Raynal, B.; Chenal, A.; Ladant, D. Calmodulin-induced conformational and hydrodynamic changes in the catalytic domain of Bordetella pertussis adenylate cyclase toxin. Biochemistry 2010, 49, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Selwa, E.; Laine, E.; Malliavin, T. Differential role of Calmodulin and Calcium ions in the stabilization of the catalytic domain of adenyl cyclase CyaA from Bordetella pertussis. Proteins 2012, 80, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar] [PubMed]
- Torchia, D.A. NMR studies of dynamic biomolecular conformational ensembles. Prog. Nucl. Magn. Reson. Spectrosc. 2015, 84–85, 14–32. [Google Scholar]
- Zhang, M.; Tanaka, T.; Ikura, M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 1995, 2, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Finn, B.; Evenas, J.; Drakenberg, T.; Waltho, J.; Thulin, E.; Forsen, S. Calcium-induced structural changes and domain autonomy in calmodulin. Nat. Struct. Biol. 1995, 2, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Jas, G.; Kuczera, K. Structure, dynamics and interaction with kinase targets: Computer simulations of calmodulin. Biochim. Biophys. Acta 2004, 1697, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Laine, E.; Blondel, A.; Malliavin, T. Dynamics and Energetics: A Consensus Analysis of the Impact of Calcium on EF-CaM Protein Complex. Biophys. J. 2009, 96, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Zhou, H.X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Hamacher, K.; Trylska, J.; McCammon, J. Dependency map of proteins in the small ribosomal subunit. PLoS Comput. Biol. 2006, 2, e10. [Google Scholar] [CrossRef] [PubMed]
- Laine, E.; Martinez, L.; Blondel, A.; Malliavin, T. Activation of the edema factor of Bacillus anthracis by calmodulin: Evidence of an interplay between the EF-calmodulin interaction and calcium binding. Biophys. J. 2010, 99, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.; Emerson, C.; Johns, C.; Finley, N. Interaction with adenylate cyclase toxin from Bordetella pertussis affects the metal binding properties of calmodulin. FEBS Open Bio 2017, 7, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Selwa, E.; Huynh, T.; Ciccotti, G.; Maragliano, L.; Malliavin, T. Temperature-accelerated molecular dynamics gives insights into globular conformations sampled in the free state of the AC catalytic domain. Proteins 2014, 82, 2483–2496. [Google Scholar] [CrossRef] [PubMed]
- Kursula, P. The many structural faces of calmodulin: A multitasking molecular jackknife. Amino Acids 2014, 46, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Aleksiev, T.; Potestio, R.; Pontiggia, F.; Cozzini, S.; Micheletti, C. PiSQRD and a web server for decomposing proteins into quasi-rigid dynamical domains. Bioinformatics 2009, 25, 2743–2744. [Google Scholar] [CrossRef] [PubMed]
- Maragliano, L.; Vanden-Eijnden, E. A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem. Phys. Lett. 2006, 426, 168–175. [Google Scholar] [CrossRef]
- Maragliano, L.; Cottone, G.; Ciccotti, G.; Vanden-Eijnden, E. Mapping the network of pathways of CO diffusion in myoglobin. J. Am. Chem. Soc. 2010, 132, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Lyman, E.; Zuckerman, D. Ensemble-based convergence analysis of biomolecular trajectories. Biophys. J. 2006, 91, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Bouvier, G.; Nilges, M.; Maragliano, L.; Malliavin, T. Temperature Accelerated Molecular Dynamics with Soft-Ratcheting Criterion Orients Enhanced Sampling by Low-Resolution Information. J. Chem. Theory Comput. 2015, 11, 3446–3454. [Google Scholar] [CrossRef] [PubMed]
- Perilla, J.; Beckstein, O.; Denning, E.; Woolf, T. Computing ensembles of transitions from stable states: Dynamic importance sampling. J. Comput. Chem. 2011, 2, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Perilla, J.; Woolf, T. Computing ensembles of transitions with molecular dynamics simulations. Methods Mol. Biol. 2015, 1215, 237–252. [Google Scholar] [PubMed]
- Resetca, D.; Wilson, D. Characterizing rapid, activity-linked conformational transitions in proteins via sub-second hydrogen deuterium exchange mass spectrometry. FEBS J. 2013, 280, 5616–5625. [Google Scholar] [CrossRef] [PubMed]
- Bizien, T.; Durand, D.; Roblina, P.; Thureau, A.; Vachette, P.; Pérez, J. A Brief Survey of State-of-the-Art BioSAXS. Protein Pept. Lett. 2016, 23, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Selwa, E.; Davi, M.; Chenal, A.; Sotomayor-Pérez, A.; Ladant, D.; Malliavin, T. Allosteric activation of Bordetella pertussis adenylyl cyclase by calmodulin: Molecular dynamics and mutagenesis studies. J. Biol. Chem. 2014, 289, 21131–21141. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.; Goebel, E.; Hariraju, D.; Finley, N. Mutation in the β-hairpin of the Bordetella pertussis adenylate cyclase toxin modulates N-lobe conformation in calmodulin. Biochem. Biophys. Res. Commun. 2014, 453, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Laine, E.; Goncalves, C.; Karst, J.; Lesnard, A.; Rault, S.; Tang, W.; Malliavin, T.; Ladant, D.; Blondel, A. Use of allostery to identify inhibitors of calmodulin- induced activation of Bacillus anthracis Edema Factor. Proc. Natl. Acad. Sci. USA 2010, 107, 11277–11282. [Google Scholar] [CrossRef] [PubMed]
- Belyy, A.; Raoux-Barbot, D.; Saveanu, C.; Namane, A.; Ogryzko, V.; Worpenberg, L.; David, V.; Henriot, V.; Fellous, S.; Merrifield, C.; et al. Actin activates Pseudomonas aeruginosa ExoY nucleotidyl cyclase toxin and ExoY-like effector domains from MARTX toxins. Nat. Commun. 2016, 7, 135–182. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malliavin, T.E. Molecular Modeling of the Catalytic Domain of CyaA Deepened the Knowledge of Its Functional Dynamics. Toxins 2017, 9, 199. https://doi.org/10.3390/toxins9070199
Malliavin TE. Molecular Modeling of the Catalytic Domain of CyaA Deepened the Knowledge of Its Functional Dynamics. Toxins. 2017; 9(7):199. https://doi.org/10.3390/toxins9070199
Chicago/Turabian StyleMalliavin, Thérèse E. 2017. "Molecular Modeling of the Catalytic Domain of CyaA Deepened the Knowledge of Its Functional Dynamics" Toxins 9, no. 7: 199. https://doi.org/10.3390/toxins9070199
APA StyleMalliavin, T. E. (2017). Molecular Modeling of the Catalytic Domain of CyaA Deepened the Knowledge of Its Functional Dynamics. Toxins, 9(7), 199. https://doi.org/10.3390/toxins9070199