
Multiplex, Quantitative, Reverse Transcription PCR Detection of Influenza Viruses Using Droplet Microfluidic Technology

Abstract
:
1. Introduction
2. Experimental Section
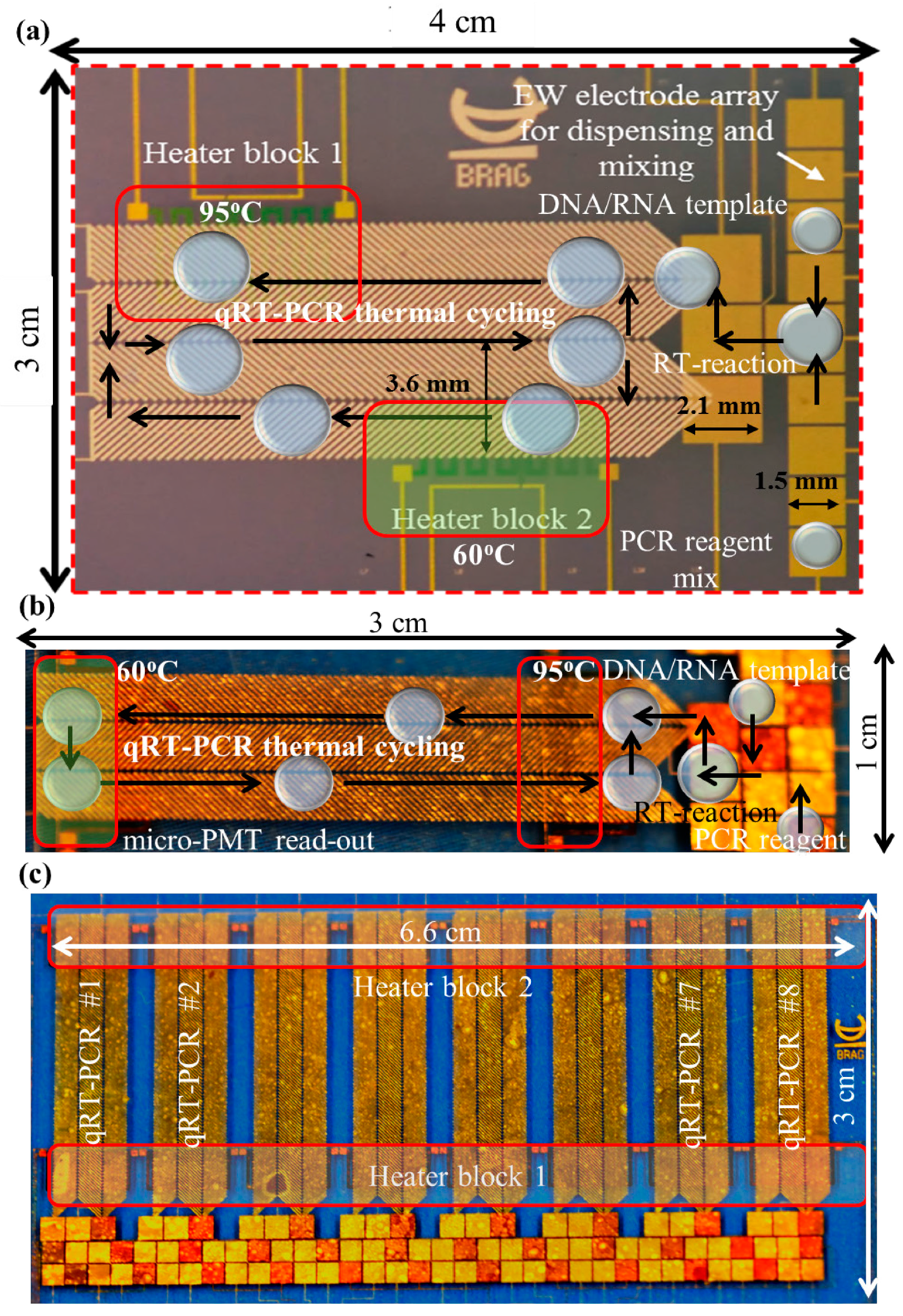
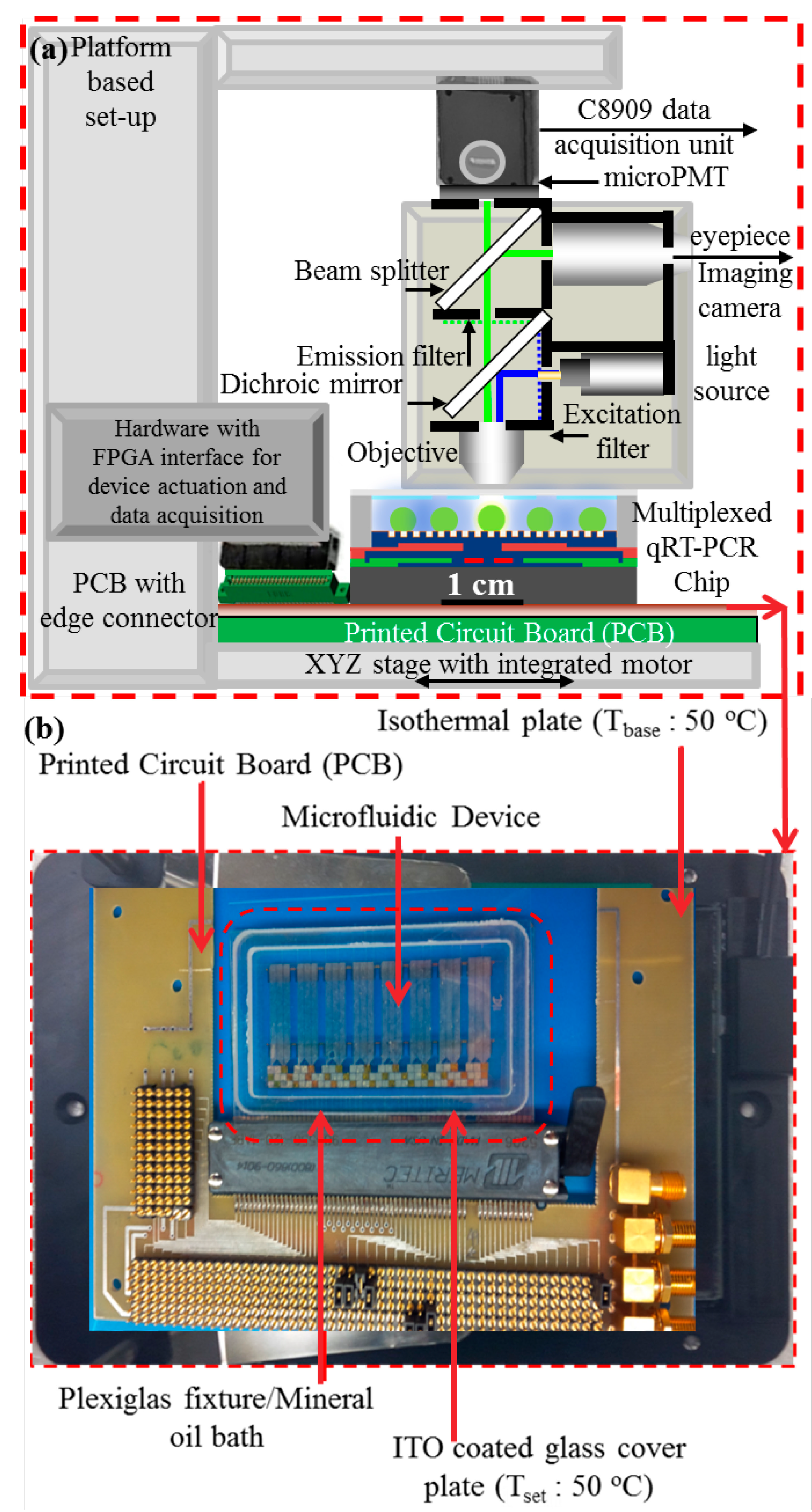
2.1. Device Fabrication

2.2. Sample Preparation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel Sample No. | Sample Style | Target |
|---|---|---|
| (a) The Influenza A panel samples (End-point PCR) | ||
| 1 | Nasopharyngeal Swab | FluA; pdm09 |
| 2 | Nasopharyngeal Swab | Respiratory negative |
| 3 | Nasopharyngeal Swab | Respiratory negative |
| 4 | Nasopharyngeal Swab | FluA; pdm09 |
| 5 | Nasopharyngeal Swab | Respiratory negative |
| 6 | Nasopharyngeal Swab | FluA; pdm09 |
| (b) The Influenza A blind panel | ||
| 1 | Nasopharyngeal Swab | FluA; pdm09 |
| 2 | Nasopharyngeal Swab | FluA; pdm09 |
| 3 | Throat Swab | FluA; pdm09 |
| 4 | Nasopharyngeal Swab | FluA; pdm09 |
| 5 | Nasopharyngeal Swab | FluA; pdm09 |
| 6 | Nasopharyngeal Swab | FluA; pdm09 |
| 7 (+ve control) | H3 M-gene In-vitro RNA | FluA; H3 |
| 8 (−ve control) | PCR water | – |
| (c) The Influenza A, Influenza B mixed blind panel | ||
| 1 | Nasopharyngeal Swab | FluA, FluB |
| 2 | Nasopharyngeal Swab | FluA, FluB |
| 3 | Nasopharyngeal Swab | FluA, FluB |
| 4 | Nasopharyngeal Swab | FluA, FluB |
2.3. Experimental Procedures


3. Results and Discussion
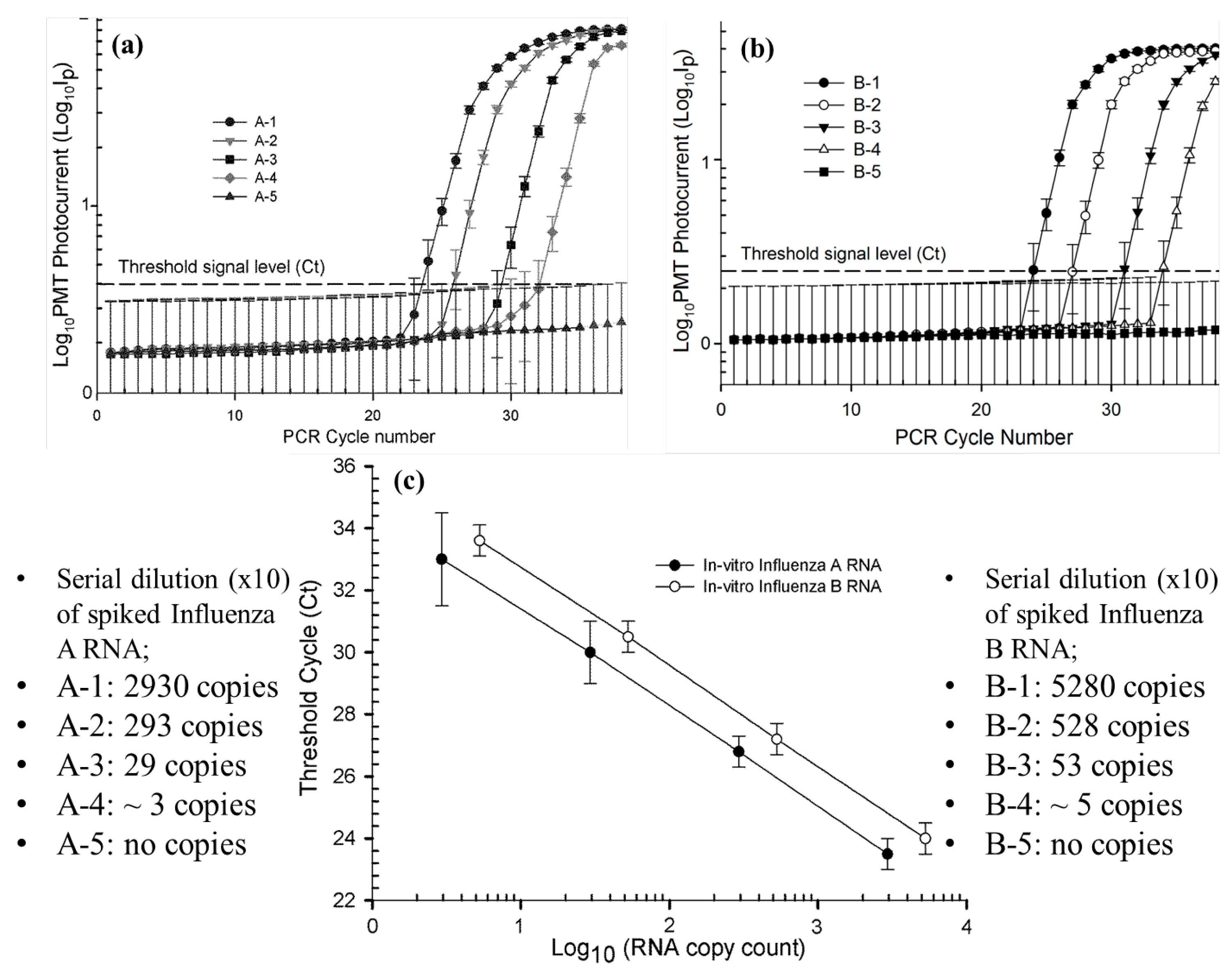
3.1. Standard Quantification Curves for qRT-PCR Amplification of Spiked Influenza A and Influenza B RNA Samples

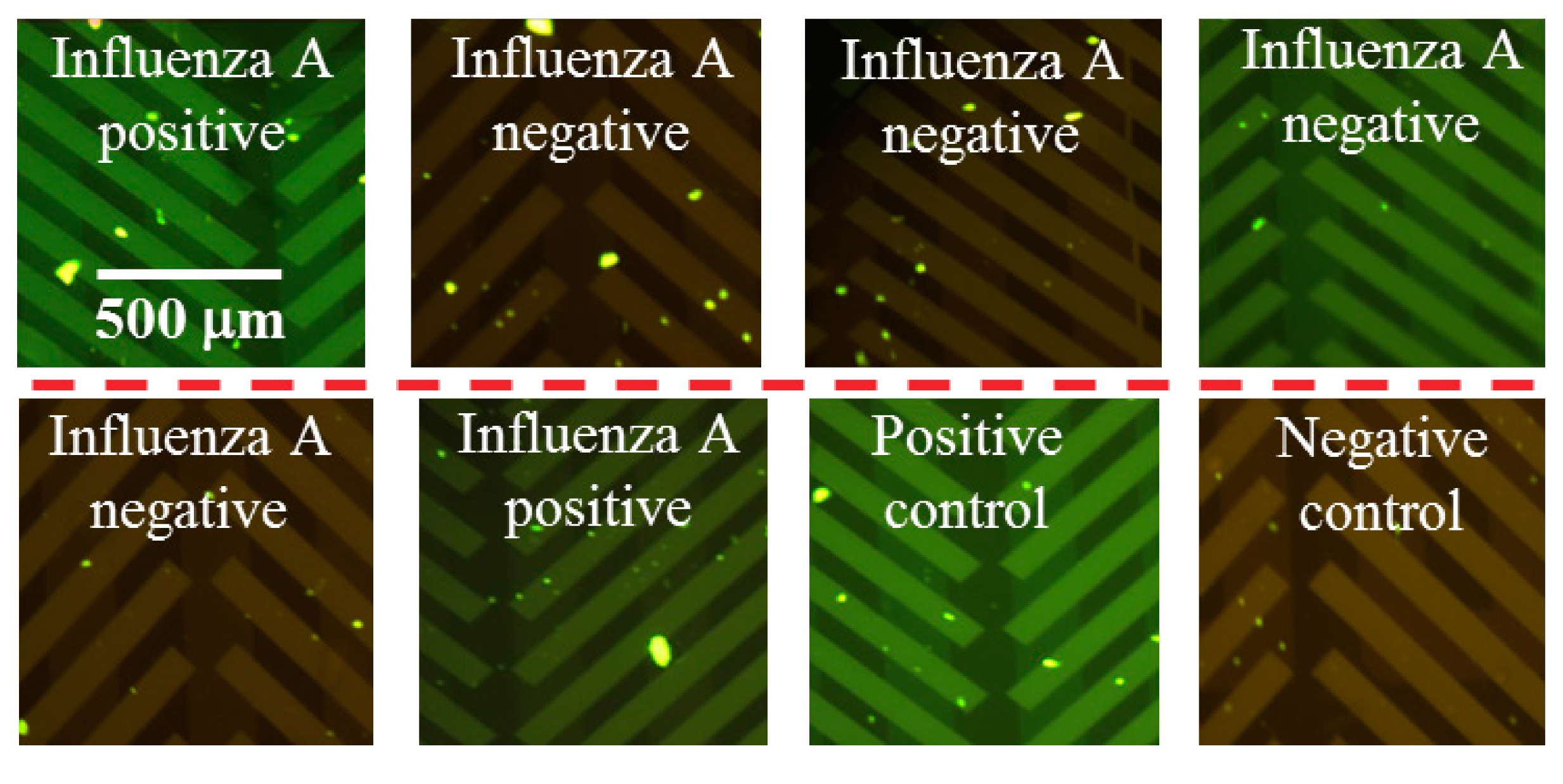
3.2. End-Point, RT-PCR Assay for a Clinical Panel of Influenza A RNA Virus

| Panel Sample No. | PMT Photocurrent at Different PCR End Points (Ip in μA) | ProvLab Ct | ||
|---|---|---|---|---|
| PCR cycle # 10 | PCR cycle # 25 | PCR cycle # 38 | ||
| 1 | 1.09 | 12.90 | 25.77 | 24 |
| 2 | 1.05 | 1.97 | 3.41 | Negative |
| 3 | 1.04 | 1.77 | 2.92 | Negative |
| 4 | 1.08 | 7.75 | 23.35 | 30 |
| 5 | 1.06 | 1.97 | 3.95 | Negative |
| 6 | 1.04 | 4.51 | 18.23 | 33 |
| 7 (+ve control) | 1.09 | 15.82 | 30.35 | 29 |
| 8 (−ve control) | 1.06 | 1.85 | 3.01 | Negative |
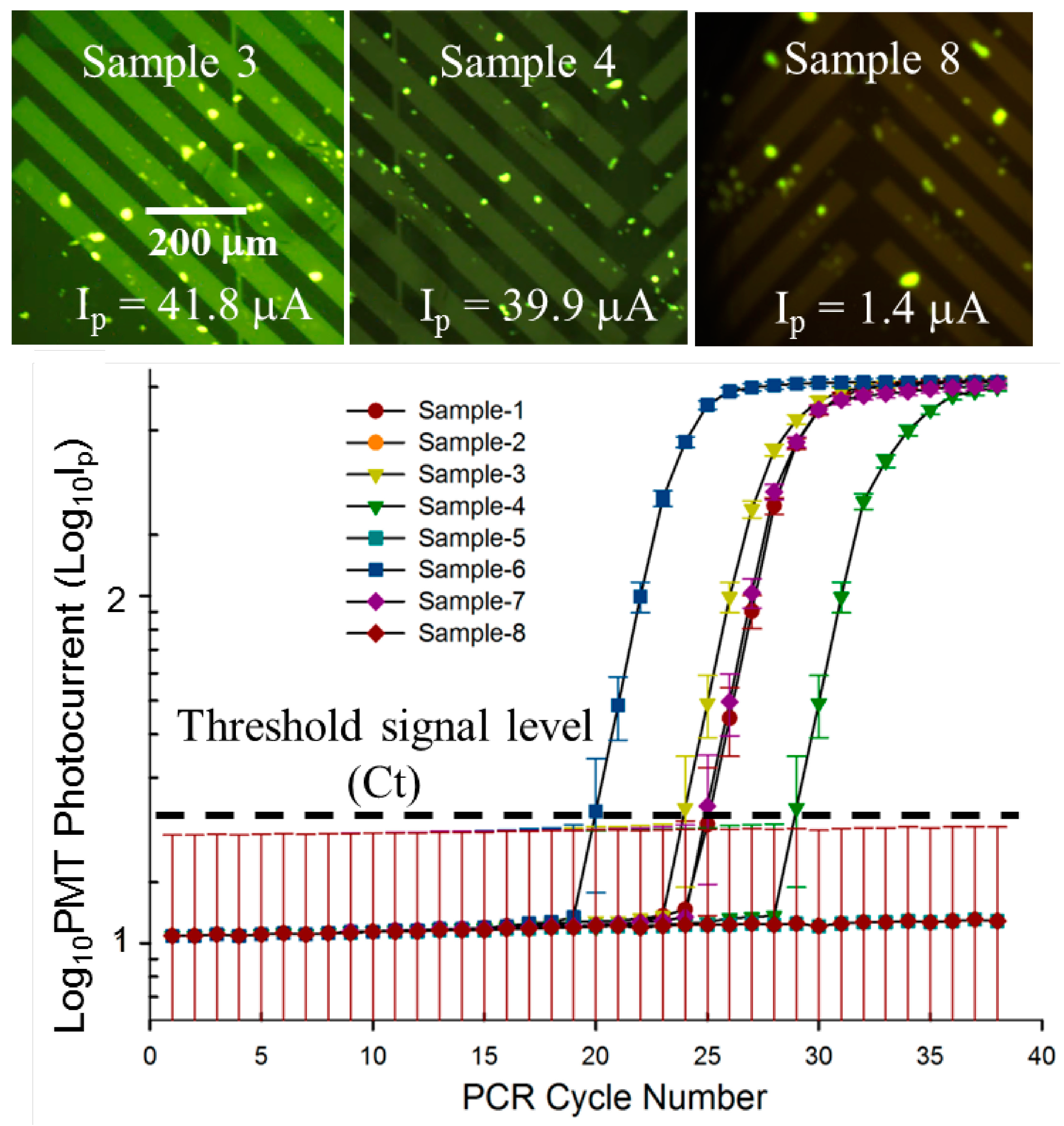
3.3. Quantitative, Multiplexed RT-PCR Assay on an Influenza A blind Panel

| Panel Sample No. | Target | ProvLab Ct | Chip Ct | Initial Copies of Template RNA |
|---|---|---|---|---|
| 1 | Flu A | 29 | 25 | ~590 |
| 2 | Flu A | Negative | Negative | Not applicable |
| 3 | Flu A | 30 | 26 | ~300 |
| 4 | Flu A | 32 | 30 | ~20 |
| 5 | Flu A | Negative | Negative | Not applicable |
| 6 | Flu A | 24 | 21 | ~3500 |
| 7 (+ve control) | Flu A | 29 | 26 | ~250 |
| 8 (−ve control) | Flu A | Negative | Negative | ~110 |
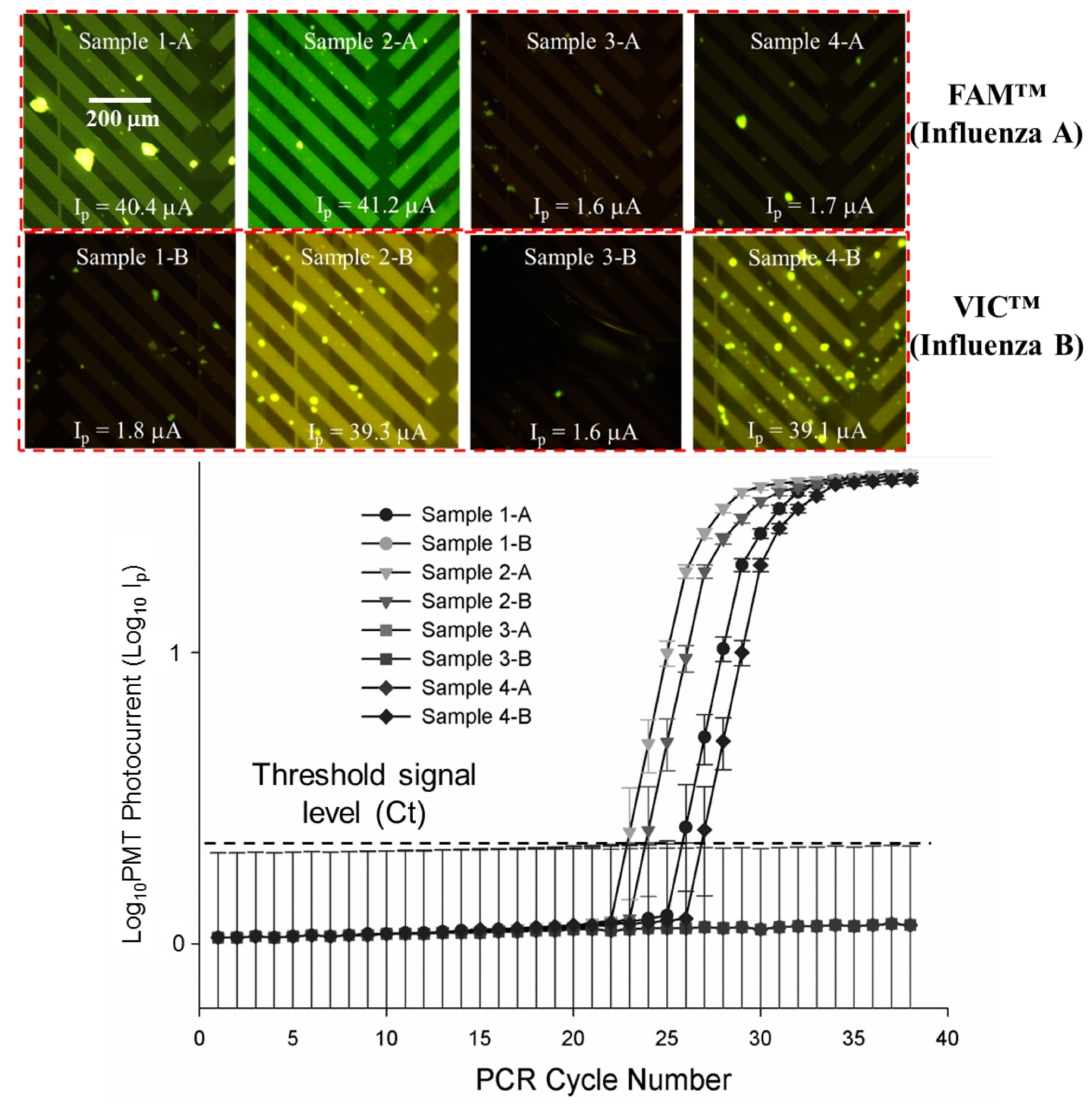
3.4. Quantitative, Multiplexed RT-PCR Assays on a Mixed, Four Sample Influenza A, Influenza B Blind Panel

| Panel Sample No. | Target | ProvLab Ct | Chip Ct | Initial Copies of Template RNA |
|---|---|---|---|---|
| 1-A | Flu A | 29 | 27 | ~290 |
| 1-B | Flu B | Negative | Negative | Not applicable |
| 2-A | Flu A | 27 | 24 | ~2900 |
| 2-B | Flu B | 28 | 25 | ~1050 |
| 3-A | Flu A | Negative | Negative | Not applicable |
| 3-B | Flu B | Negative | Negative | Not applicable |
| 4-A | Flu A | Negative | Negative | Not applicable |
| 4-B | Flu B | 30 | 28 | ~110 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saiki, D.H.; Gelfand, S.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef]
- Langin, T.; Robert, T. Mullis, K. B., Ferré, F. and Gibbs, R. A. (Eds.). 1994. The polymerase chain reaction (PCR). Birkhäuser Verlag AG, Basel, Switzerland. ISBN 3-7643-3607-2 (H.b.). ISBN 3-7643-3750-8 (P.b.). J. Evol. Biol. 1994, 8, 399–401. [Google Scholar] [CrossRef]
- Markoulatos, P.; Siafakas, N.; Moncany, M. Multiplex polymerase chain reaction: A practical approach. J. Clin. Lab. Anal. 2002, 16, 47–51. [Google Scholar] [CrossRef]
- Logan, J.; Edwards, K.J.; Saunders, N. Real-Time PCR: Current Technology and Applications; Caister Academic Press: Norfolk, UK, 2009. [Google Scholar]
- Wittmer, C.T.; Kusukawa, N. Real-time PCR and melting analysis. In Molecular Microbiology Diagnostic Principles and Practice, 2nd ed.; ASM Press: Washington, DC, USA, 2011. [Google Scholar]
- Kopp, M.U.; de Mello, A.J.; Manz, A. Chemical amplification: Continuous-flow PCR on a chip. Science 1998, 280, 1046–1048. [Google Scholar] [CrossRef]
- Manz, A.; Becker, H. Microsystem Technology in Chemistry and Life Sciences; Springer: Berlin, Germany, 1998. [Google Scholar]
- Lagally, E.T.; Emrich, C.A.; Mathies, R.A. Fully integrated PCR-capillary electrophoresis microsystem for DNA analysis. Lab Chip 2001, 1–2, 102–107. [Google Scholar] [CrossRef]
- Liu, R.H.; Yang, J.N.; Lenigk, R.; Bonanno, J.; Grodzinski, P. Self-contained, fully integrated biochip for sample preparation, polymerase chain reaction amplification, and DNA microarray detection. J. Anal. Chem. 2004, 76, 1824–1831. [Google Scholar] [CrossRef]
- Tewhey, R.; Warner, J.B.; Nakano, M.; Libby, B.; Medkova, M.; David, P.H.; Kotsopoulos, S.K.; Samuels, M.L.; Hutchison, J.B.; Larson, J.W.; Topol, E.J.; Weiner, M.P.; Harismendy, O.; Olson, J.; Link, D.R.; Frazer, K.A. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat. Biotechnol. 2009, 27, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Pabbaraju, K.; Wong, S.; Wong, A.; Tellier, R.; Kaler, K.V.I.S. Droplet microfluidic chip based nucleic acid amplification and real-time detection of influenza viruses. J. Electrochem. Soc. 2014, 161, 3083–3093. [Google Scholar] [CrossRef]
- Dahl, A.; Sultan, M.; Jung, A.; Schwartz, R.; Lange, M.; Steinwand, M.; Livak, K.; Lehrach, H.; Nyarsik, L. Quantitative PCR based expression analysis on a nanoliter scale using polymer nano-well chips. Biomed. Microdev. 2007, 9, 307–314. [Google Scholar] [CrossRef]
- Freire, V.S.; Ebert, A.D.; Kalisky, T.; Quake, S.R.; Wu, J.C. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns. Nat. Protoc. 2012, 7, 829–838. [Google Scholar] [CrossRef]
- Saunders, D.C.; Holst, G.L.; Phaneuf, C.R.; Pak, N.; Marchese, M.; Sondej, N.; McKinnon, M.; Forest, C.R. Rapid, quantitative, reverse transcription PCR in a polymer microfluidic chip. Biosens. Bioelec. 2013, 44, 222–228. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, S.W.; Kang, J.Y.; Ahn, C.H. A polymer lab-on-a-chip for reverse transcription (RT)-PCR based point-of-care clinical diagnostics. Lab Chip 2008, 8, 2121–2127. [Google Scholar] [CrossRef]
- Roberts, C.H.; Jiang, W.; Jayaraman, J.; Trowsdale, J.; Holland, M.J.; Traherne, J.A. Killer-cell Immunoglobulin-like receptor gene linkage and copy number variation analysis by droplet digital PCR. Genome Med. 2014, 6, 1–9. [Google Scholar] [CrossRef]
- Pollack, M.G.; Fair, R.B.; Shenderov, A.D. Electrowetting-based actuation of liquid droplets for microfluidic applications. Appl. Phys. Lett. 2000, 77, 1725–1726. [Google Scholar] [CrossRef]
- Cho, S.K.; Moon, H.; Kim, C.J. Creating, Transporting, Cutting, and Merging Liquid Droplets by Electrowetting-Based Actuation for Digital Microfluidic Circuits. J. Microelectromech. Syst. 2003, 12, 70–80. [Google Scholar] [CrossRef]
- Sista, R.; Hua, Z.; Thwar, P.; Sudarsan, A.; Srinivasan, V.; Eckhardt, A.E.; Pollock, M.; Pamula, V. Development of a digital microfluidic platform for point of care testing. Lab Chip 2008, 8, 2091–2104. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Rouse, J.L.; Eckhardt, A.E.; Srinivasan, V.; Pamula, V.K.; Schell, W.A.; Benton, J.L.; Mitchell, T.G.; Pollack, M.G. Multiplexed Real-Time Polymerase Chain Reaction on a Digital Microfluidic Platform. Anal. Chem. 2010, 82, 2310–2316. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Pamula, V.K.; Fair, R.B. An integrated digital microfluidic lab-on-a-chip for clinical diagnostics on human physiological fluids. Lab Chip 2004, 4, 310–315. [Google Scholar] [CrossRef]
- Chang, Y.H.; Lee, G.B.; Huang, F.C.; Chen, Y.Y.; Lin, J.L. Integrated polymerase chain reaction chips utilizing digital microfluidics. Biomed. Microdev. 2006, 8, 215–225. [Google Scholar] [CrossRef]
- Gunji, M.; Nakanishi, H.; Washizu, M. Droplet actuation based on single-phase electrostatic excitation. In Proceedings of 8th International Conf. on Miniaturized Systems in Chemistry and Life Sciences (μ-TAS2004), Malmö, Sweden, September 26–30 2004; Volume 1, pp. 168–170.
- Kaler, K.V.I.S.; Prakash, R.; Chugh, D. Liquid Dieletrophoresis and Surface Microfluidics. Biomicrofluidics 2010, 4, 022805. [Google Scholar] [CrossRef]
- Prakash, R.; Papageorgiou, D.P.; Papathanasiou, A.G.; Kaler, K.V.I.S. Dielectrophoretic liquid actuation on nano-textured super hydrophobic surfaces. Sens. Actuators B Chem. 2013, 182, 351–361. [Google Scholar] [CrossRef]
- Lagally, E.T.; Simpson, P.C.; Mathies, R.A. Monolithic integrated microfluidic DNA amplification and capillary electrophoresis analysis system. Sens. Actuators B Chem. 2006, 63, 138–146. [Google Scholar] [CrossRef]
- Zhong, R.; Pan, X.; Jiang, L.; Dai, Z.; Qin, J.; Lin, B. Simply and reliably integrating micro heaters/sensors in a monolithic PCR-CE microfluidic genetic analysis system. Electrophoresis 2009, 30, 1297–1305. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, 2002–2007. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prakash, R.; Pabbaraju, K.; Wong, S.; Wong, A.; Tellier, R.; Kaler, K.V.I.S. Multiplex, Quantitative, Reverse Transcription PCR Detection of Influenza Viruses Using Droplet Microfluidic Technology. Micromachines 2015, 6, 63-79. https://doi.org/10.3390/mi6010063
Prakash R, Pabbaraju K, Wong S, Wong A, Tellier R, Kaler KVIS. Multiplex, Quantitative, Reverse Transcription PCR Detection of Influenza Viruses Using Droplet Microfluidic Technology. Micromachines. 2015; 6(1):63-79. https://doi.org/10.3390/mi6010063
Chicago/Turabian StylePrakash, Ravi, Kanti Pabbaraju, Sallene Wong, Anita Wong, Raymond Tellier, and Karan V. I. S. Kaler. 2015. "Multiplex, Quantitative, Reverse Transcription PCR Detection of Influenza Viruses Using Droplet Microfluidic Technology" Micromachines 6, no. 1: 63-79. https://doi.org/10.3390/mi6010063
APA StylePrakash, R., Pabbaraju, K., Wong, S., Wong, A., Tellier, R., & Kaler, K. V. I. S. (2015). Multiplex, Quantitative, Reverse Transcription PCR Detection of Influenza Viruses Using Droplet Microfluidic Technology. Micromachines, 6(1), 63-79. https://doi.org/10.3390/mi6010063