Single-Cell Point Constrictions for Reagent-Free High-Throughput Mechanical Lysis and Intact Nuclei Isolation

Abstract
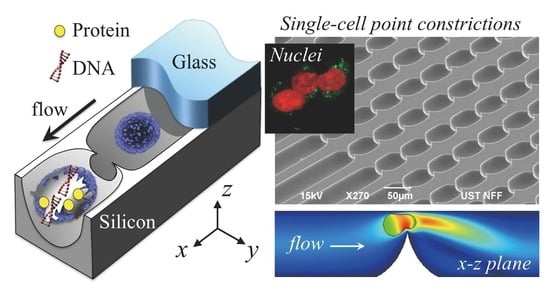
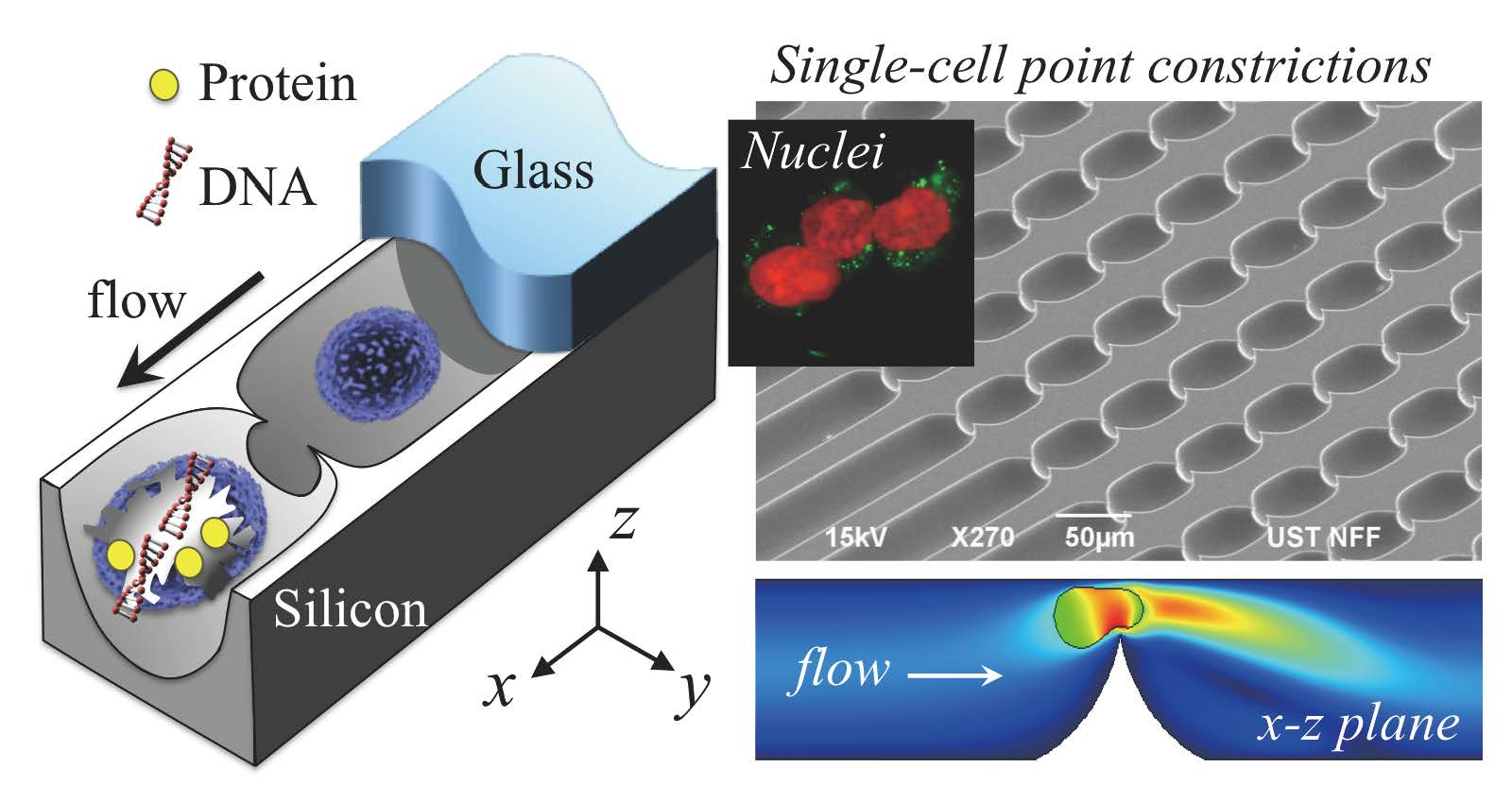
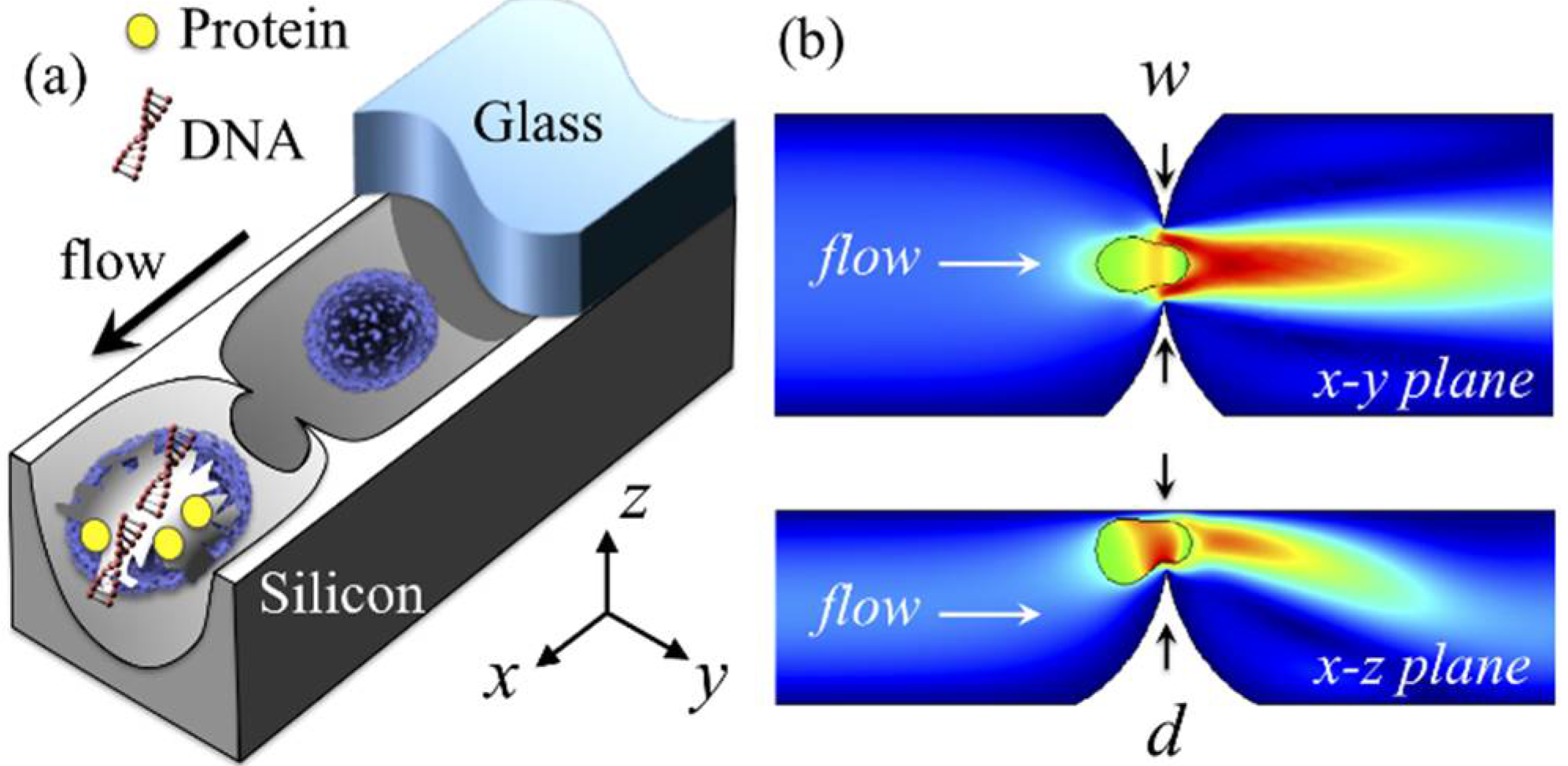
:
1. Introduction
2. Materials and Methods
2.1. Device Fabrication
2.2. Device Preparation
2.3. Cells
2.4. Cell Treatment
2.5. Microscopy
2.6. Protein Yield
2.7. DNA Yield
2.8. Flow Cytometry
3. Results
3.1. Device
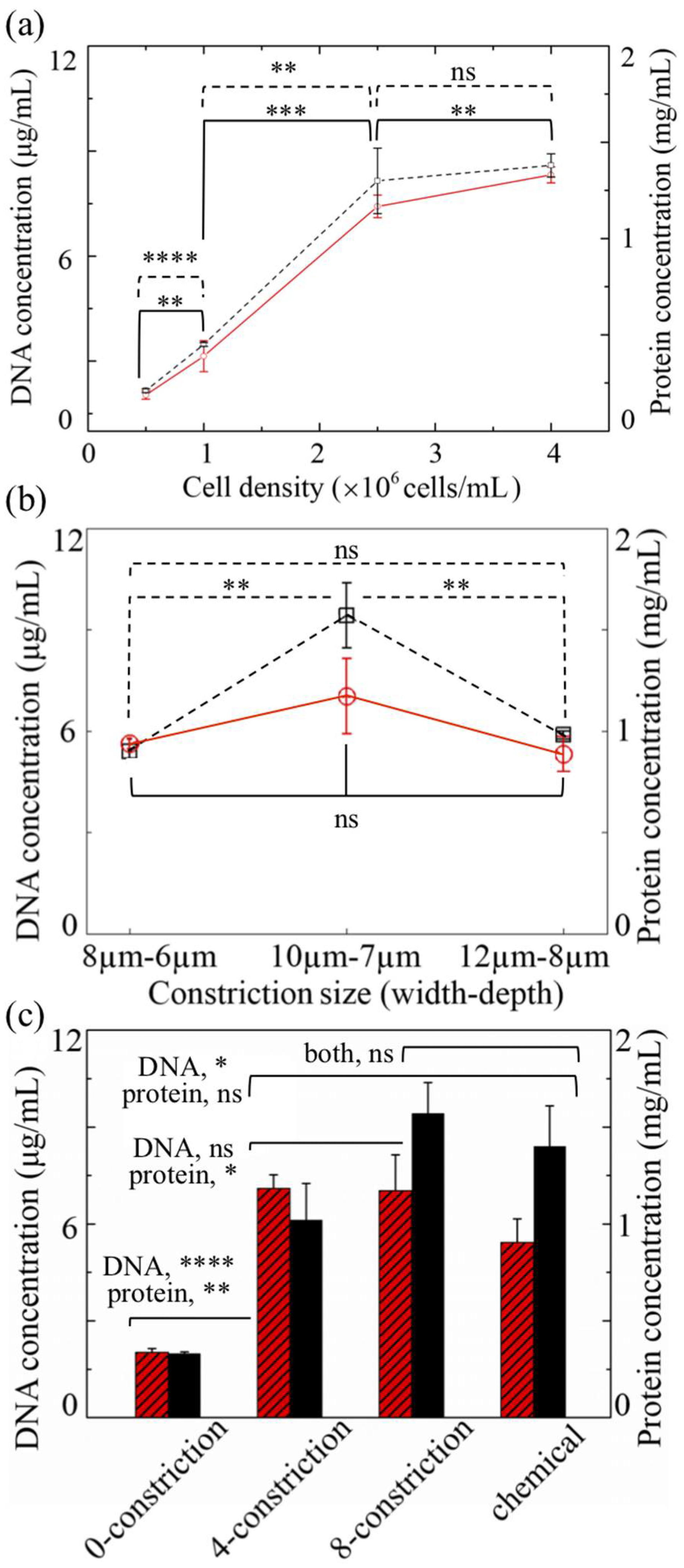
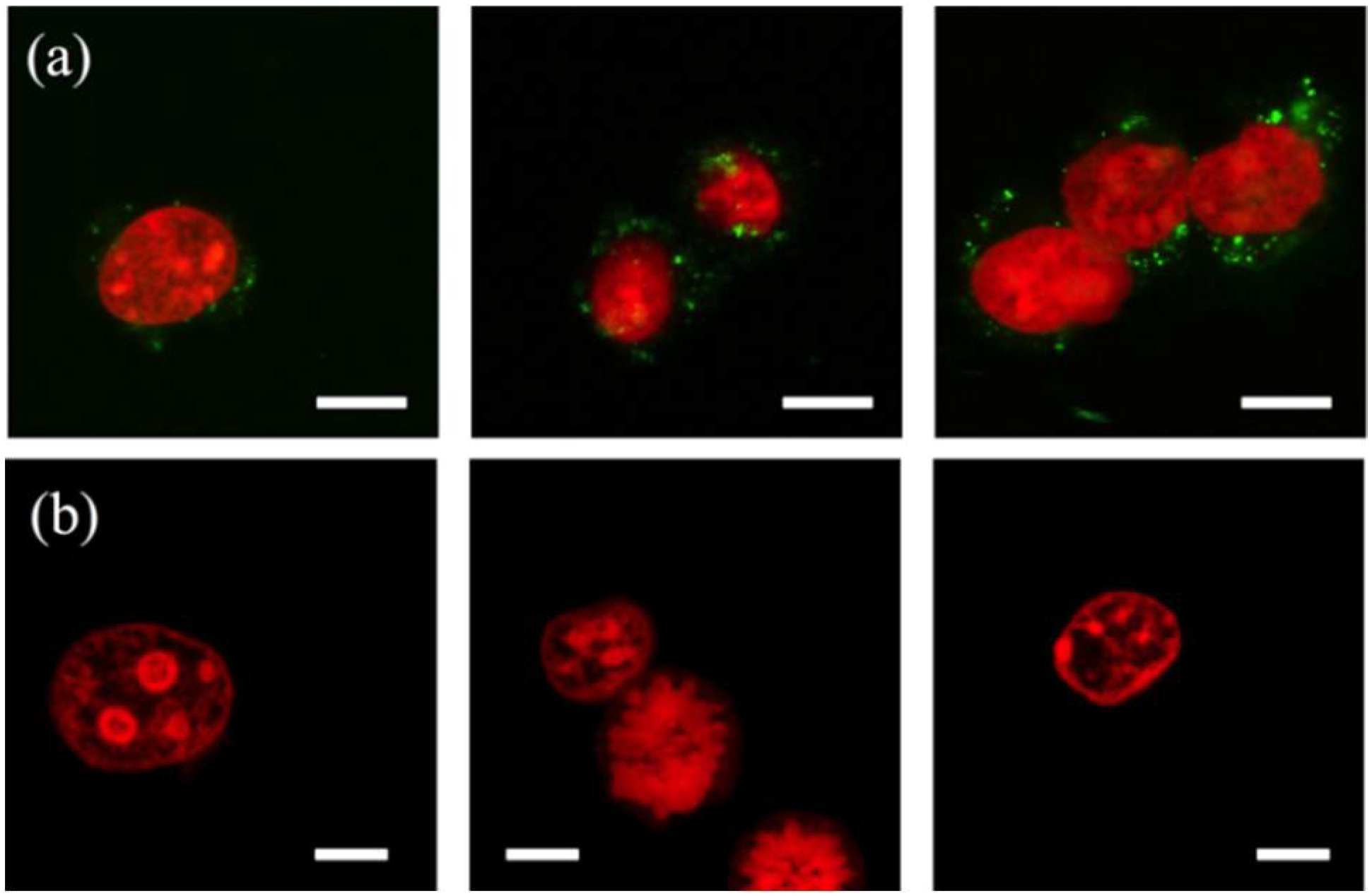
3.2. Cell Lysis
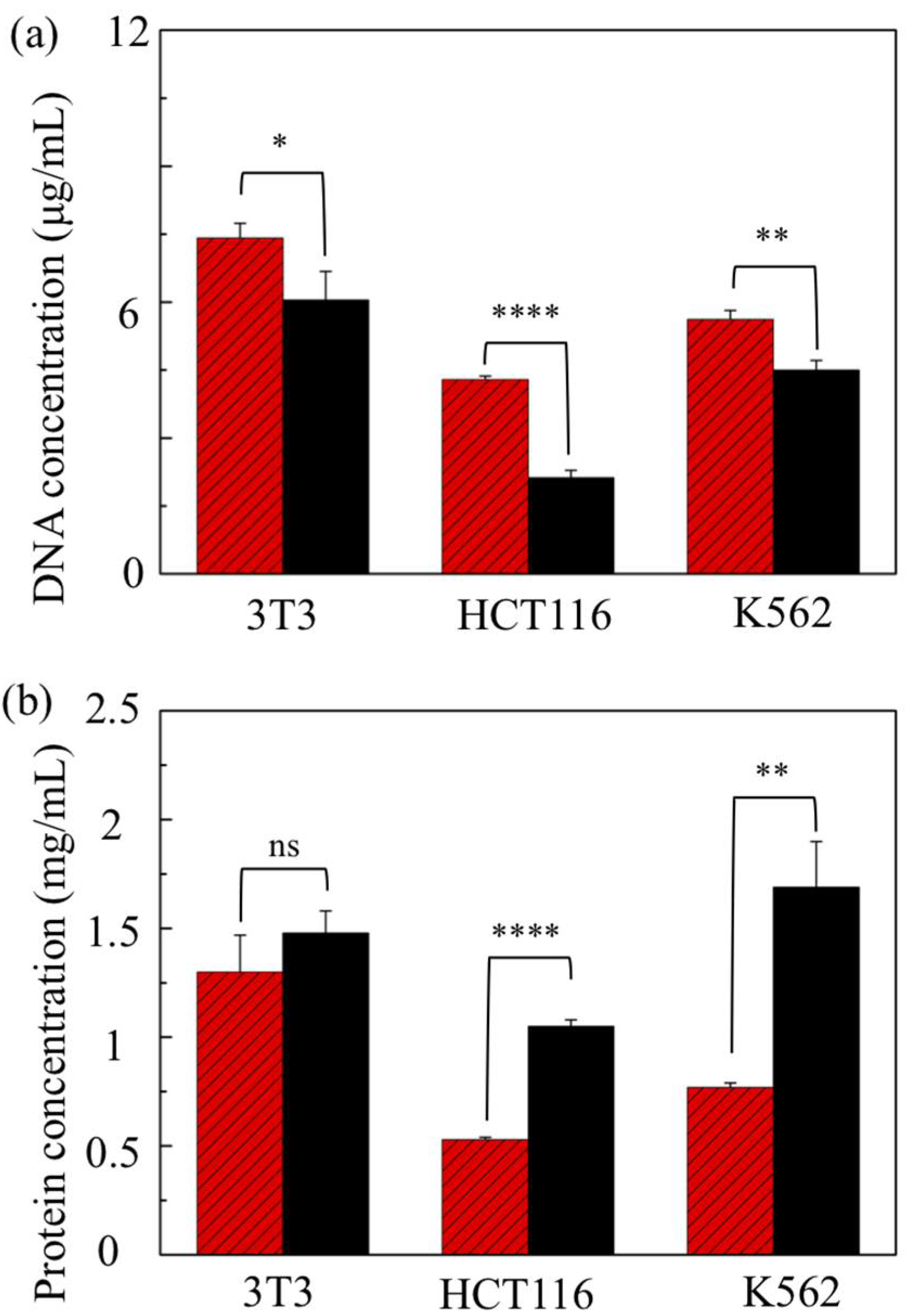
3.3. Cell Types
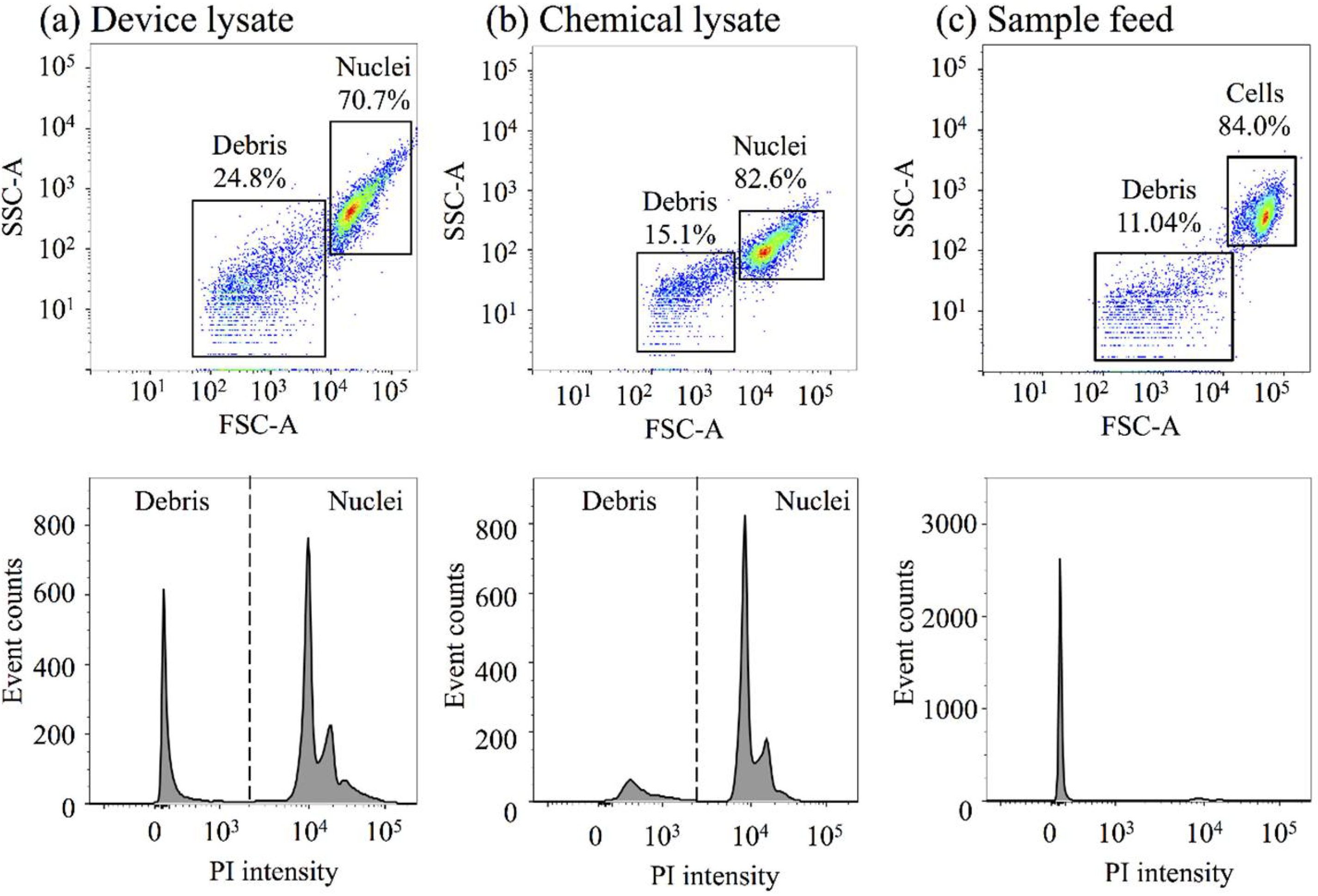
3.4. Nuclei Isolation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bisen, P.S. Chapter 19. Genetic Engineerig. In Laboratory Protocols in Applied Life Sciences, 1st ed.; CRC Press: Boca Rato, FL, USA, 2014. [Google Scholar]
- Saunders, G.C.; Rossi, J.M. Chapter 4. DNA Extraction. In Essentials of Nucleic Acid Analysis: A Robust Approach; RSC: Cambridge, UK, 2010. [Google Scholar]
- Porter, J.; Edwards, C.; Morgan, J.A.W.; Pickup, R.W. Rapid, Automated Separation of Specific Bacteria from Lake Water and Sewage by Flow Cytometry and Cell Sorting. Appl. Environ. Microbiol. 1993, 59, 3327–3333. [Google Scholar] [PubMed]
- Sethu, P.; Anahtar, M.; Moldawer, L.L.; Tompkins, R.G.; Toner, M. Continuous Flow Microfluidic Device for Rapid Erythrocyte Lysis. Anal. Chem. 2004, 76, 6247–6253. [Google Scholar] [CrossRef] [PubMed]
- Morton, K.J.; Loutherback, K.; Inglis, D.W.; Tsui, O.K.; Sturm, J.C.; Chou, S.Y.; Austin, R.H. Crossing Microfluidic Streamlines to Lyse, Label and Wash Cells. Lab Chip 2008, 8, 1448–1453. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Schmidt, M.A.; Jensen, K.F. A Microfluidic Electroporation Device for Cell Lysis. Lab Chip 2005, 5, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Cho, Y.H. A Continuous Electrical Cell Lysis Device Using a Low Dc Voltage for a Cell Transport and Rupture. Sens. Actuators B Chem. 2007, 124, 84–89. [Google Scholar] [CrossRef]
- Wang, H.Y.; Bhunia, A.K.; Lu, C. A Microfluidic Flow-through Device for High Throughput Electrical Lysis of Bacterial Cells Based on Continuous Dc Voltage. Biosens. Bioelectron. 2006, 22, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, D.; Ionescu-Zanetti, C.; Zhang, Y.; Hung, P.; Lee, L.P. On-Chip Cell Lysis by Local Hydroxide Generation. Lab Chip 2005, 5, 171–178. [Google Scholar] [CrossRef]
- Privorotskaya, N.; Liu, Y.S.; Lee, J.; Zeng, H.; Carlisle, J.A.; Radadia, A.; Millet, L.; Bashir, R.; King, W.P. Rapid Thermal Lysis of Cells Using Silicon-Diamond Microcantilever Heaters. Lab Chip 2010, 10, 1135–1141. [Google Scholar] [CrossRef]
- Ke, C.; Kelleher, A.M.; Berney, H.; Sheehan, M.; Mathewson, A. Single Step Cell Lysis/PCR Detection of Escherichia Coli in an Independently Controllable Silicon Microreactor. Sens. Actuators B Chem. 2007, 120, 538–544. [Google Scholar] [CrossRef]
- Baek, S.K.; Min, J.; Park, J.H. Wireless Induction Heating in a Microfluidic Device for Cell Lysis. Lab Chip 2010, 10, 909–917. [Google Scholar] [CrossRef]
- Cheong, K.H.; Yi, D.K.; Lee, J.G.; Park, J.M.; Kim, M.J.; Edel, J.B.; Ko, C. Gold Nanoparticles for One Step DNA Extraction and Real-Time PCR of Pathogens in a Single Chamber. Lab Chip 2008, 8, 810–813. [Google Scholar] [CrossRef]
- Taylor, M.T.; Belgrader, P.; Furman, B.J.; Pourahmadi, F.; Kovacs, G.T.A.; Northrup, M.A. Lysing Bacterial Spores by Sonication through a Flexible Interface in a Microfluidic System. Anal. Chem. 2001, 73, 492–496. [Google Scholar] [CrossRef]
- Rau, K.R.; Quinto-Su, P.A.; Hellman, A.N.; Venugopalan, V. Pulsed Laser Microbeam-Induced Cell Lysis: Time-Resolved Imaging and Analysis of Hydrodynamic Effects. Biophys. J. 2006, 91, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, M.D.; Wise, F.; Baeumner, A.J. Development of a Laser-Induced Cell Lysis System. Anal. Bioanal. Chem. 2002, 374, 421–426. [Google Scholar] [CrossRef]
- Kido, H.; Micic, M.; Smith, D.; Zoval, J.; Norton, J.; Madou, M. A Novel, Compact Disk-like Centrifugal Microfluidics System for Cell Lysis and Sample Homogenization. Colloids Surf. B Biointerfaces 2007, 58, 44–51. [Google Scholar] [CrossRef]
- Siegrist, J.; Gorkin, R.; Bastien, M.; Stewart, G.; Peytavi, R.; Kido, H.; Bergeron, M.; Madou, M. Validation of a Centrifugal Microfluidic Sample Lysis and Homogenization Platform for Nucleic Acid Extraction with Clinical Samples. Lab Chip 2010, 10, 363–371. [Google Scholar] [CrossRef]
- Kim, Y.C.; Kang, J.H.; Park, S.J.; Yoon, E.S.; Park, J.K. Microfluidic Biomechanical Device for Compressive Cell Stimulation and Lysis. Sens. Actuators B Chem. 2007, 128, 108–116. [Google Scholar] [CrossRef]
- Di Carlo, D.; Jeong, K.H.; Lee, L.P. Reagentless Mechanical Cell Lysis by Nanoscale Barbs in Microchannels for Sample Preparation. Lab Chip 2003, 3, 287–291. [Google Scholar] [CrossRef]
- Kim, J.; Hong, J.W.; Kim, D.P.; Shin, J.H.; Park, I. Nanowire-Integrated Microfluidic Devices for Facile and Reagent-Free Mechanical Cell Lysis. Lab Chip 2012, 12, 2914–2921. [Google Scholar] [CrossRef]
- Yun, S.S.; Yoon, S.Y.; Song, M.K.; Im, S.H.; Kim, S.; Lee, J.H.; Yang, S. Handheld Mechanical Cell Lysis Chip with Ultra-Sharp Silicon Nano-Blade Arrays for Rapid Intracellular Protein Extraction. Lab Chip 2010, 10, 1442–1446. [Google Scholar] [CrossRef]
- So, H.; Lee, K.; Seo, Y.H.; Murthy, N.; Pisano, A.P. Hierarchical Silicon Nanospikes Membrane for Rapid and High-Throughput Mechanical Cell Lysis. ACS Appl. Mater. Interfaces 2014, 6, 6993–6997. [Google Scholar] [CrossRef]
- So, H.; Lee, K.; Murthy, N.; Pisano, A.P. All-in-One Nanowire-Decorated Multifunctional Membrane for Rapid Cell Lysis and Direct DNA Isolation. ACS Appl. Mater. Interfaces 2014, 6, 20693–20699. [Google Scholar] [CrossRef] [Green Version]
- Ziv, R.; Steinhardt, Y.; Pelled, G.; Gazit, D.; Rubinsky, B. Micro-Electroporation of Mesenchymal Stem Cells with Alternating Electrical Current Pulses. Biomed. Microdevices 2009, 11, 95–101. [Google Scholar] [CrossRef]
- Sharei, A.; Zoldan, J.; Adamo, A.; Sim, W.Y.; Cho, N.; Jackson, E.; Mao, S.; Schneider, S.; Han, M.-J.; Lytton-Jean, A.; et al. A Vector-Free Microfluidic Platform for Intracellular Delivery. Proc. Natl. Acad. Sci. USA 2013, 110, 2082–2087. [Google Scholar] [CrossRef]
- Xing, X.; Pan, Y.; Yobas, L. A Low-Backpressure Single-Cell Point Constriction for Cytosolic Delivery Based on Rapid Membrane Deformations. Anal. Chem. 2018, 90, 1836–1844. [Google Scholar] [CrossRef]
- Toyama, K.; Yamada, M.; Seki, M. Isolation of Cell Nuclei in Microchannels by Short-Term Chemical Treatment via Two-Step Carrier Medium Exchange. Biomed. Microdevices 2012, 14, 751–757. [Google Scholar] [CrossRef]
- Huang, S.H.; Hung, L.Y.; Lee, G. Bin. Continuous Nucleus Extraction by Optically-Induced Cell Lysis on a Batch-Type Microfluidic Platform. Lab Chip 2016, 16, 1447–1456. [Google Scholar] [CrossRef]
- Edstrom, J.-E. Microchemical Deoxyribonucleic Acid Determination in Individual Cells. J. Cell Biol. 2004, 9, 619–626. [Google Scholar] [CrossRef]
- Segal, D.J.; McCoy, E.E. Studies on Down’s Syndrome in Tissue Culture. J. Cell. Physiol. 1973, 83, 85–90. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “Proteomic Ruler” for Protein Copy Number and Concentration Estimation without Spike-in Standards. Mol. Cell. Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef]
- Morton, N.E. Parameters of the Human Genome. Proc. Natl. Acad. Sci. USA 1991, 88, 7474–7476. [Google Scholar] [CrossRef]
- English, C.A.; Merson, S.; Keer, J.T. Use of Elemental Analysis to Determine Comparative Performance of Established DNA Quantification Methods. Anal. Chem. 2006, 78, 4630–4633. [Google Scholar] [CrossRef]
- Warburg, O.; Christian, W. Isolierung Und Kristallisation Des Gärungsferments Enolase. Naturwissenschaften 1941. [Google Scholar] [CrossRef]
- Teare, J.M.; Islam, R.; Flanagan, R.; Gallagher, S.; Davies, M.G.; Grabau, C. Measurement of Nucleic Acid Concentrations Using the DyNA Quant(TM) and the GeneQuant(TM). Biotechniques 1997, 22, 1170–1174. [Google Scholar] [CrossRef]
- Manchester, K.L. Use of UV Methods for Measurement of Protein and Nucleic Acid Concentrations. Biotechniques 1996, 20, 968–970. [Google Scholar] [CrossRef]
- Noble, J.E.; Bailey, M.J.A. Chapter 8 Quantitation of Protein. In Methods in Enzymology; Academic press: Cambridge, MA, USA, 2009. [Google Scholar]
- Simbolo, M.; Gottardi, M.; Corbo, V.; Fassan, M.; Mafficini, A.; Malpeli, G.; Lawlor, R.T.; Scarpa, A. DNA Qualification Workflow for Next Generation Sequencing of Histopathological Samples. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Cross, S.E.; Jin, Y.S.; Rao, J.; Gimzewski, J.K. Nanomechanical Analysis of Cells from Cancer Patients. Nat. Nanotechnol. 2007, 2, 780–783. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell. In Biochemistry and Molecular Biology Education, 5th ed.; Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., Walter, P., Eds.; Garland Science: New York, NY, USA, 2008. [Google Scholar]
- Pollard, T.D.; Earnshaw, W.C.; Lippincott-Schwartz, J.; Johnson, G.T. Chapter 20: Endoplasmic Reticulum. In Cell Biology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Hetzer, M.W. The Nuclear Envelope. In Cold Spring Harbor Perspectives in Biology; Cold Spring Harbor Lab: Woodbury, NY, USA, 2010. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Subpopulation Fractions (%) | n | (s−1) | (s−1) | (s−1) | (%) | ||
|---|---|---|---|---|---|---|---|---|
| Device | 83.5 | 10.2 | 6.3 | 1.23 | 133 | 164 | 227 | 72.24 |
| Chemical | 92.6 | 4.8 | 2.6 | 1.10 | 184 | 202 | 227 | 88.98 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Xing, X.; Ng, C.N.; Yobas, L. Single-Cell Point Constrictions for Reagent-Free High-Throughput Mechanical Lysis and Intact Nuclei Isolation. Micromachines 2019, 10, 488. https://doi.org/10.3390/mi10070488
Huang X, Xing X, Ng CN, Yobas L. Single-Cell Point Constrictions for Reagent-Free High-Throughput Mechanical Lysis and Intact Nuclei Isolation. Micromachines. 2019; 10(7):488. https://doi.org/10.3390/mi10070488
Chicago/Turabian StyleHuang, Xiaomin, Xiaoxing Xing, Chun Ning Ng, and Levent Yobas. 2019. "Single-Cell Point Constrictions for Reagent-Free High-Throughput Mechanical Lysis and Intact Nuclei Isolation" Micromachines 10, no. 7: 488. https://doi.org/10.3390/mi10070488
APA StyleHuang, X., Xing, X., Ng, C. N., & Yobas, L. (2019). Single-Cell Point Constrictions for Reagent-Free High-Throughput Mechanical Lysis and Intact Nuclei Isolation. Micromachines, 10(7), 488. https://doi.org/10.3390/mi10070488