
Metabolism of the Marine Phycotoxin PTX-2 and Its Effects on Hepatic Xenobiotic Metabolism: Activation of Nuclear Receptors and Modulation of the Phase I Cytochrome P450

 ,
,
Abstract
:1. Introduction
2. Results
2.1. PTX-2 Metabolism in Rat and Human S9 Fractions
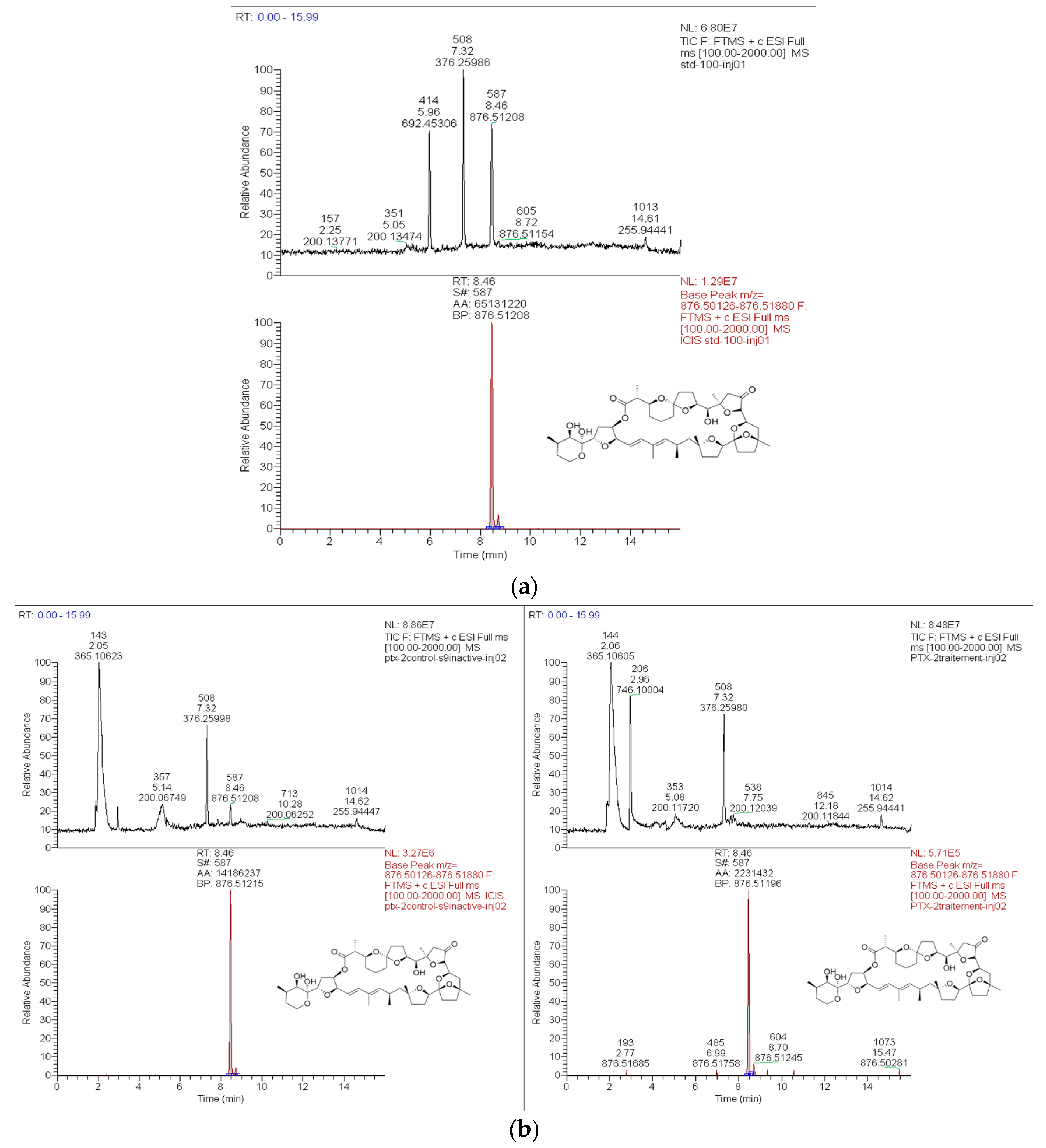
2.1.1. High Resolution Mass Spectrometry (HRMS) Method for PTX-2 Quantification and the Detection of Metabolites
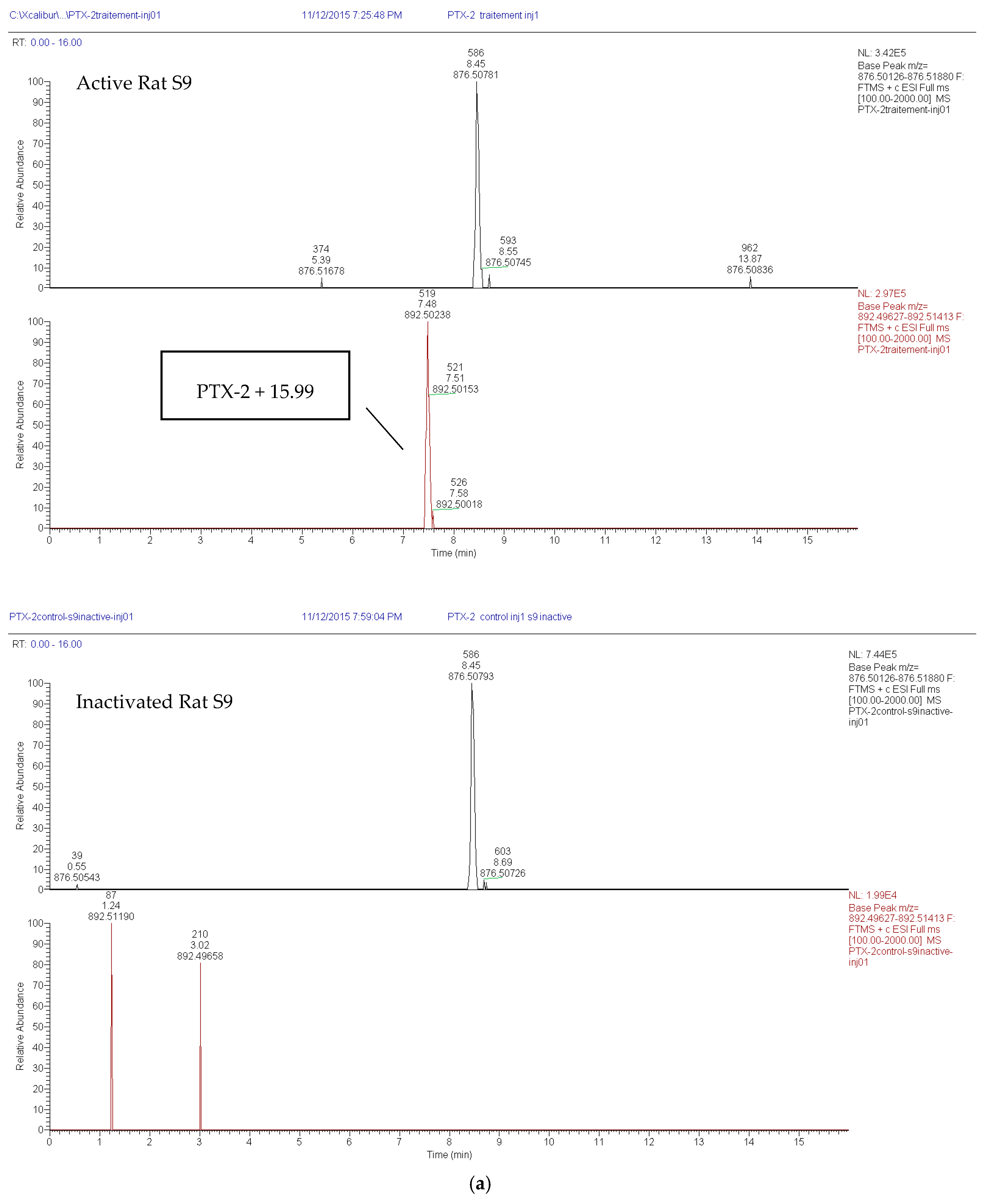
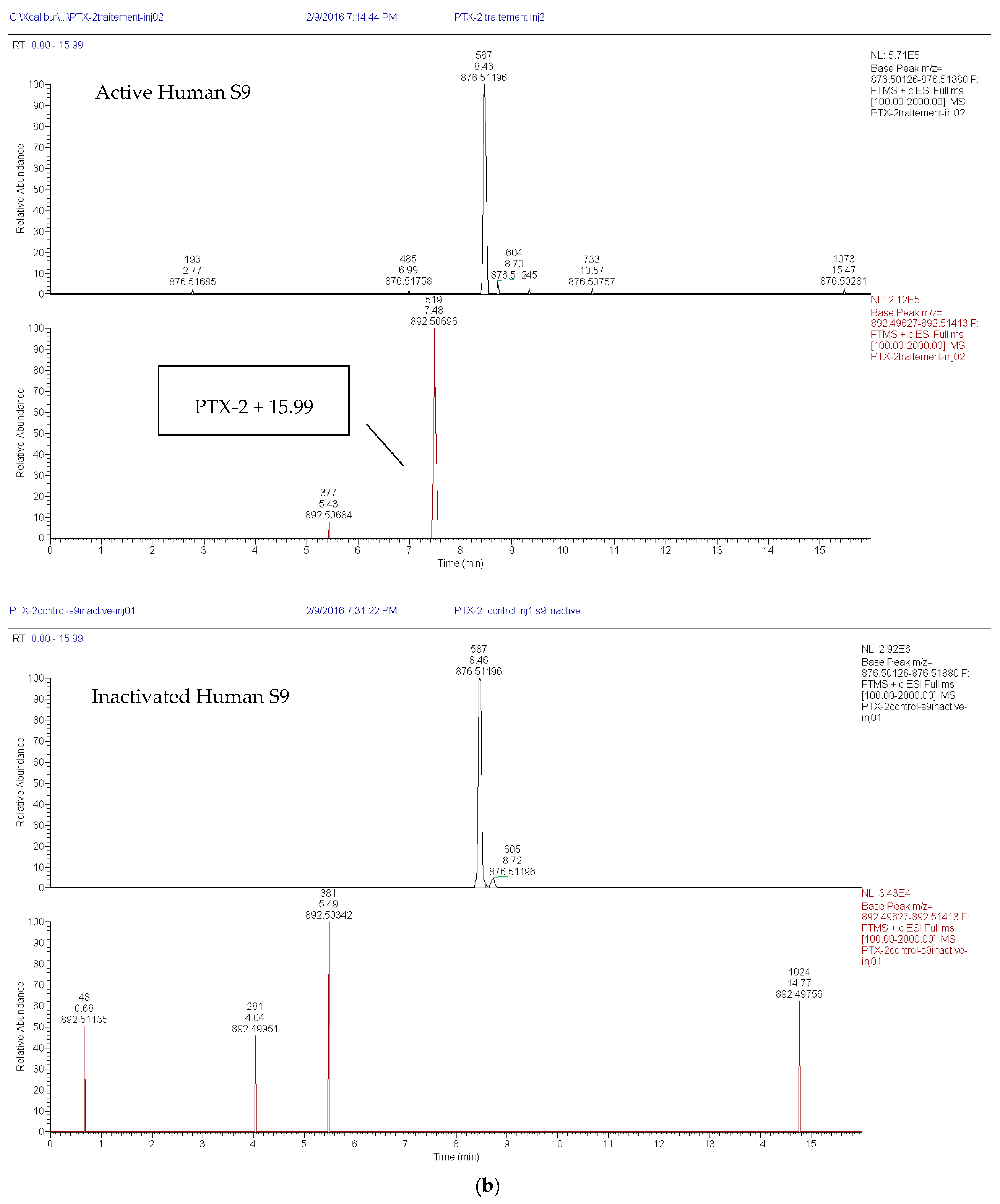
2.1.2. Loss of PTX-2 and Metabolite Formation in Active S9 Fractions
2.2. Effects of PTX-2 on the Expression of Phase 0, I, II, and III Metabolism Genes in HepaRG Cells by qRT-PCR
2.3. Induction of CYP1A2 Proteins in HepaRG Cells
2.4. CYP1A1 Reporter Gene Assay in HepG2 Cells
2.5. Induction of CYP1A Proteins in HepaRG Cells
2.6. Effects of PTX-2 on CYP1A Activities in HepaRG Cells
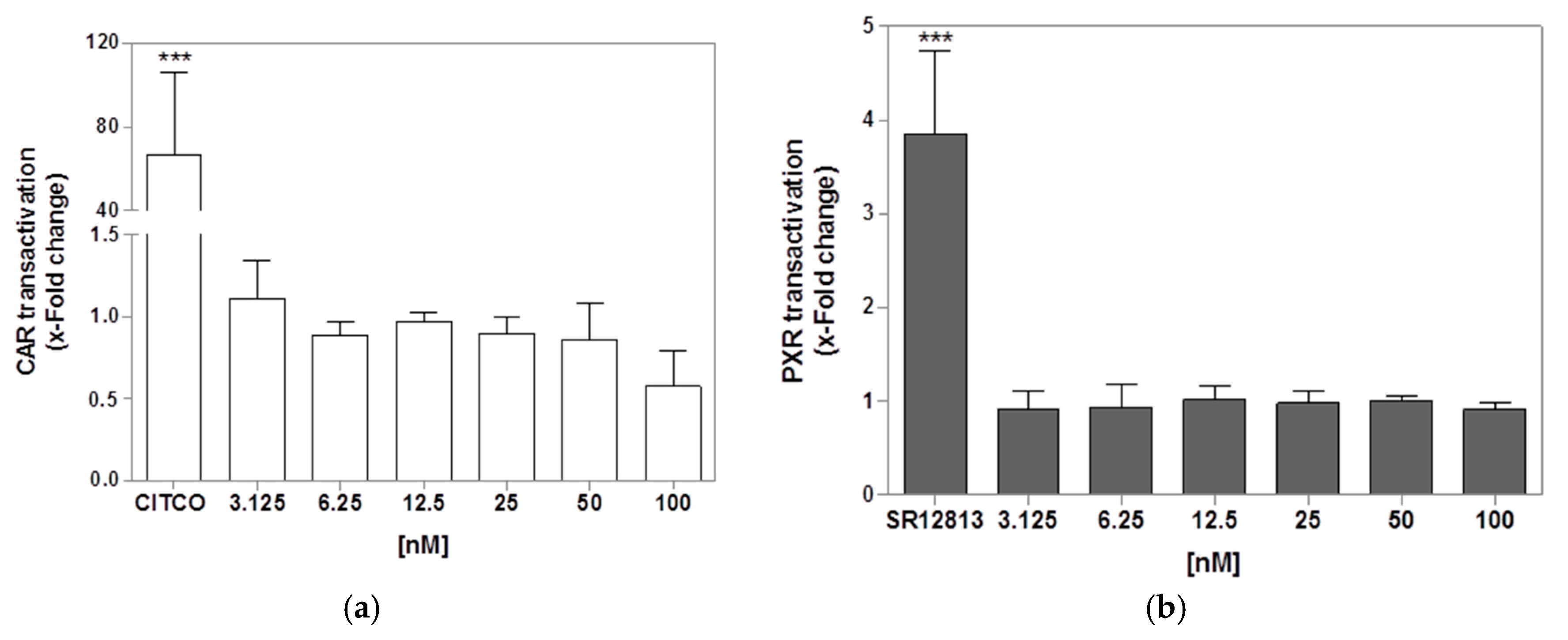
2.7. CAR and PXR Transactivation Assay in Transfected HepG2 and HEK-T Cells
2.8. AhR Reporter Gene Assay in HepG2 Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals
5.2. S9 Phase I Metabolism
5.3. LC–HRMS Analysis
5.4. Cell Culture
5.4.1. HepaRG Cells
5.4.2. HepG2 and HEK-T Cells
5.5. Cytotoxicity Assays
5.6. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) Analysis
5.7. CYP1A2 Protein Expression
5.8. Western Blot for CYP1A Expression
5.9. EROD Activity
5.10. CAR and PXR Transactivation Assays
5.11. AhR and CYP1A1 Reporter Gene Assays
5.12. Statistics/Data Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yasumoto, T.; Murata, M.; Oshima, Y.; Sano, M.; Matsumoto, G.K.; Clardy, J. Diarrhetic shellfish toxins. Tetrahedron 1985, 41, 1019–1025. [Google Scholar] [CrossRef]
- Yasumoto, T.; Murata, M.; Lee, J. Polyether toxins produced by dinoflagellates. In Mycotoxins andPhycotoxins ’88; Natori, S., Hashimoto, K., Ueno, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 1989; pp. 375–382. [Google Scholar]
- Draisci, R.; Lucentini, L.; Giannetti, L.; Boria, P.; Poletti, R. First report of pectenotoxin-2 (PTX-2) in algae (Dinophysis fortii) related to seafood poisoning in Europe. Toxicon 1996, 34, 923–935. [Google Scholar] [CrossRef]
- Miles, C.O.; Wilkins, A.L.; Munday, R.; Dines, M.H.; Hawkes, A.D.; Briggs, L.R.; Sandvik, M.; Jensen, D.J.; Cooney, J.M.; Holland, P.T.; et al. Isolation of pectenotoxin-2 from Dinophysis acuta and its conversion to pectenotoxin-2 seco acid, and preliminary assessment of their acute toxicities. Toxicon 2004, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ito, E.; Suzuki, T.; Oshima, Y.; Yasumoto, T. Studies of diarrhetic activity on pectenotoxin-6 in the mouse and rat. Toxicon 2008, 51, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Ishige, M.; Satoh, N.; Yasumoto, T. Pathological studies on mice administered with the causative agent of diarrhetic shellfish poisoning (okadaic acid and pectenotoxin-2). Hokkaidoritsu Eisei Kenkyushoho 1988, 38, 15–18. [Google Scholar]
- Terao, K.; Ito, E.; Yanagi, T.; Yasumoto, T. Histopathological studies on experimental marine toxin poisoning. I. Ultrastructural changes in the small intestine and liver of suckling mice induced by dinophysistoxin-1 and pectenotoxin-1. Toxicon 1986, 24, 1141–1151. [Google Scholar] [CrossRef]
- Espina, B.; Louzao, M.C.; Ares, I.R.; Cagide, E.; Vieytes, M.R.; Vega, F.V.; Rubiolo, J.A.; Miles, C.O.; Suzuki, T.; Yasumoto, T.; et al. Cytoskeletal toxicity of pectenotoxins in hepatic cells. Br. J. Pharmacol. 2008, 155, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.-O.; Kim, M.-O.; Kang, S.-H.; Lee, K.-J.; Heo, M.-S.; Choi, K.-S.; Choi, Y.-H.; Kim, G.-Y. Induction of G2/M arrest, endoreduplication, and apoptosis by actin depolymerization agent pextenotoxin-2 in human leukemia cells, involving activation of ERK and JNK. Biochem. Pharmacol. 2008, 76, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-O.; Moon, D.-O.; Heo, M.-S.; Lee, J.-D.; Jung, J.H.; Kim, S.-K.; Choi, Y.H.; Kim, G.-Y. Pectenotoxin-2 abolishes constitutively activated NF-κB, leading to suppression of NF-κB related gene products and potentiation of apoptosis. Cancer Lett. 2008, 271, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Ferron, P.J.; Hogeveen, K.; De Sousa, G.; Rahmani, R.; Dubreil, E.; Fessard, V.; Le Hegarat, L. Modulation of CYP3A4 activity alters the cytotoxicity of lipophilic phycotoxins in human hepatic HepaRG cells. Toxicol. In Vitro 2016, 33, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.Y.; Kim, Y.C. Acute toxicity of pectenotoxin-2 and its effects on hepatic metabolising enzyme system in mice. Korean J. Toxicol. 1997, 13, 183–186. [Google Scholar]
- Kittler, K.; Preiss-Weigert, A.; These, A. Identification Strategy Using Combined Mass Spectrometric Techniques for Elucidation of Phase I and Phase II in Vitro Metabolites of Lipophilic Marine Biotoxins. Anal. Chem. 2010, 82, 9329–9335. [Google Scholar] [CrossRef] [PubMed]
- Ramadoss, P.; Marcus, C.; Perdew, G.H. Role of the aryl hydrocarbon receptor in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2005, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Beischlag, T.V.; Morales, J.L.; Hollingshead, B.D.; Perdew, G.H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-M.; Ong, S.S.; Chai, S.C.; Chen, T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin. Drug Metab. Toxicol. 2012, 8, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Omiecinski, C.J.; Vanden Heuvel, J.P.; Perdew, G.H.; Peters, J.M. Xenobiotic Metabolism, Disposition, and Regulation by Receptors: From Biochemical Phenomenon to Predictors of Major Toxicities. Toxicol. Sci. 2011, 120 (Suppl. 1), S49–S75. [Google Scholar] [CrossRef] [PubMed]
- Nishibe, Y.; Hirata, M. Effect of phenobarbital and other model inducers on cytochrome P450 isoenzymes in primary culture of dog hepatocytes. Xenobiotica 1993, 23, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P.; Rasmussen, A.; Xu, L.; Kaminski, D.L. Rifampicin induction of lidocaine metabolism in cultured human hepatocytes. J. Pharmacol. Exp. Ther. 1995, 274, 673–677. [Google Scholar] [PubMed]
- Masubuchi, N.; Li, A.P.; Okazaki, O. An evaluation of the cytochrome P450 induction potential of pantoprazole in primary human hepatocytes. Chem. Biol. Interact. 1998, 114, 1–13. [Google Scholar] [CrossRef]
- Fux, E.; Rode, D.; Bire, R.; Hess, P. Approaches to the evaluation of matrix effects in the liquid chromatography-mass spectrometry (LC-MS) analysis of three regulated lipophilic toxin groups in mussel matrix ( Mytilus edulis). Food Addit. Contam. Part A 2008, 25, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Zendong, Z.; McCarron, P.; Herrenknecht, C.; Sibat, M.; Amzil, Z.; Cole, R.B.; Hess, P. High resolution mass spectrometry for quantitative analysis and untargeted screening of algal toxins in mussels and passive samplers. J. Chromatogr. A 2015, 1416, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Genies, C.; Maître, A.; Lefèbvre, E.; Jullien, A.; Chopard-Lallier, M.; Douki, T. The Extreme Variety of Genotoxic Response to Benzo[a]pyrene in Three Different Human Cell Lines from Three Different Organs. PLoS ONE 2013, 8, e78356. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; An, T.; Rein, K.S. The algal hepatoxoxin okadaic acid is a substrate for human cytochromes CYP3A4 and CYP3A5. Toxicon 2010, 55, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Le Hegarat, L.; Dumont, J.; Josse, R.; Huet, S.; Lanceleur, R.; Mourot, A.; Poul, J.M.; Guguen-Guillouzo, C.; Guillouzo, A.; Fessard, V. Assessment of the genotoxic potential of indirect chemical mutagens in HepaRG cells by the comet and the cytokinesis-block micronucleus assays. Mutagenesis 2010, 25, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, T.; Yoshitomi, S.; Asahi, S.; Matsumura, S.; Chatani, F.; Oda, H. In vitro micronucleus test in HepG2 transformants expressing a series of human cytochrome P450 isoforms with chemicals requiring metabolic activation. Mutat. Res. Toxicol. Environ. Mutagen. 2009, 677, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Berthou, F.; Goasduff, T.; Dreano, Y.; Ménez, J.-F. Caffeine increases its own metabolism through cytochrome P4501A induction in rats. Life Sci. 1998, 57, 541–549. [Google Scholar] [CrossRef]
- Aimova, D.; Svobodova, L.; Kotrbova, V.; Mrazova, B.; Hodek, P.; Hudecek, J.; Václavíková, R.; Frei, E.; Stiborová, M. The Anticancer Drug Ellipticine Is a Potent Inducer of Rat Cytochromes P450 1A1 and 1A2, Thereby Modulating Its Own Metabolism. Drug Metab. Dispos. 2007, 35, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Bazin, E.; Mourot, A.; Humpage, A.R.; Fessard, V. Genotoxicity of a freshwater cyanotoxin, cylindrospermopsin, in two human cell lines: Caco-2 and HepaRG. Environ. Mol. Mutagen. 2010, 51, 251–259. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information (NCBI). GenBank Sequence Database. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on 11 January 2016).
- National Center for Biotechnology Information (NCBI). Primer Designing Tool. Available online: http://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome (accessed on 15 January 2016).
- Luckert, C.; Hessel, S.; Lampen, A.; Braeuning, A. Utility of an appropriate reporter assay: Heliotrine interferes with GAL4/upstream activation sequence-driven reporter gene systems. Anal. Biochem. 2015, 487, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Hampf, M.; Gossen, M. A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal. Biochem. 2006, 356, 94–99. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolism Phases | Gene | (nM) | OME | RIF | Gene | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 16 | 32 | 64 | ||||||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |||
| Nuclear receptors | AHR | 1.0 | 0.4 | 1.2 | 0.6 | 1.5 | 0.7 | 0.8 | 0.3 | 0.9 | 0.3 | AHR |
| NR1I2 | 1.3 | 0.2 | 1.5 | 0.6 | 1.4 | 0.3 | 1.0 | / | 0.7 | 0.4 | NR1I2 | |
| Phase 0 influx transporters | SLC22A1 | 1.4 | 0.6 | 1.4 | 0.2 | 1.8 | 1.1 | 1.3 | 1.2 | 1.1 | 0.3 | SLC22A1 |
| SLC22A3 | 1.6 | 0.3 | 1.8 | 0.4 | 2.4 ** | 0.3 | 1.0 | 0.4 | 1.1 | 0.5 | SLC22A3 | |
| SLCO1A2 | 0.9 | 0.6 | 1.2 | 1.1 | 1.1 | 0.6 | 0.4 | 0.3 | 0.2 | 0.2 | SLCO1A2 | |
| SLCO1B1 | 1.0 | 0.2 | 1.2 | 0.6 | 1.0 | 0.3 | 0.6 | 0.1 | 1.0 | 0.3 | SLCO1B1 | |
| Phase I mono-oxygenases | CYP1A1 | 4.2 | 2.6 | 10.1 | 9.1 | 18.7 | 21.8 | 127.0 *** | 67.1 | 0.4 | 0.4 | CYP1A1 |
| CYP1A2 | 4.4 | 0.6 | 8.3 | 2.9 | 8.8 | 4.8 | 245.2 ** | 160.1 | 1.4 | 0.2 | CYP1A2 | |
| CYP2B6 | 1.6 | 0.5 | 3.1 | 2.1 | 3.5 | 2.6 | 8.0 | 8.0 | 4.3 | 2.5 | CYP2B6 | |
| CYP2C9 | 1.2 | 0.1 | 1.7 | 0.5 | 1.9 | 0.6 | 1.4 | 0.3 | 2.2 ** | 0.3 | CYP2C9 | |
| CYP2C19 | 1.3 | 0.3 | 1.4 | 0.2 | 1.8 * | 0.3 | 1.2 | 0.2 | 1.6 | 0.4 | CYP2C19 | |
| CYP3A4 | 1.2 | 0.2 | 1.2 | 0.2 | 1.3 | 0.2 | 13.9 ** | 6.7 | 29.2 *** | 3.6 | CYP3A4 | |
| CYP3A5 | 1.0 | 0.1 | 1.1 | 0.3 | 1.3 | 0.3 | 1.1 | 0.2 | 1.5* | 0.2 | CYP3A5 | |
| Phase II transferases | GSTM1 | 0.8 | 0.3 | 1.0 | 0.1 | 0.8 | 0.1 | 1.2 | 0.3 | 1.0 | 0.1 | GSTM1 |
| NAT1 | 1.2 | 0.5 | 1.2 | 0.6 | 1.3 | 0.5 | 1.0 | 0.2 | 1.0 | 0.2 | NAT1 | |
| NAT2 | 1.1 | 0.0 | 1.2 | 0.1 | 1.3 | 0.3 | 0.8 | 0.2 | 0.9 | 0.3 | NAT2 | |
| SULT1A1 | 1.2 | 0.1 | 1.2 | 0.2 | 1.2 | 0.2 | 0.9 | 0.1 | 1.0 | 0.1 | SULT1A1 | |
| SULT1E1 | 1.7 | 1.2 | 2.4 | 2.0 | 3.5 | 3.4 | 0.4 | 0.2 | 0.7 | 0.4 | SULT1E1 | |
| UGT1A1 | 1.2 | 0.4 | 1.8 | 0.8 | 2.3 | 0.8 | 2.8 | 1.5 | 1.7 | 0.2 | UGT1A1 | |
| UGT1A9 | 1.3 | 0.3 | 2.1* | 0.7 | 1.8 | 0.3 | 1.2 | 0.1 | 1.1 | 0.3 | UGT1A9 | |
| UGT2B4 | 1.6 | 0.6 | 2.1 | 0.7 | 1.7 | 0.5 | 1.3 | 0.2 | 1.4 | 0.5 | UGT2B4 | |
| Phase III efflux transporters | ABCB1 | 1.4 | 0.2 | 1.9 | 0.6 | 2.1 * | 0.6 | 1.4 | 0.5 | 1.7 | 0.4 | ABCB1 |
| ABCC2 | 1.2 | 0.3 | 1.4 | 0.4 | 1.5 | 0.5 | 1.2 | 0.5 | 1.2 | 0.2 | ABCC2 | |
| ABCC3 | 1.0 | 0.1 | 1.3 | 0.3 | 1.4 | 0.3 | 1.0 | 0.3 | 0.9 | 0.0 | ABCC3 | |
| ABCG2 | 1.2 | 0.3 | 1.5 | 0.3 | 2.0 | 0.9 | 2.6 | 1.4 | 1.1 | 0.1 | ABCG2 | |
| 0.4 | 0.9 | 2.5 | 8 | 250 | ||||||||
| x-Fold change compared to solvent control | ||||||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alarcan, J.; Dubreil, E.; Huguet, A.; Hurtaud-Pessel, D.; Hessel-Pras, S.; Lampen, A.; Fessard, V.; Le Hegarat, L. Metabolism of the Marine Phycotoxin PTX-2 and Its Effects on Hepatic Xenobiotic Metabolism: Activation of Nuclear Receptors and Modulation of the Phase I Cytochrome P450. Toxins 2017, 9, 212. https://doi.org/10.3390/toxins9070212
Alarcan J, Dubreil E, Huguet A, Hurtaud-Pessel D, Hessel-Pras S, Lampen A, Fessard V, Le Hegarat L. Metabolism of the Marine Phycotoxin PTX-2 and Its Effects on Hepatic Xenobiotic Metabolism: Activation of Nuclear Receptors and Modulation of the Phase I Cytochrome P450. Toxins. 2017; 9(7):212. https://doi.org/10.3390/toxins9070212
Chicago/Turabian StyleAlarcan, Jimmy, Estelle Dubreil, Antoine Huguet, Dominique Hurtaud-Pessel, Stefanie Hessel-Pras, Alfonso Lampen, Valérie Fessard, and Ludovic Le Hegarat. 2017. "Metabolism of the Marine Phycotoxin PTX-2 and Its Effects on Hepatic Xenobiotic Metabolism: Activation of Nuclear Receptors and Modulation of the Phase I Cytochrome P450" Toxins 9, no. 7: 212. https://doi.org/10.3390/toxins9070212
APA StyleAlarcan, J., Dubreil, E., Huguet, A., Hurtaud-Pessel, D., Hessel-Pras, S., Lampen, A., Fessard, V., & Le Hegarat, L. (2017). Metabolism of the Marine Phycotoxin PTX-2 and Its Effects on Hepatic Xenobiotic Metabolism: Activation of Nuclear Receptors and Modulation of the Phase I Cytochrome P450. Toxins, 9(7), 212. https://doi.org/10.3390/toxins9070212