Size Distribution Analysis with On-Chip Multi-Imaging Cell Sorter for Unlabeled Identification of Circulating Tumor Cells in Blood


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
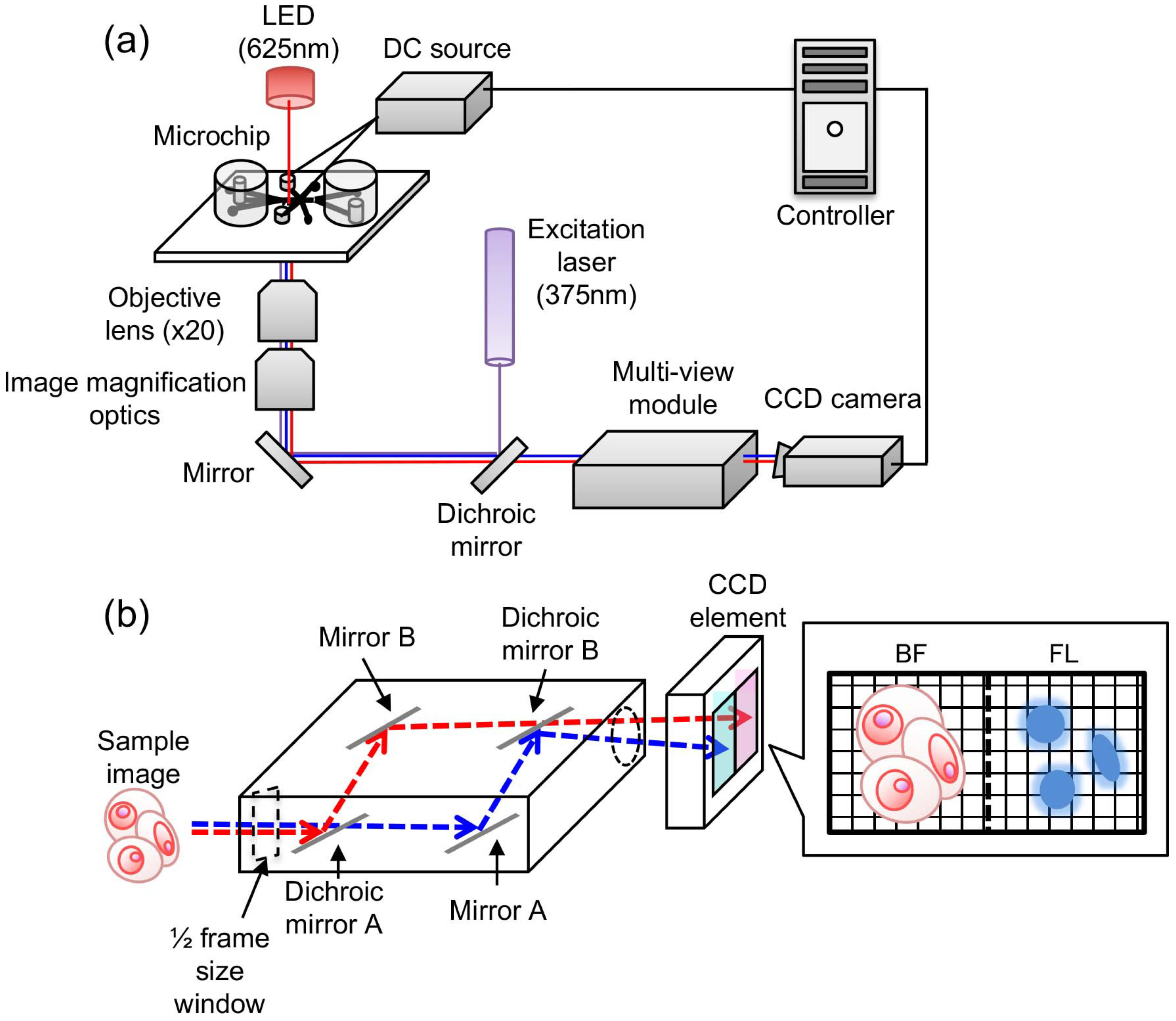
2.1. On-Chip Multi-Imaging Flow Cytometry System with Agarose Gel Electrodes
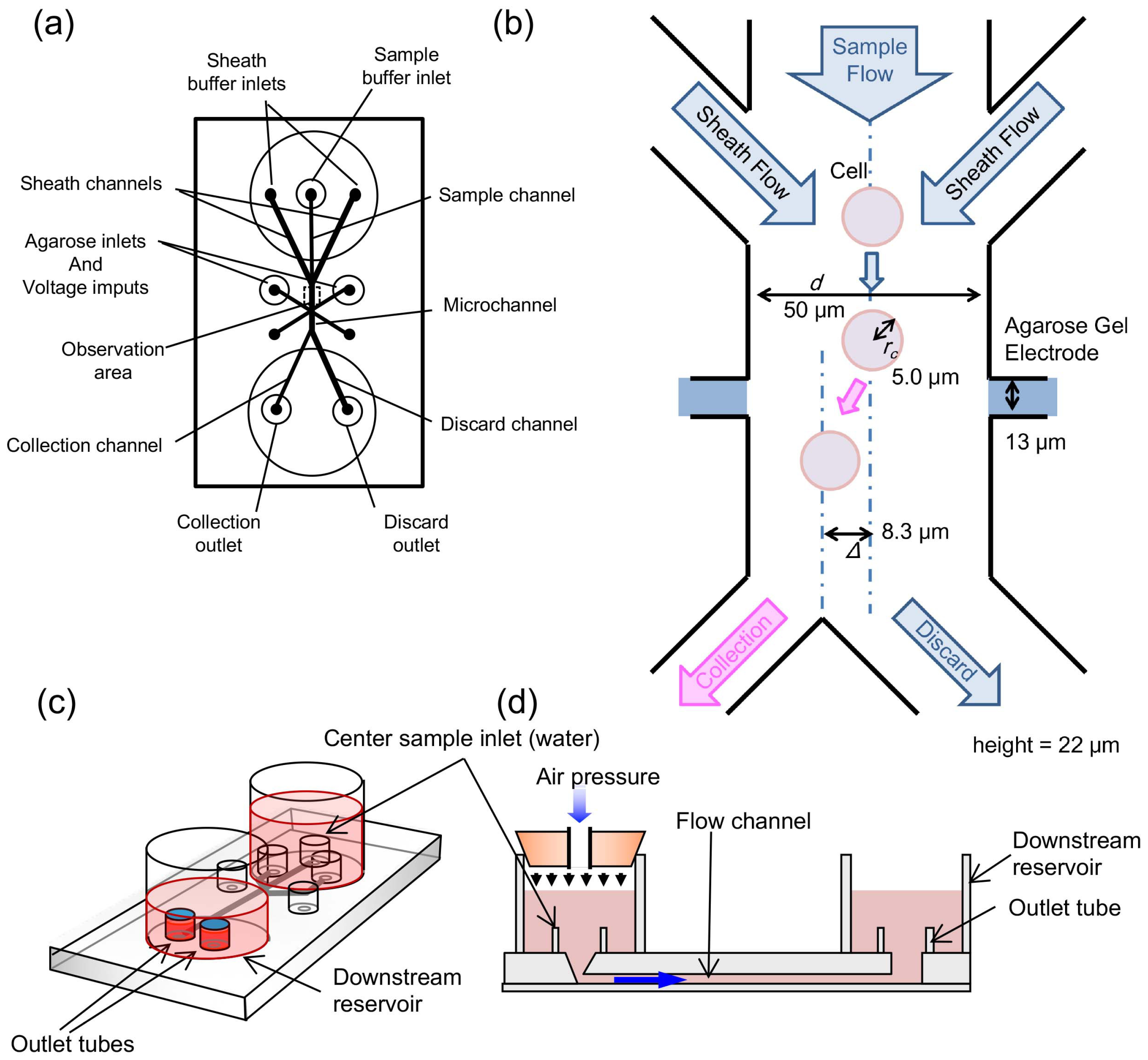
2.2. Microfluidic Chip
2.3. Agarose Gel Electrodes for Cell Sorting
2.4. Imaging Biomarker Detection
2.5. Preparation of Sample Blood
2.6. Procedure of Imaging Flow Cytometry
3. Results and Discussion
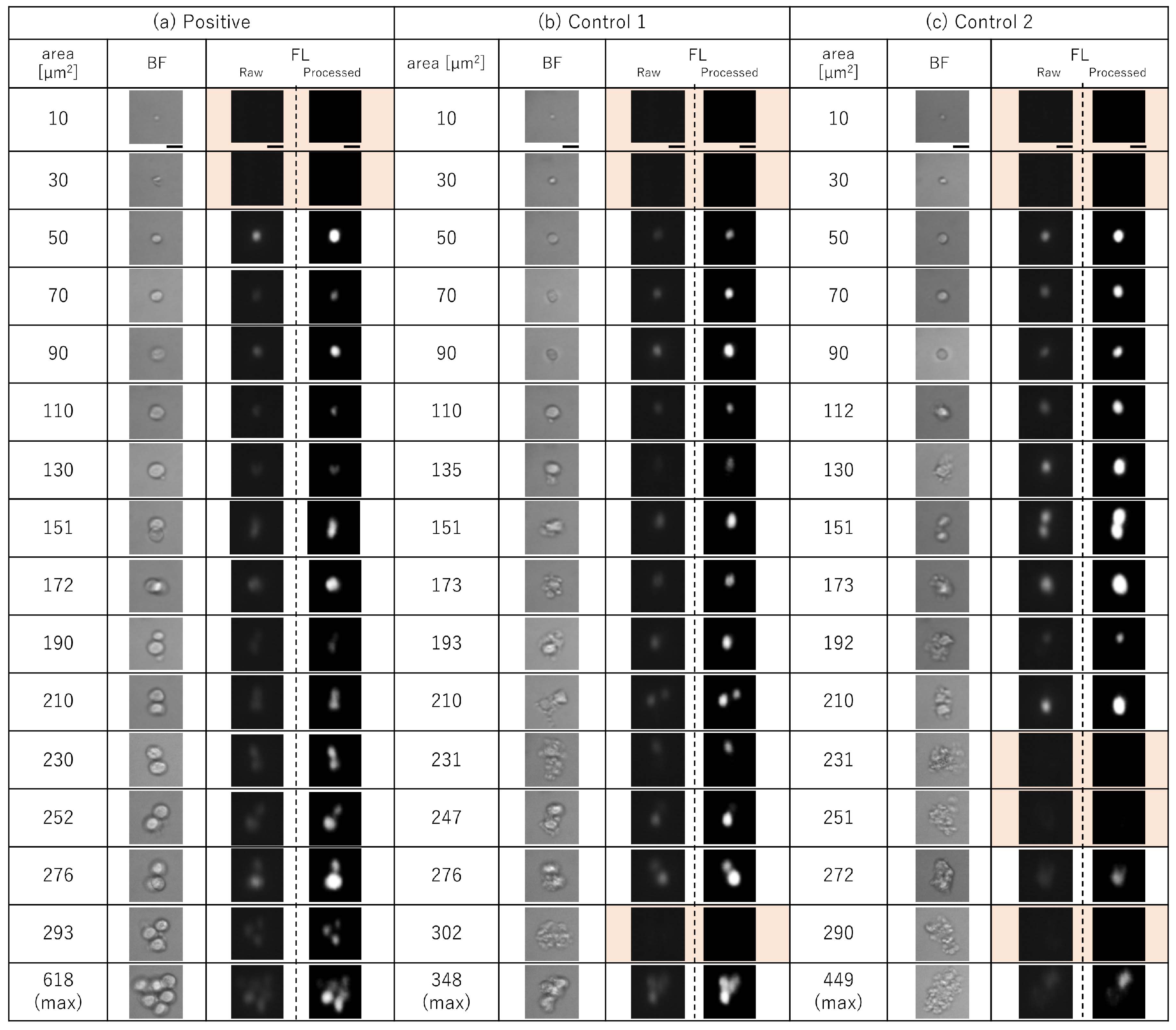
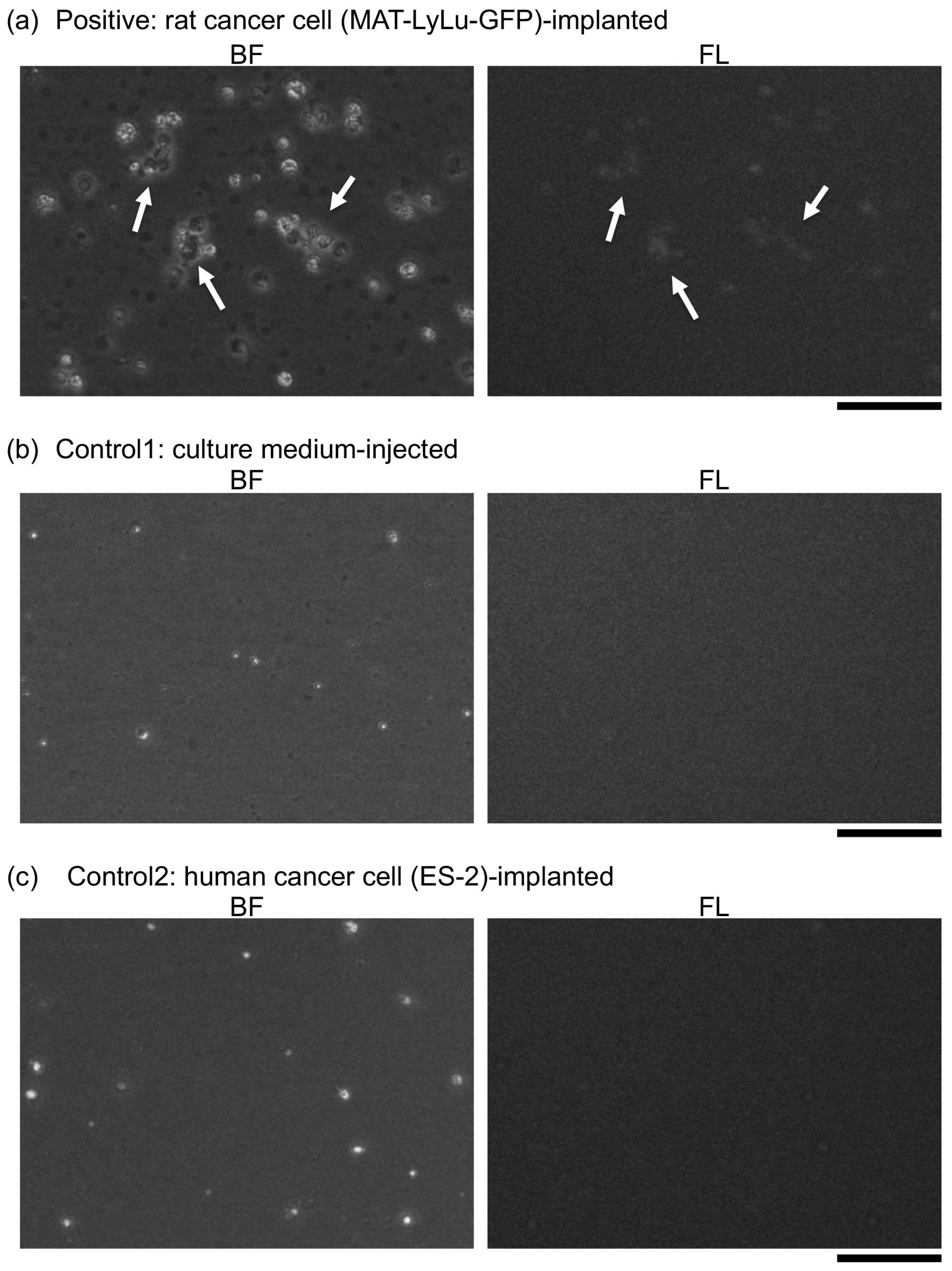
3.1. Detection of Time-Course Change of Imaging Biomarkers of Cancer-Implanted Rat Blood
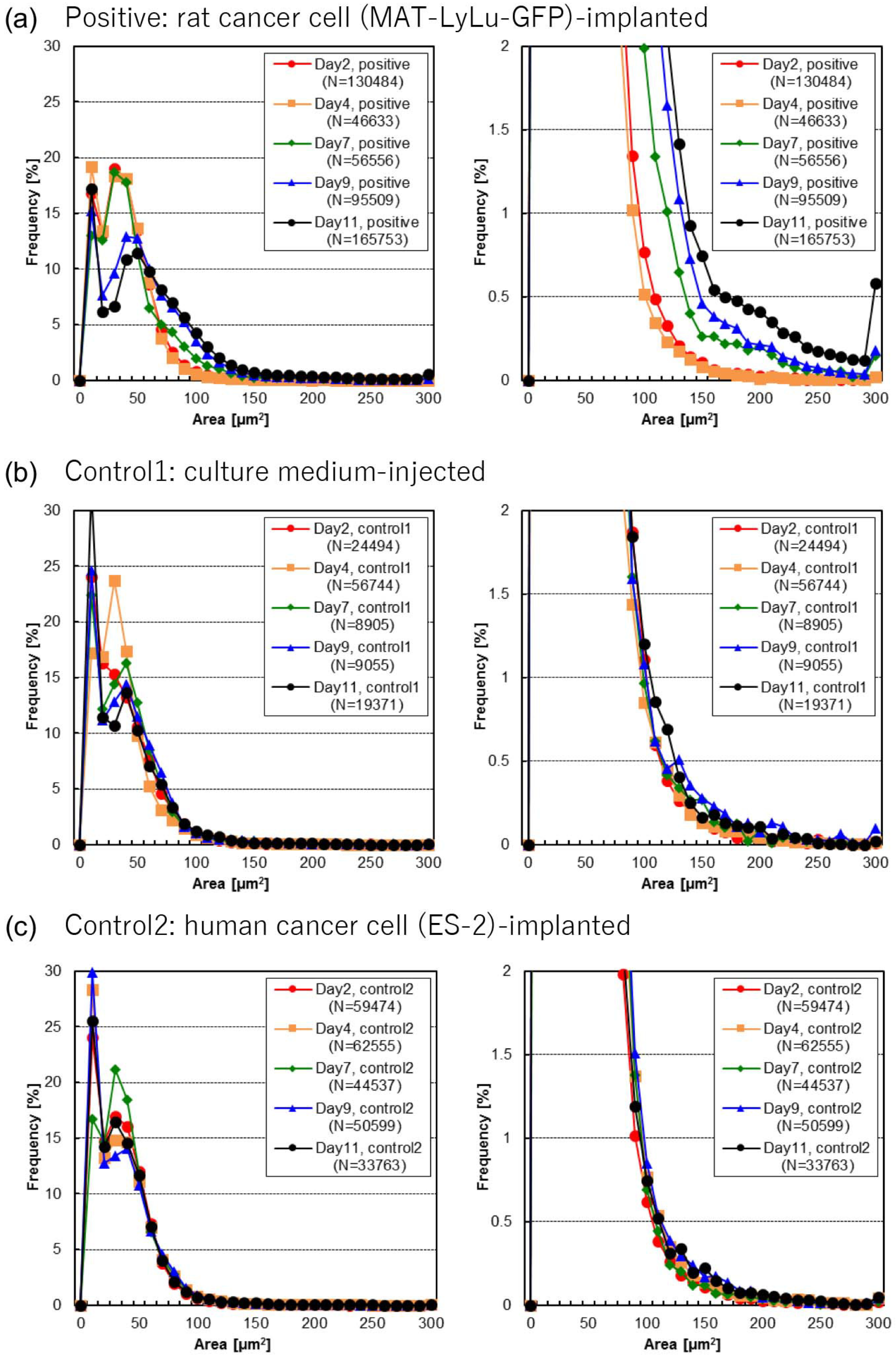
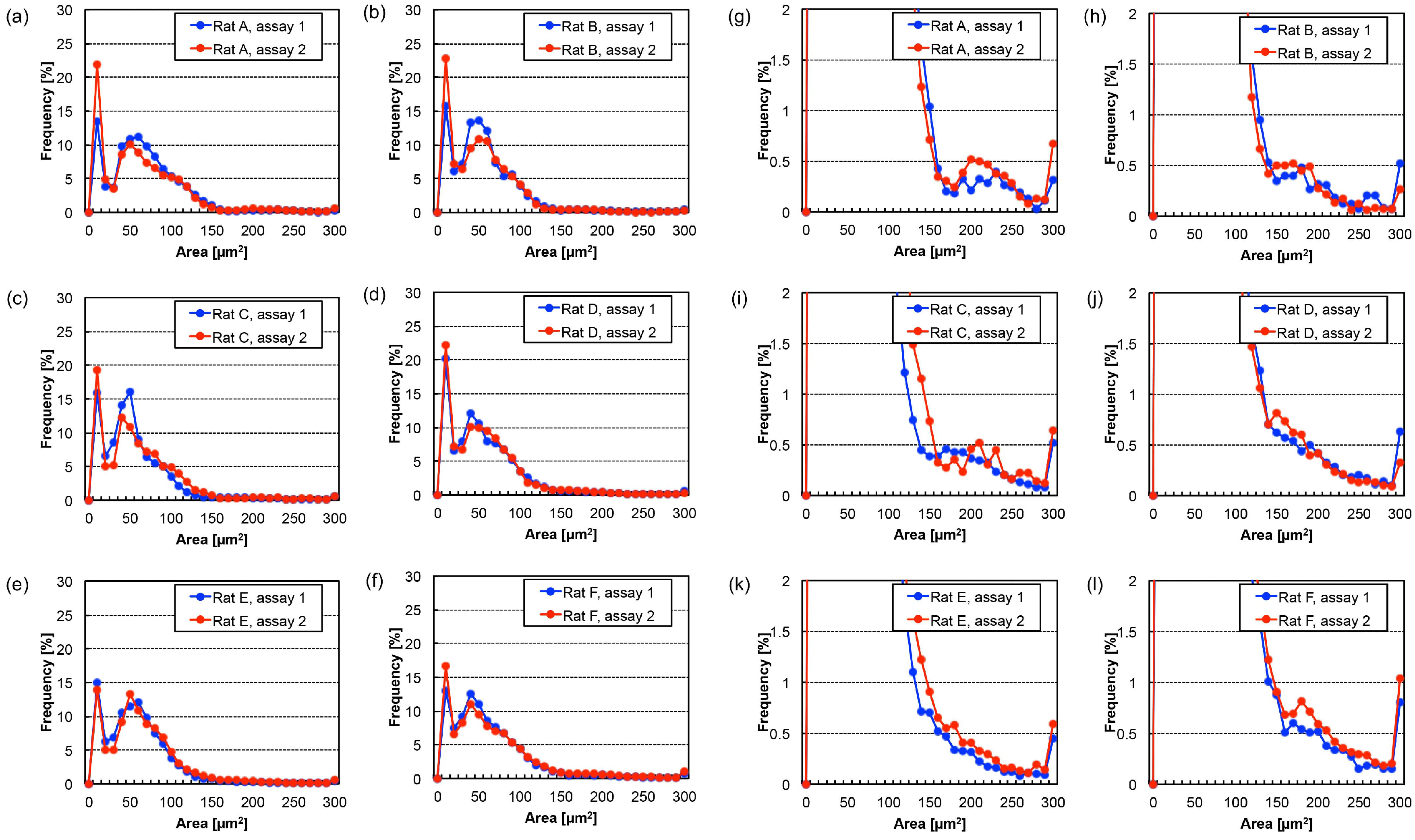
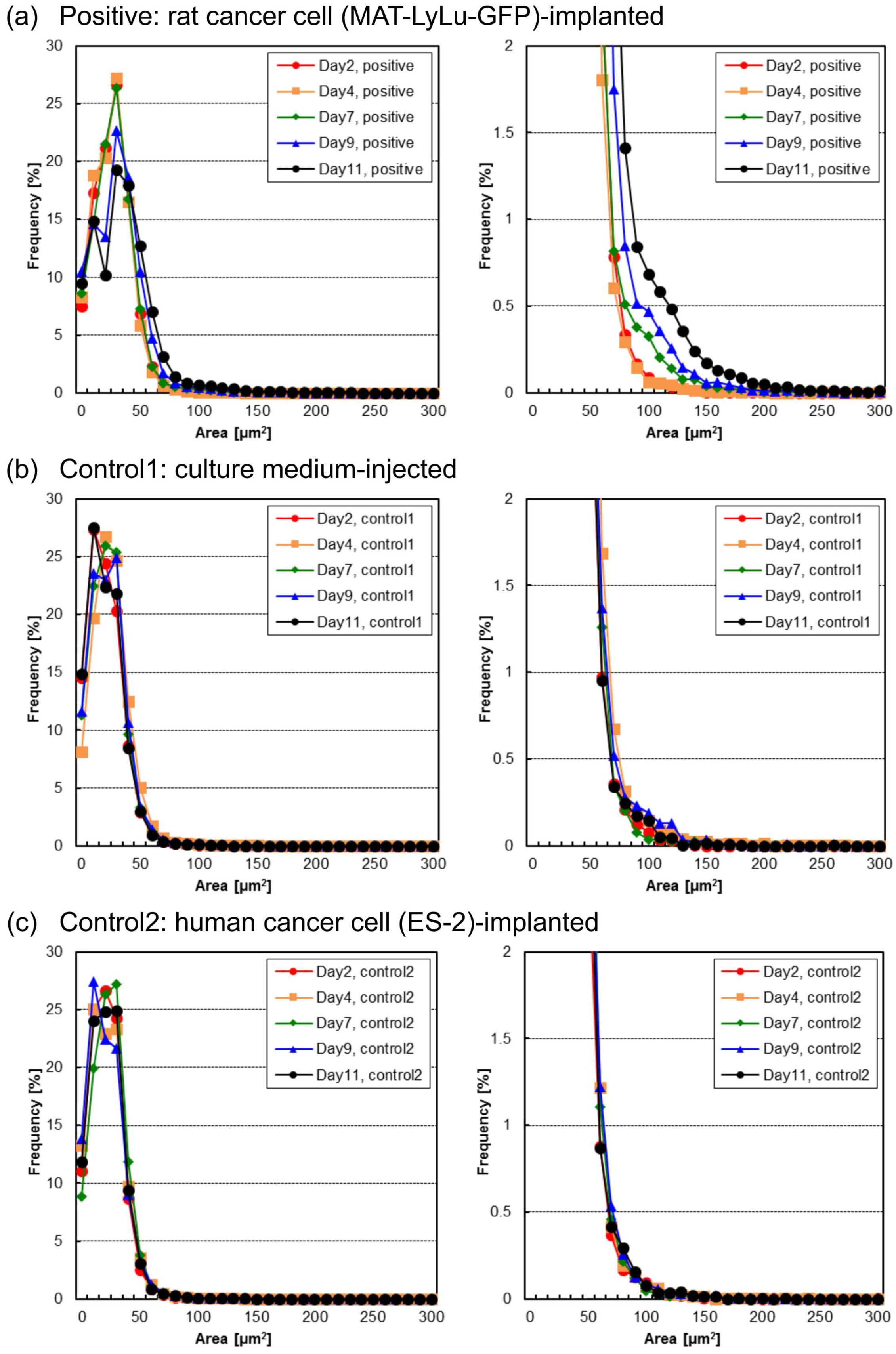
3.2. Time-Course Change of Cell Area
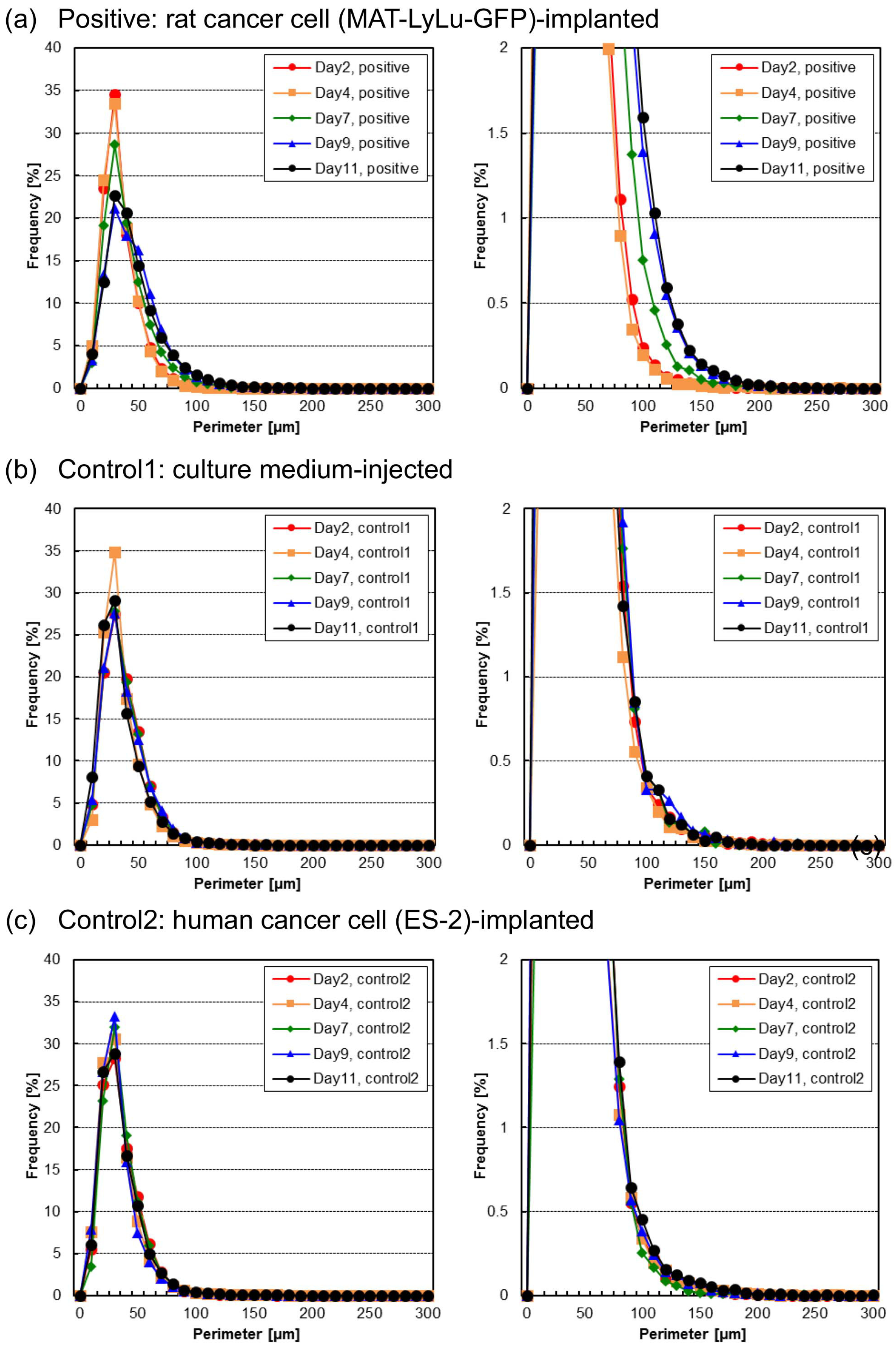
3.3. Time-Course Change of Cell and Cell Cluster Perimeter Lengths
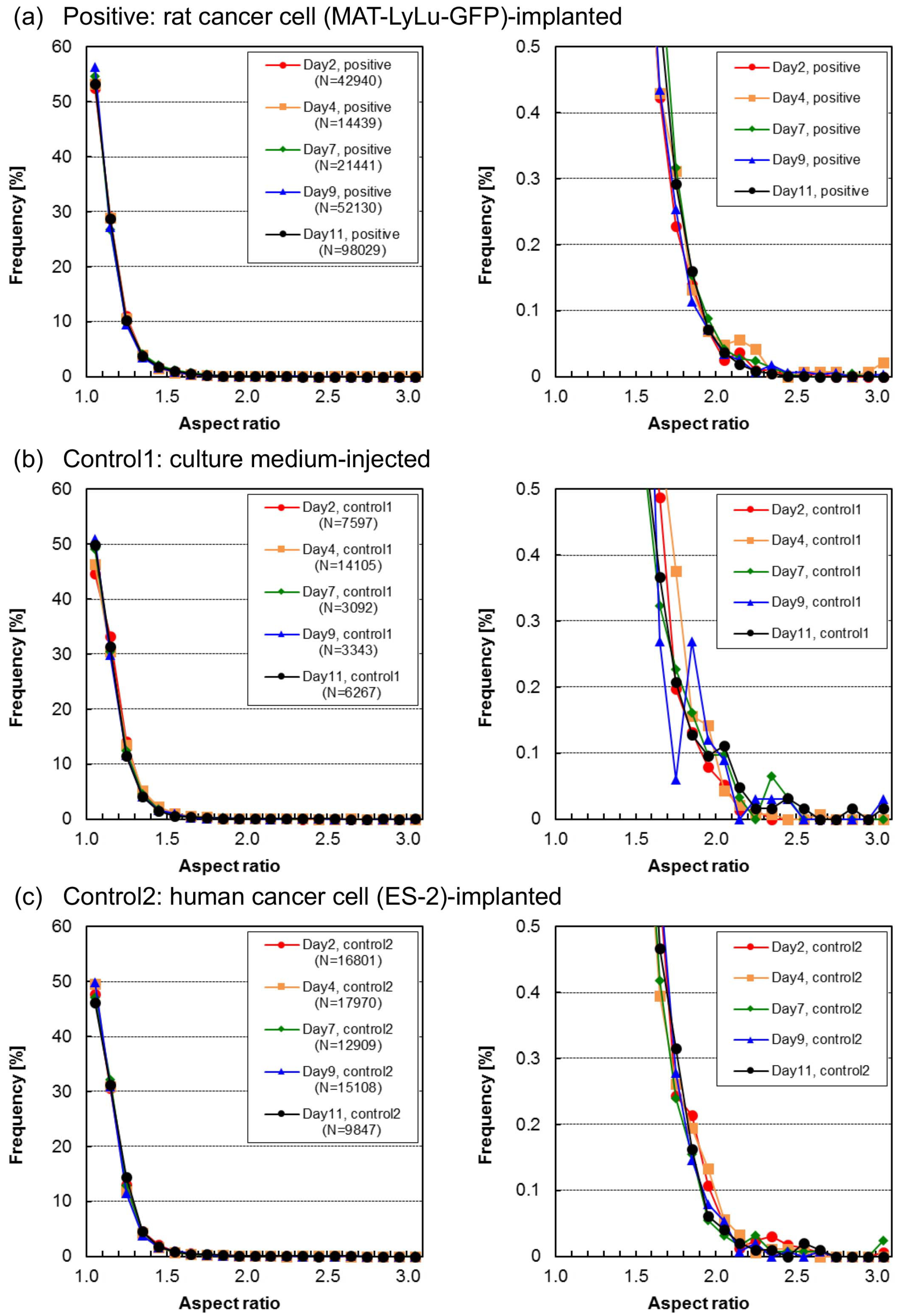
3.4. Time-Course Change of Aspect Ratio in Cells and Cell Clusters
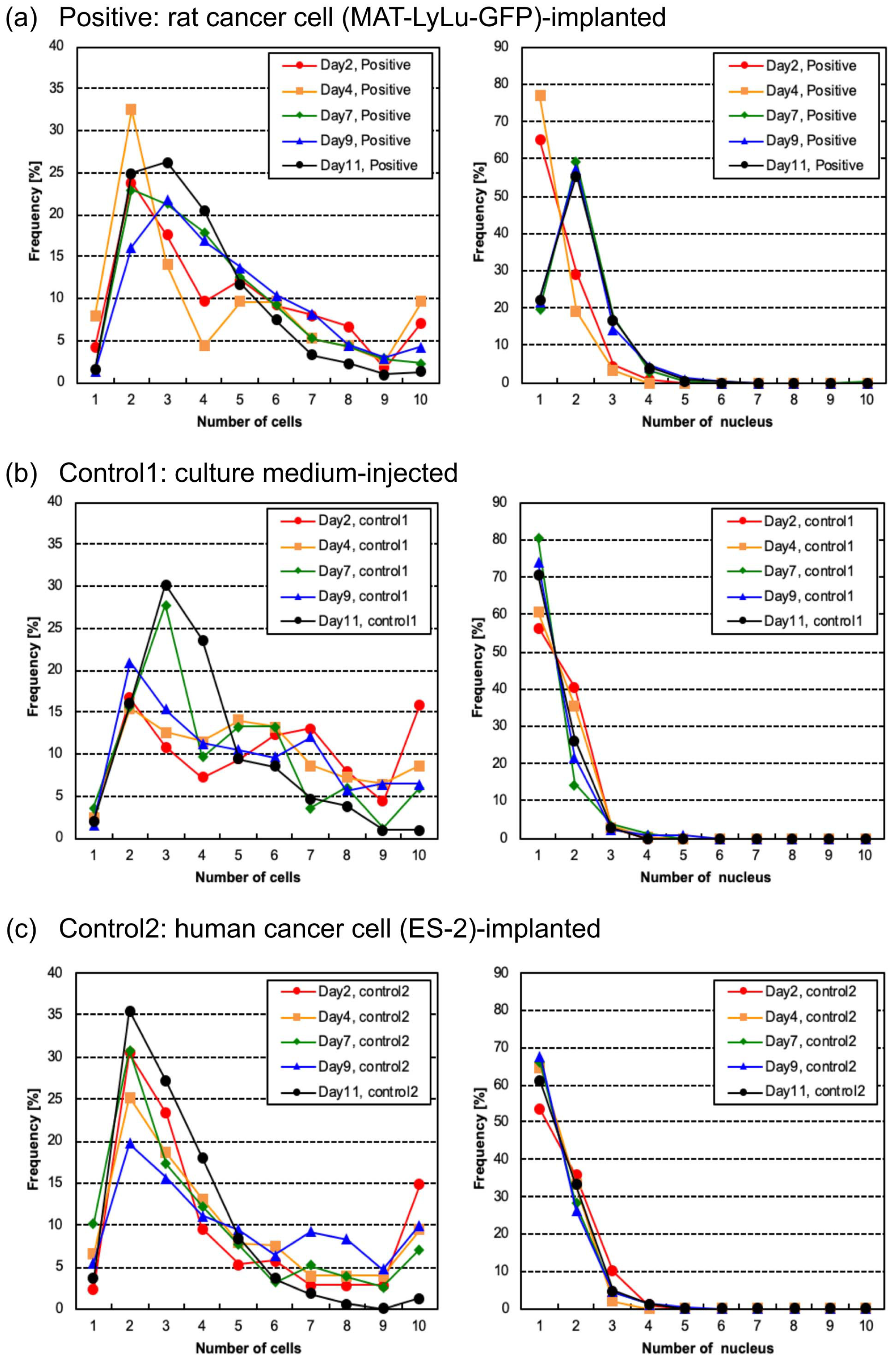
3.5. Time-Course Changes of Cell Number, Nucleus Number, and Nucleus Area
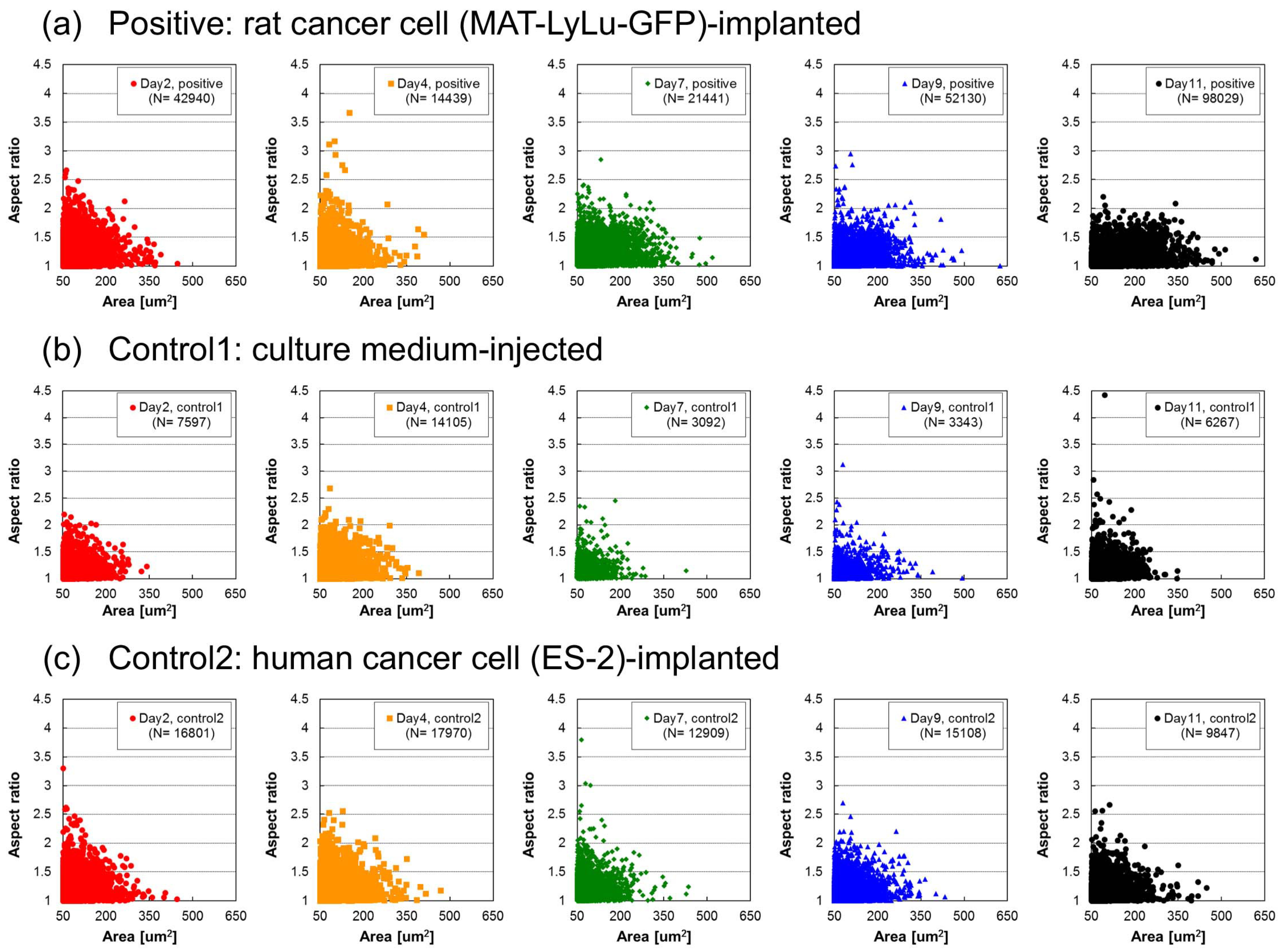
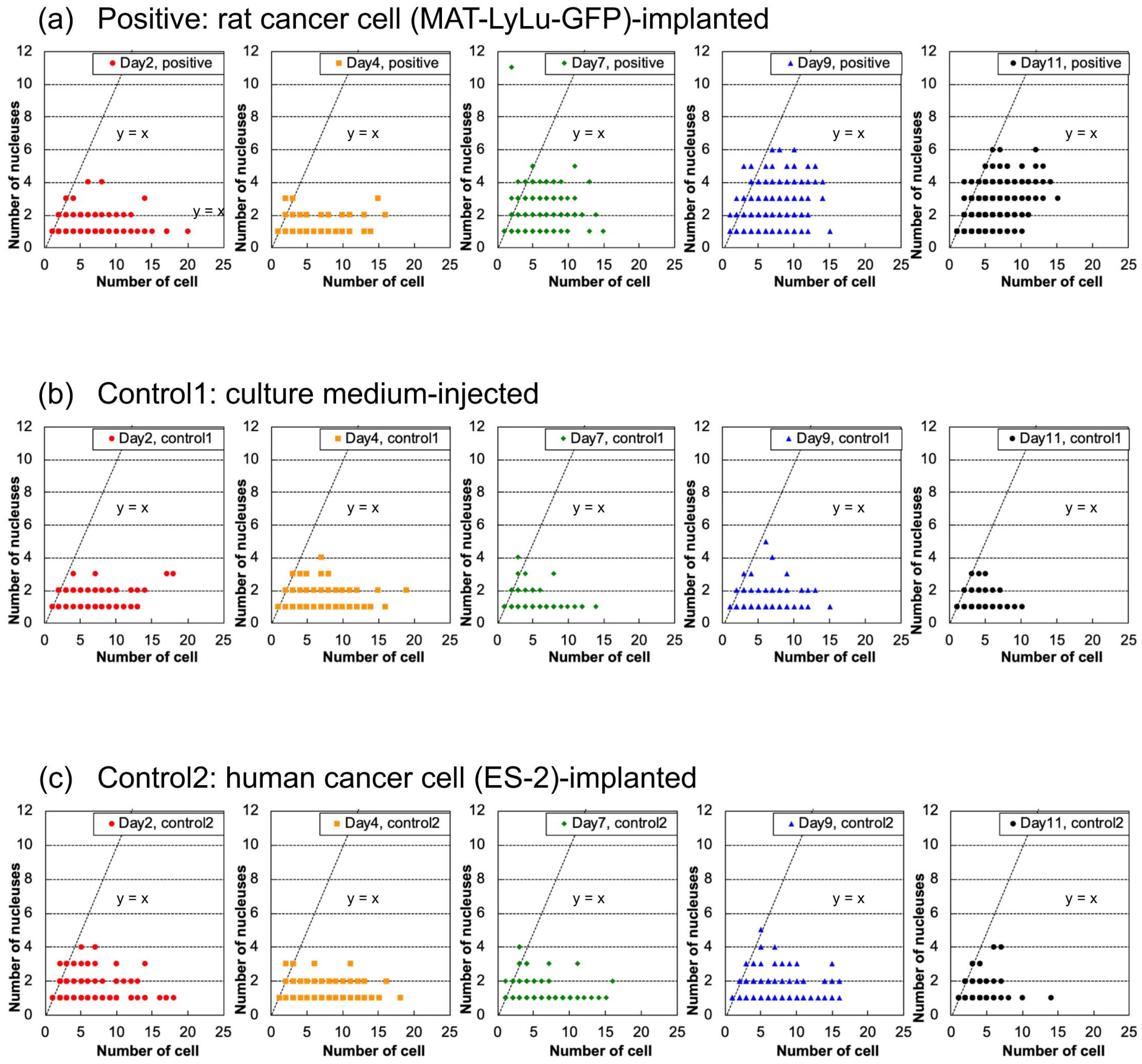
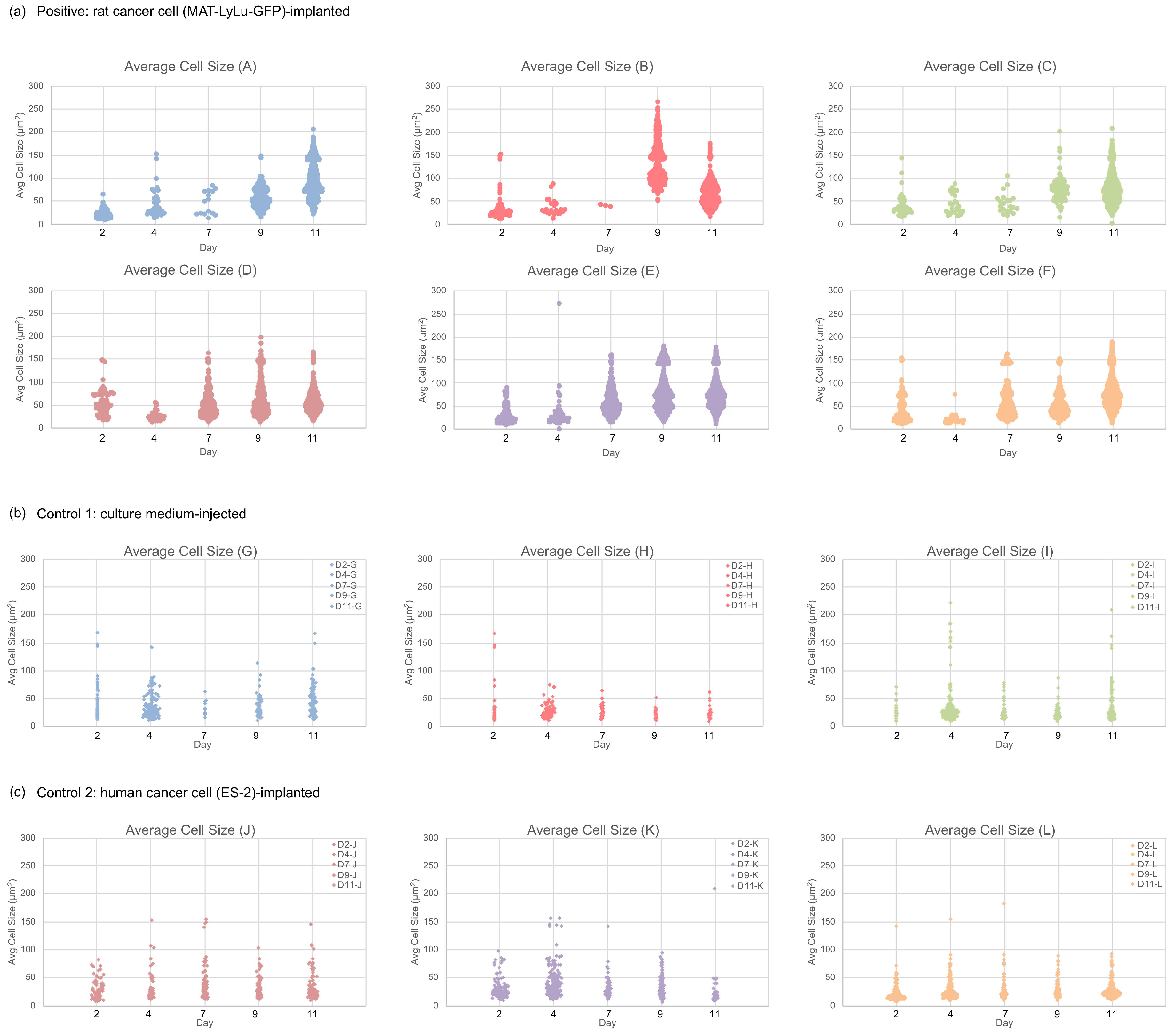
3.6. Correlation of Cell Size Distribution with Cell Number in Clusters
3.7. Ability and Limitation of On-Chip Imaging Cell Sorter for CTC Screening
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- Sethi, N.; Kang, Y. Unravelling the complexity of metastasis—Molecular understanding and targeted therapies. Nat. Rev. Cancer 2011, 11, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Stott, S.; Toner, M.; Maheswaran, S.; Haber, D.A. Circulating tumor cells: Approaches to isolation and characterization. J. Cell Biol. 2011, 192, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.A.; Inglis, D.W.; Morton, K.J.; Lawrence, D.A.; Huang, L.R.; Chou, S.Y.; Sturm, J.C.; Austin, R.H. Deterministic hydrodynamics: Taking blood apart. Proc. Natl. Acad. Sci. USA 2006, 103, 14779–14784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascoyne, P.R.; Noshari, J.; Anderson, T.J.; Becker, F.F. Isolation of rare cells from cell mixtures by dielectrophoresis. Electrophoresis 2009, 30, 1388–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stott, S.L.; Hsu, C.H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Lin, H.K.; Lu, B.; Williams, A.; Datar, R.; Cote, R.J.; Tai, Y.C. 3D microfilter device for viable circulating tumor cell (CTC) enrichment from blood. Biomed. Microdevices 2011, 13, 203–213. [Google Scholar] [CrossRef]
- Budd, G.T.; Cristofanilli, M.; Ellis, M.J.; Stopeck, A.; Borden, E.; Miller, M.C.; Matera, J.; Repollet, M.; Doyle, G.V.; Terstappen, L.W.; et al. Circulating tumor cells versus imaging–predicting overall survival in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 6403–6409. [Google Scholar] [CrossRef]
- Danila, D.C.; Heller, G.; Gignac, G.A.; Gonzalez-Espinoza, R.; Anand, A.; Tanaka, E.; Lilja, H.; Schwartz, L.; Larson, S.; Fleisher, M.; et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin. Cancer Res. 2007, 13, 7053–7058. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Diepenbruck, M.; Christofori, G. Epithelial–Mesenchymal Transition (EMT) and Metastasis: Yes, No, Maybe? CTC Metastasis. Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Mong, J.; Tan, M.H. Size-Based Enrichment Technologies for Non-cancerous Tumor-Derived Cells in Blood. Trends Biotechnol. 2018, 36, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.J.; Wan, Y.; Xia, Y.Q.; Zou, X.; Zheng, S.Y. Size-based separation methods of circulating tumor cells. Adv. Drug Deliv. Rev. 2018, 125, 3–20. [Google Scholar] [CrossRef]
- Che, J.; Yu, V.; Garon, E.B.; Goldman, J.W.; Di Carlo, D. Biophysical isolation and identification of circulating tumor cells. Lab Chip 2017, 17, 1452–1461. [Google Scholar] [CrossRef] [Green Version]
- Jan, Y.J.; Chen, J.F.; Zhu, Y.; Lu, Y.T.; Chen, S.H.; Chung, H.; Smalley, M.; Huang, Y.W.; Dong, J.; Chen, L.C.; et al. NanoVelcro rare-cell assays for detection and characterization of circulating tumor cells. Adv. Drug Deliv. Rev. 2018, 125, 78–93. [Google Scholar] [CrossRef]
- Huang, X.; Tang, J.; Hu, L.; Bian, R.; Liu, M.; Cao, W.; Zhang, H. Arrayed microfluidic chip for detection of circulating tumor cells and evaluation of drug potency. Anal. Biochem. 2019, 564–565, 64–71. [Google Scholar] [CrossRef]
- Takahashi, K.; Hattori, A.; Suzuki, I.; Ichiki, T.; Yasuda, K. Non-destructive on-chip cell sorting system with real-time microscopic image processing. J. Nanobiotechnol. 2004, 2, 5. [Google Scholar] [CrossRef]
- Hayashi, M.; Hattori, A.; Kim, H.; Terazono, H.; Kaneko, T.; Yasuda, K. Fully automated on-chip imaging flow cytometry system with disposable contamination-free plastic re-cultivation chip. Int. J. Mol. Sci. 2011, 12, 3618–3634. [Google Scholar] [CrossRef]
- Kim, H.; Terazono, H.; Nakamura, Y.; Sakai, K.; Hattori, A.; Odaka, M.; Girault, M.; Arao, T.; Nishio, K.; Miyagi, Y.; et al. Development of on-chip multi-imaging flow cytometry for identification of imaging biomarkers of clustered circulating tumor cells. PLoS ONE 2014, 9, e104372. [Google Scholar] [CrossRef]
- Hattori, A.; Kim, H.; Terazono, H.; Odaka, M.; Girault, M.; Matsuura, K.; Yasuda, K. Identification of cells using morphological information of bright field/fluorescent multi-imaging flow cytometer images. Jpn. J. Appl. Phys. 2014, 53, 06JL03. [Google Scholar] [CrossRef]
- Vona, G.; Sabile, A.; Louha, M.; Sitruk, V.; Romana, S.; Schutze, K.; Capron, F.; Franco, D.; Pazzagli, M.; Vekemans, M.; et al. Isolation by size of epithelial tumor cells—A new method for the immunomorphological and molecular characterization of circulating tumor cells. Am. J. Pathol. 2000, 156, 57–63. [Google Scholar] [CrossRef]
- Desitter, I.; Guerrouahen, B.S.; Benali-Furet, N.; Wechsler, J.; Janne, P.A.; Kuang, Y.A.; Yanagita, M.; Wang, L.L.; Berkowitz, J.A.; Distel, R.J.; et al. A new device for rapid isolation by size and characterization of rare circulating tumor cells. Anticancer Res. 2011, 31, 427–441. [Google Scholar]
- Hosokawa, M.; Kenmotsu, H.; Koh, Y.; Yoshino, T.; Yoshikawa, T.; Naito, T.; Takahashi, T.; Murakami, H.; Nakamura, Y.; Tsuya, A.; et al. Size-based isolation of circulating tumor cells in lung cancer patients using a microcavity array system. PLoS ONE 2013, 8, e67466. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Yoshikawa, T.; Negishi, R.; Yoshino, T.; Koh, Y.; Kenmotsu, H.; Naito, T.; Takahashi, T.; Yamamoto, N.; Kikuhara, Y.; et al. Microcavity array system for size-based enrichment of circulating tumor cells from the blood of patients with small-cell lung cancer. Anal. Chem. 2013, 85, 5692–5698. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, F.; Boder, J.; Markus, R.; Hashmi, H.; Buhmeida, A.; Collan, Y. Correlation of nuclear morphometry of breast cancer in histological sections with clinicopathological features and prognosis. Anticancer Res. 2009, 29, 1771–1776. [Google Scholar] [PubMed]
- Buhmeida, A.; Algars, A.; Ristamaki, R.; Collan, Y.; Syrjanen, K.; Pyrhonen, S. Nuclear size as prognostic determinant in stage II and stage III colorectal adenocarcinoma. Anticancer Res. 2006, 26, 455–462. [Google Scholar] [PubMed]
- De Andrea, C.E.; Petrilli, A.S.; Jesus-Garcia, R.; Bleggi-Torres, L.F.; Alves, M.T. Large and round tumor nuclei in osteosarcoma: Good clinical outcome. Int. J. Clin. Exp. Pathol. 2011, 4, 169–174. [Google Scholar] [PubMed]
- Deans, G.T.; Hamilton, P.W.; Watt, P.C.; Heatley, M.; Williamson, K.; Patterson, C.C.; Rowlands, B.J.; Parks, G.; Spence, R. Morphometric analysis of colorectal cancer. Dis. Colon Rectum 1993, 36, 450–456. [Google Scholar] [CrossRef]
- Dundas, S.A.; Laing, R.W.; O’Cathain, A.; Seddon, I.; Slater, D.N.; Stephenson, T.J.; Underwood, J.C. Feasibility of new prognostic classification for rectal cancer. J. Clin. Pathol. 1988, 41, 1273–1276. [Google Scholar] [CrossRef]
- Meachem, M.D.; Burgess, H.J.; Davies, J.L.; Kidney, B.A. Utility of nuclear morphometry in the cytologic evaluation of canine cutaneous soft tissue sarcomas. J. Vet. Diagn. Investig. 2012, 24, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokmen, S.; Sarioglu, S.; Fuzun, M.; Terzi, C.; Kupelioglu, A.; Aslan, B. Prognostic significance of angiogenesis in rectal cancer: A morphometric investigation. Anticancer Res. 2001, 21, 4341–4348. [Google Scholar] [PubMed]
- Chou, H.P.; Spence, C.; Scherer, A.; Quake, S. A microfabricated device for sizing and sorting DNA molecules. Proc. Natl. Acad. Sci. USA 1999, 96, 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.; Gawad, S.; Renaud, P. Impedance spectroscopy flow cytometry: On-chip label-free cell differentiation. Cytom. Part A 2005, 65A, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Gu, W.; Kamotani, Y.; Grotberg, J.B.; Takayama, S. Microfluidics for flow cytometric analysis of cells and particles. Physiol. Meas. 2005, 26, R73–R98. [Google Scholar] [CrossRef]
- Cheung, K.C.; Di Berardino, M.; Schade-Kampmann, G.; Hebeisen, M.; Pierzchalski, A.; Bocsi, J.; Mittag, A.; Tárnok, A. Microfluidic impedance-based flow cytometry. Cytom. Part A 2010, 77A, 648–666. [Google Scholar] [CrossRef]
- Bow, H.; Pivkin, I.V.; Diez-Silva, M.; Goldfless, S.J.; Dao, M.; Niles, J.C.; Suresh, S.; Han, J. A microfabricated deformability-based flow cytometer with application to malaria. Lab Chip 2011, 11, 1065–1073. [Google Scholar] [CrossRef]
- Karabacak, N.M.; Spuhler, P.S.; Fachin, F.; Lim, E.J.; Pai, V.; Ozkumur, E.; Martel, J.M.; Kojic, N.; Smith, K.; Chen, P.I.; et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protocols 2014, 9, 694–710. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.Y.; Elbuken, C.; Shen, C.; Huissoon, J.P.; Ren, C.L. An integrated microfluidic device for the sorting of yeast cells using image processing. Sci. Rep. 2018, 8, 3550. [Google Scholar] [CrossRef] [Green Version]
- Utharala, R.; Tseng, Q.; Furlong, E.E.; Merten, C.A. A versatile, low-cost, multiway microfluidic sorter for droplets, cells, and embryos. Anal. Chem. 2018, 90, 5982–5988. [Google Scholar] [CrossRef]
- Nitta, N.; Sugimura, T.; Isozaki, A.; Mikami, H.; Hiraki, K.; Sakuma, S.; Iino, T.; Arai, F.; Endo, T.; Fujiwaki, Y.; et al. Intelligent image-activated cell sorting. Cell 2018, 175, 266–276.e13. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Yasuda, K. Extended depth of field optics for precise image analysis in microfluidic flow cytometry. Jpn. J. Appl. Phys. 2012, 51, 06FK05. [Google Scholar] [CrossRef]
- Kinosita, K., Jr.; Itoh, H.; Ishiwata, S.; Hirano, K.; Nishizaka, T.; Hayakawa, T. Dual-view microscopy with a single camera: Real-time imaging of molecular orientations and calcium. J. Cell Biol. 1991, 115, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Kaneko, T.; Yasuda, K. Improvement of particle alignment control and precise image acquisition for on-chip high-speed imaging cell sorter. Jpn. J. Appl. Phys. 2011, 50, 06GL06. [Google Scholar] [CrossRef]
- Tennant, T.R.; Kim, H.; Sokoloff, M.; Rinker-Schaeffer, C.W. The Dunning model. Prostate 2000, 43, 295–302. [Google Scholar] [CrossRef]
- Yasuda, K.; Hattori, A.; Kim, H.; Terazono, H.; Hayashi, M.; Takei, H.; Kaneko, T.; Nomura, F. Non-destructive on-chip imaging flow cell-sorting system for on-chip cellomics. Microfluid. Nanofluid. 2013, 14, 907–931. [Google Scholar] [CrossRef]















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odaka, M.; Kim, H.; Nakamura, Y.; Hattori, A.; Matsuura, K.; Iwamura, M.; Miyagi, Y.; Yasuda, K. Size Distribution Analysis with On-Chip Multi-Imaging Cell Sorter for Unlabeled Identification of Circulating Tumor Cells in Blood. Micromachines 2019, 10, 154. https://doi.org/10.3390/mi10020154
Odaka M, Kim H, Nakamura Y, Hattori A, Matsuura K, Iwamura M, Miyagi Y, Yasuda K. Size Distribution Analysis with On-Chip Multi-Imaging Cell Sorter for Unlabeled Identification of Circulating Tumor Cells in Blood. Micromachines. 2019; 10(2):154. https://doi.org/10.3390/mi10020154
Chicago/Turabian StyleOdaka, Masao, Hyonchol Kim, Yoshiyasu Nakamura, Akihiro Hattori, Kenji Matsuura, Moe Iwamura, Yohei Miyagi, and Kenji Yasuda. 2019. "Size Distribution Analysis with On-Chip Multi-Imaging Cell Sorter for Unlabeled Identification of Circulating Tumor Cells in Blood" Micromachines 10, no. 2: 154. https://doi.org/10.3390/mi10020154
APA StyleOdaka, M., Kim, H., Nakamura, Y., Hattori, A., Matsuura, K., Iwamura, M., Miyagi, Y., & Yasuda, K. (2019). Size Distribution Analysis with On-Chip Multi-Imaging Cell Sorter for Unlabeled Identification of Circulating Tumor Cells in Blood. Micromachines, 10(2), 154. https://doi.org/10.3390/mi10020154