We now consider the system response under the action of an applied AC electric field with amplitude and frequency .
3.2.1. Dielectric Response
The dielectric response of the system is determined computing the frequency behavior of the dipolar coefficient: the amplitude (divided by
) of the field induced dipolar field far away from the particle:
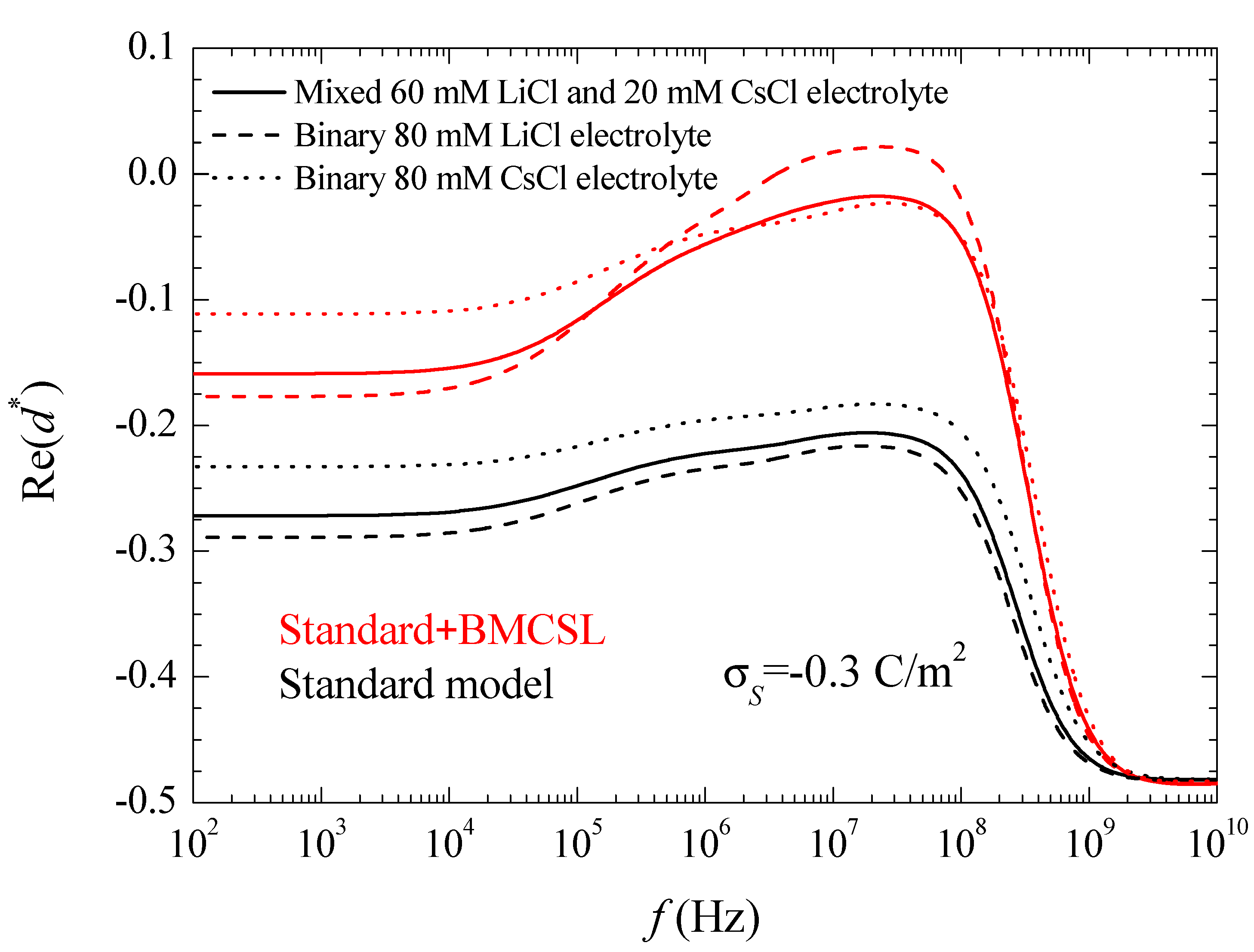
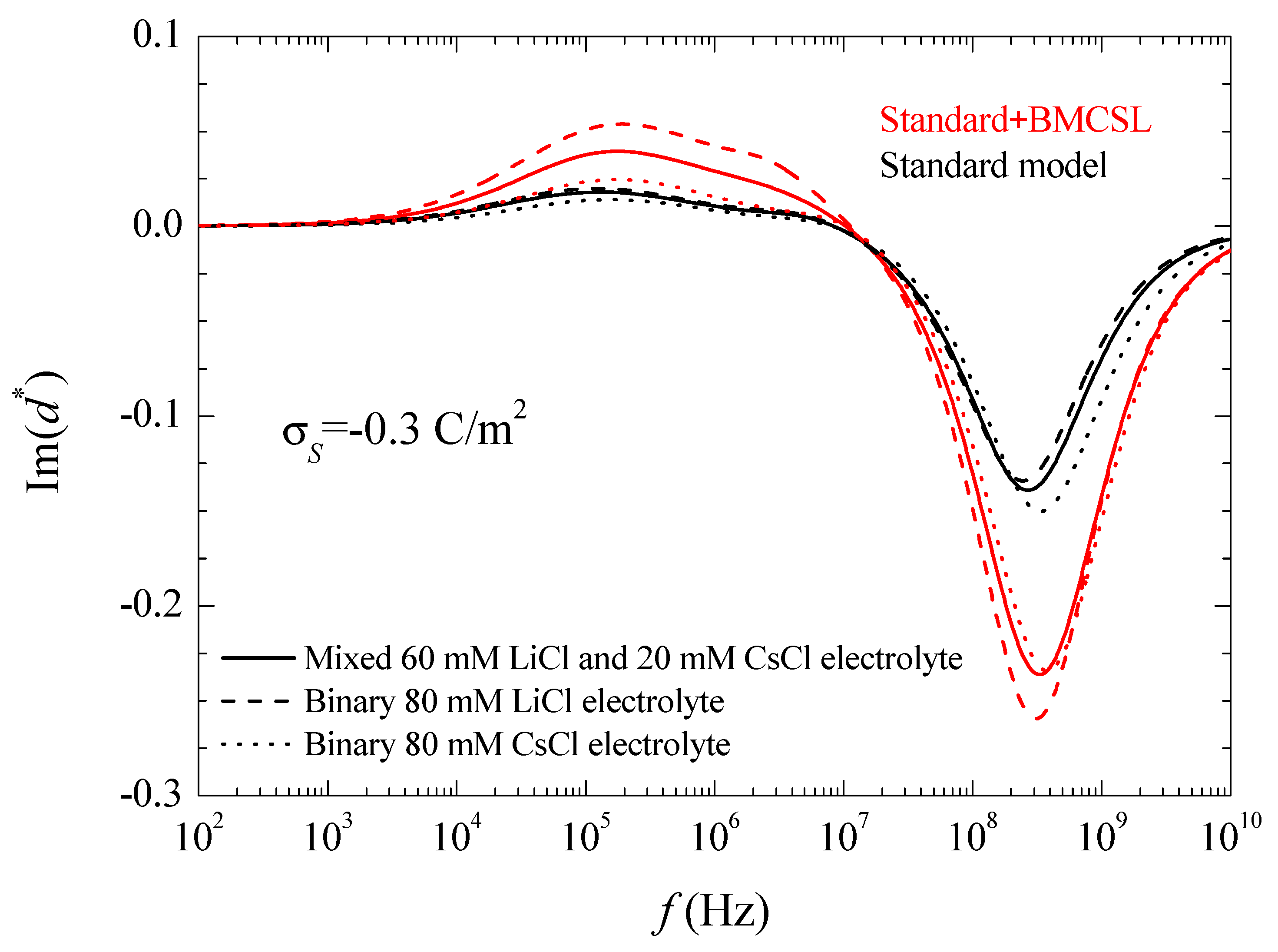
where the asterisk denotes a complex magnitude. The spectra of the real and imaginary parts of this coefficient are represented in
Figure 3 and
Figure 4.
For uncharged particles, the low-frequency dipolar coefficient value is simply equal to −1/2 (this is rigorous for point ions but also holds to high approximation for finite size ions because the ionic concentrations remain everywhere low when particles are uncharged). For increasing (in absolute value) surface charge values, the standard model behavior can be analyzed using the results presented in Reference [
24]. When the counterion and co-ion diffusion coefficients differ from one another, volume charge density clouds buildup outside the equilibrium double layer boundaries. This charge that has the same sign as
increases the dipolar coefficient value which, for equal diffusion coefficients, is solely due to the charge density distribution inside the double layer. This explains why
in
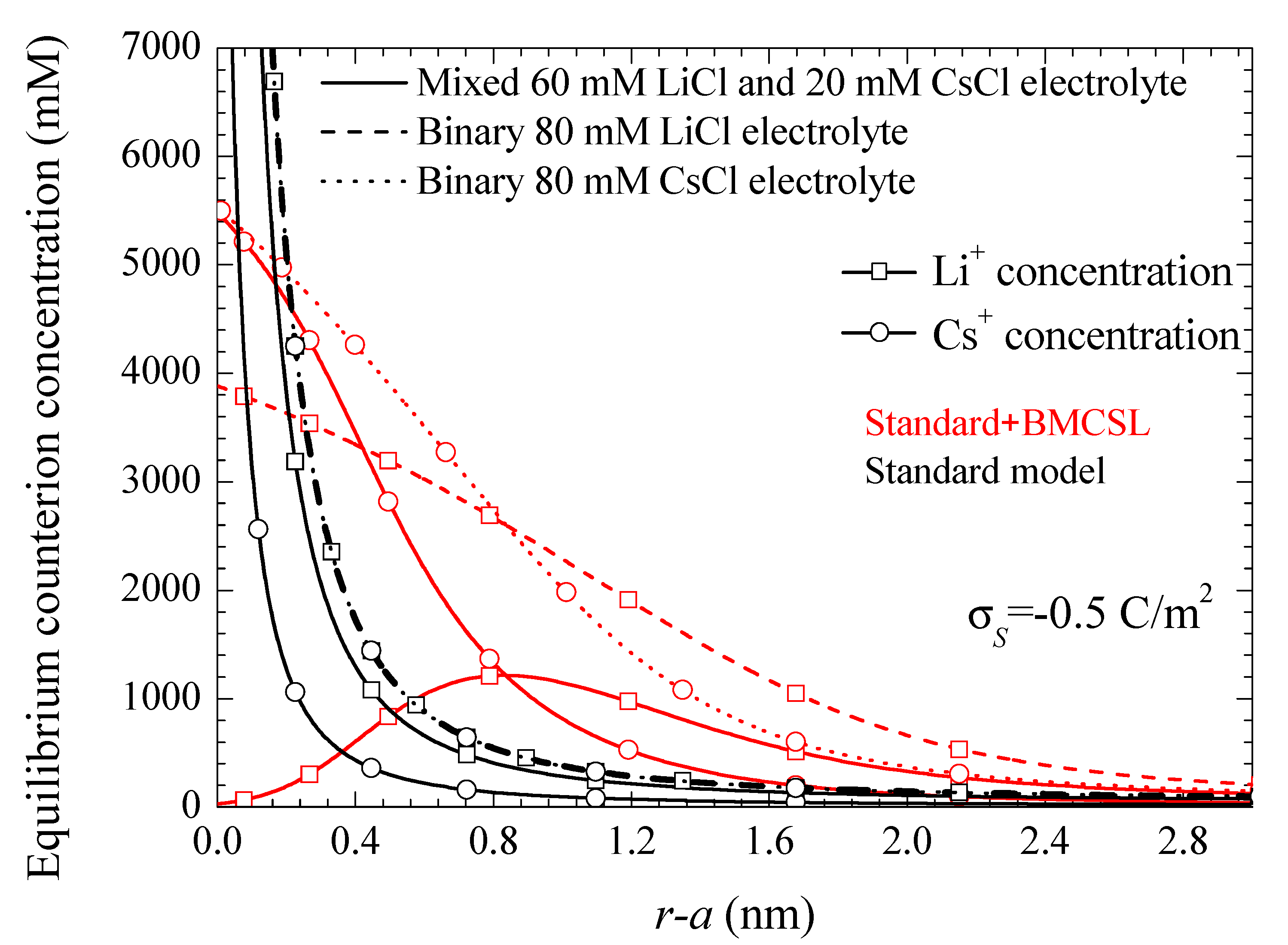
Figure 3. As for the dipolar coefficient corresponding to the mixed electrolyte, it always lies in between the two binary cases closer to LiCl than to CsCl as expected.
The Standard + BMCSL theory predicts a very strong increase of the low frequency dipolar coefficient value as compared to the standard model. The main reason for this is the increment of the diffuse double layer thickness,
Figure 1, that increases the distance of the charged fluid away from the zero-velocity boundary condition at the particle surface. This leads to an enhancement of the convective flow contribution to its dipolar coefficient [
25]. Besides this difference, the Standard + BMCSL low-frequency dipolar coefficient behavior is similar to that of the standard model.
At higher frequencies (10
6–10
7 Hz), above the low-frequency dielectric dispersion region, the concentration polarization vanishes, and the charged particle behaves essentially as a conductive sphere inside a conductive medium with different conductivity value. Its dipolar coefficient tends to the classical value:
where
is the equivalent conductivity of the insulating particle of radius
surrounded by a conducting layer with surface conductivity
[
26]. For highly-charged particles, the dipolar coefficient tends to a maximum value
d → 1, that is far from being attained in the considered case,
Figure 3. The standard model curves in this figure clearly show the increment of the surface conductivity with the counterion diffusion coefficient. As for the BMCSL model, it predicts a much stronger increment that further depends on the finite ionic sizes that increase the diffuse double layer thickness.
Figure 3 clearly shows that the highest surface conductivity value corresponds to the binary LiCl electrolyte in which the lowest diffusion coefficient of the Li
+ counterion is compensated by the largest diffuse double layer thickness,
Figure 1.
At even higher frequencies (10
9–10
10 Hz), the field induced surface charge densities can no longer build up so that only polarization charges contribute to the dipolar coefficient. The charged particle behaves, therefore, as a low permittivity insulating sphere in a high permittivity medium. Its classical dipole coefficient value is:
As expected, the imaginary part of the dipolar coefficient tends to zero at low frequencies independently of the considered model. Moreover, another feature that can only be appreciated in a Log-Log plot is that becomes proportional to the frequency when . This property will be used in the interpretation of the suspension permittivity. At higher frequencies, each curve attains a maximum with a value that is proportional to the relaxation amplitude of the real part of the dipolar coefficient, as expected. At high frequencies, the standard model predicts the highest relaxation frequency for CsCl and the lowest for LiCl as expected in view of the higher diffusion coefficient of Cs+ as compared to Li+. On the contrary, according to the Standard + BMCSL model, this qualitative behavior is reversed showing again that the surface conductivity of the particle immersed in binary LiCl is higher than in CsCl because the effect of the greater double layer thickness in the former case outweighs that of the higher diffusion coefficient in the latter. Finally, at even higher frequencies, the imaginary part of the dipolar coefficient tends to zero for both considered models, as expected.
The complex conductivity of the suspension can now be deduced from the dipolar coefficient spectra:
where
is the complex conductivity of the electrolyte solution and
is the volume fraction occupied by the particles in the suspension (assumed to be low for Equation (12) to be valid). Combining Equations (8), (12), and (13) leads to the suspension permittivity and conductivity expressions:
These equations make it possible to define the permittivity and conductivity increments, which are independent of the particle volume fraction:
The corresponding spectra are represented in
Figure 5a,b and
Figure 6.
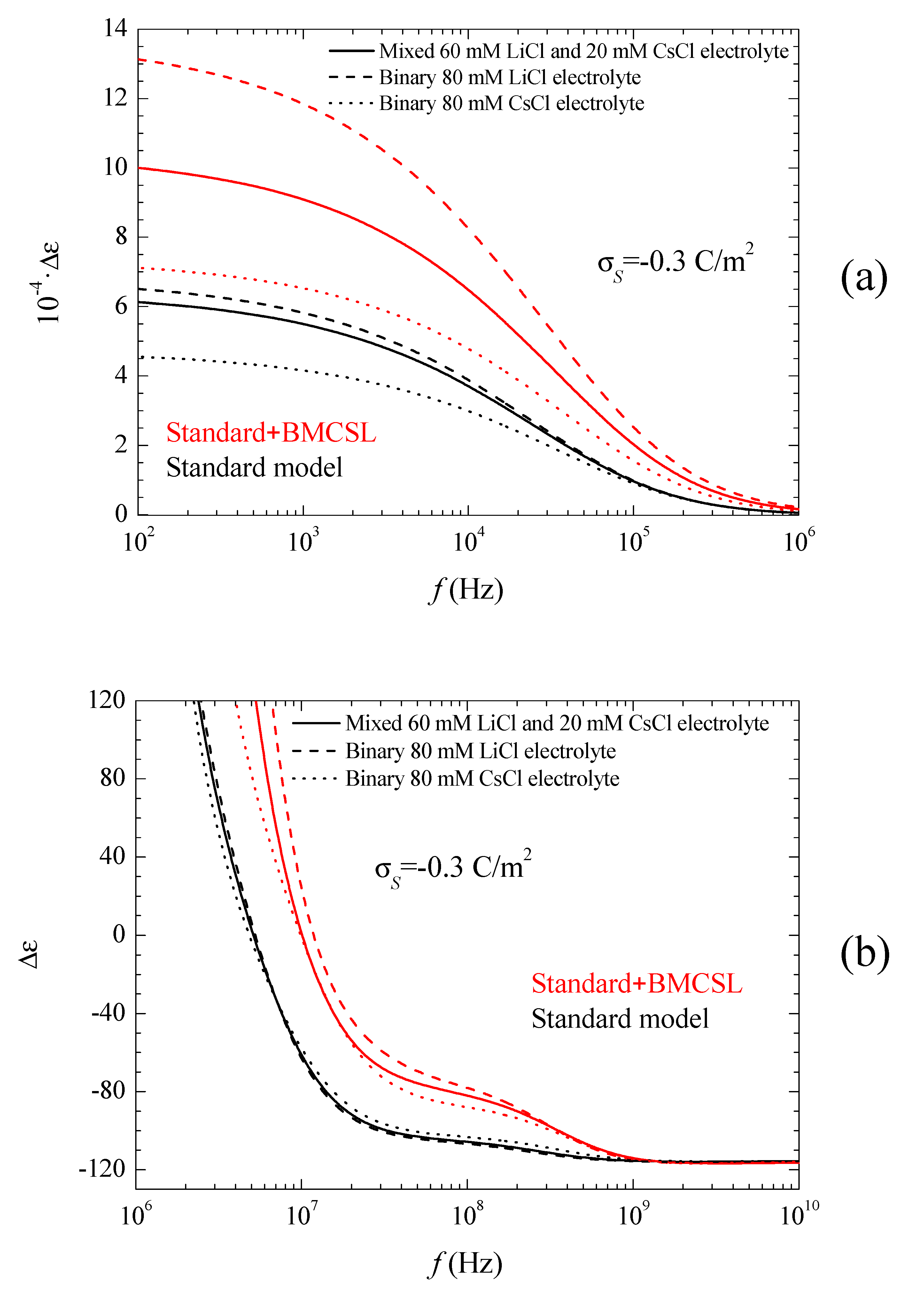
Figure 5a shows the permittivity increment spectra for low frequencies. Obviously, the huge values of the permittivity increment imply that they are mainly due to the second addend in Equation (16): quotient of the imaginary part of the dipolar coefficient and frequency (the imaginary part of the dipolar coefficient is proportional to the frequency for
). As can be seen, according to the Standard model the dielectric increment is highest (lowest) when the counterion diffusion coefficient is lower (higher) than that of the co-ion: LiCl (CsCl). The main reason for this behavior is the increase of the second addend in Equation (16) with the characteristic time of the low-frequency dielectric dispersion, which is determined by the ion with the lowest diffusion coefficient: Li
+ [
27]. As for the Standard + BMCSL predictions, they show the same qualitative behavior as for the standard model except for a much higher amplitude.
This difference is mainly due to the value of the characteristic time of the low-frequency dielectric dispersion that increases with the convective fluid flow around the particle (Equation (38) in Reference [
25]). This is also the reason why the permittivity increment of the mixed electrolyte tends to that of the binary CsCl electrolyte for large surface charges: under these conditions the smaller Cs
+ ions expel the larger Li
+ from the diffuse double layer,
Figure 1.
Figure 5b shows the permittivity increment spectra for high frequencies, for which the dispersion amplitude is so small as compared to the low-frequency dielectric dispersion that they would be invisible if
Figure 5a were extended up to 10
10 Hz. This Maxwell–Wagner dispersion confirms the comments following
Figure 3. For the standard model, Equations (9) and (10) show that the highest (lowest) dispersion amplitude corresponds to the CsCl (LiCl) binary electrolyte in view of the high (low) diffusion coefficient of the Cs
+ (Li
+) ion. For the Standard + BMCSL model, the dispersion amplitude is much greater because steric forces among ions increase the thickness of the diffuse double layer leading to a large increase of the surface conductivity. This effect is so strong that it outweighs that determined by the diffusion coefficient value: the dispersion amplitude is highest (lowest) for LiCl (CsCl) binary electrolyte. Finally,
Figure 5b clearly shows the increase of the relaxation frequency predicted by the Standard + BMCSL as compared to the standard model, which is due to the increment of the surface conductivity.
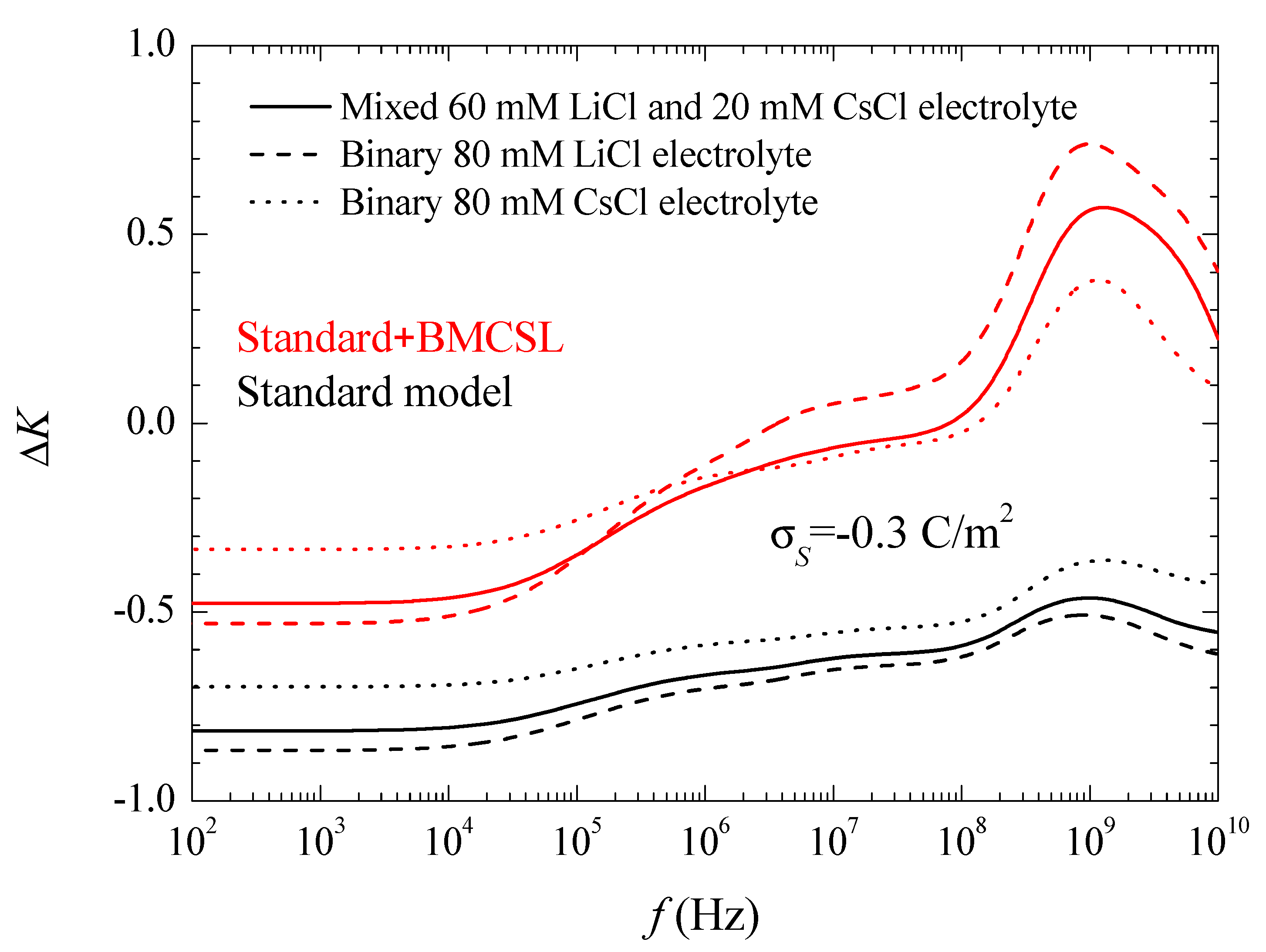
Figure 6 shows the conductivity increment spectra for the considered systems and models. At low frequencies, the imaginary part of the dipolar coefficient in Equation (17) vanishes so that the conductivity increment is just proportional to the real part of the dipolar coefficient,
Figure 3. This dependence remains practically throughout the low-frequency dielectric dispersion range because the second addend in Equation (17) contains the frequency as a factor that is still small. On the contrary, in the high-frequency range corresponding to the Maxwell–Wagner dispersion this factor becomes decisive. The huge increment of the conductivity at high frequencies is almost entirely due to the imaginary part of the dipolar coefficient. Classically, in this frequency range its value should decrease as 1/
so that its product by the frequency leads to a constant high amplitude conductivity increment value:
Actually, all the conductivity increment curves suddenly decrease on the right-hand side of
Figure 6. This is due to inertial effects: the last addend in Equation (4), which contains the fluid mass density and opposes convective fluid velocity changes, becomes non-negligible at the highest frequencies.
3.2.2. Electrophoretic Mobility
The dimensionless electrophoretic mobility is determined calculating the field induced fluid velocity
far away from the suspended particle:
where
is the electrophoretic velocity of the particle.
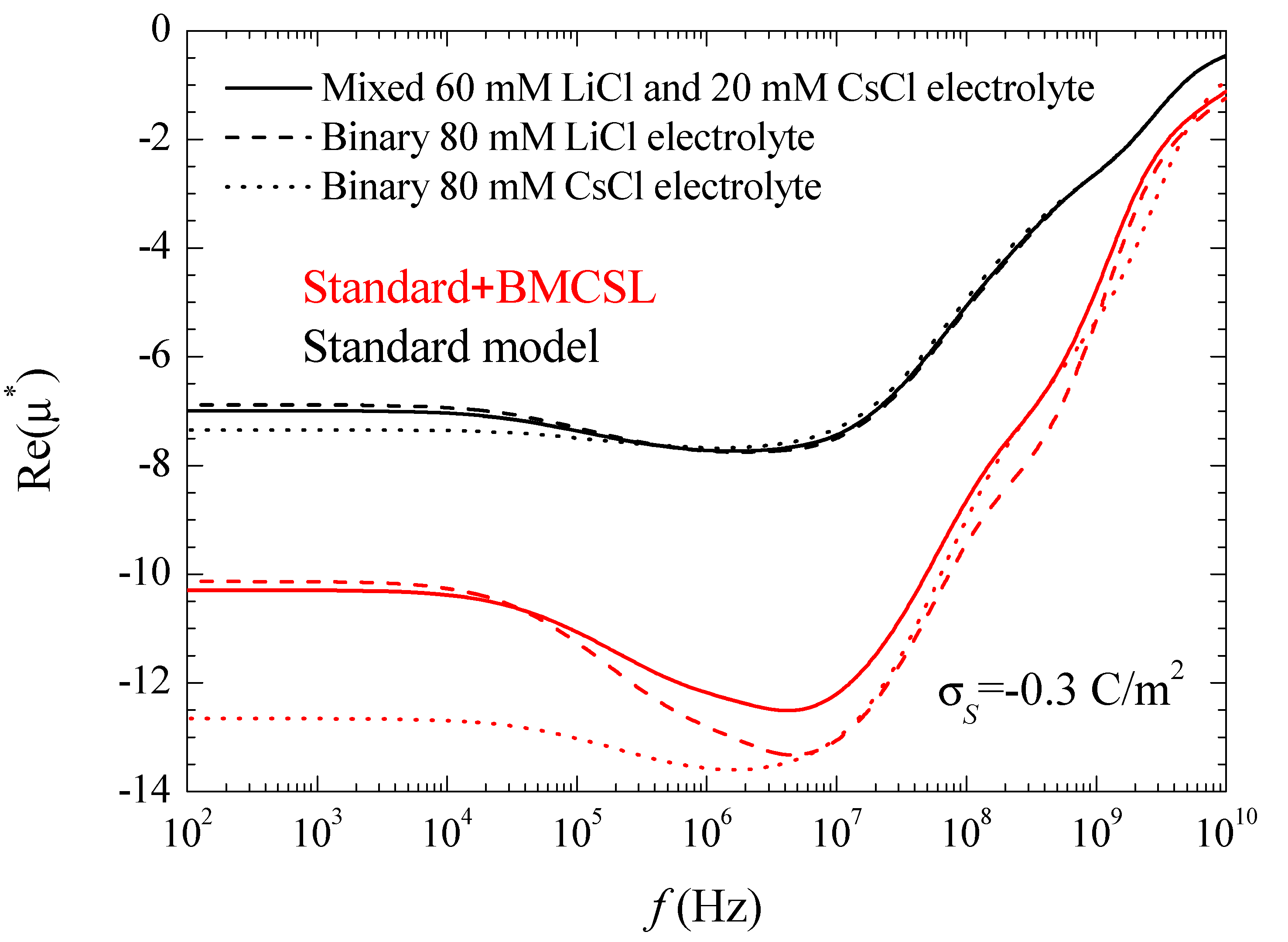
Figure 7 represents the spectra corresponding to the real part of the electrophoretic mobility. This magnitude is determined by the total fluid flow in the diffuse double layer which, at low frequencies, is caused by electroosmosis and capillary osmosis. The first is proportional to the total tangential field in the double layer,
so that, in the considered case, it slightly decreases with frequency,
Figure 3. The second is proportional to the tangential gradient of the electrolyte concentration and opposes the fluid flow [
27]. It decreases with frequency over the low-frequency dielectric dispersion range and vanishes at around 10
6–10
7 Hz.
The increase (in modulus) of the electrophoretic mobility at low frequencies shows that in the considered case the electroosmotic contribution outweighs that of the dipolar coefficient. At higher frequencies, 10
8–10
9 Hz, the dipolar coefficient strongly diminishes,
Figure 3, which should lead to an increase the electrophoretic mobility. This behavior does not appear in
Figure 7, since at these high-frequency values the fluid flow behavior is dominated by inertial effects that lead to a monotonic decrease of the electrophoretic mobility.
As for the different considered electrolytes,
Figure 7 shows the expected behavior predicted by the standard model. At low frequencies, the electrophoretic mobility modulus is highest (lowest) for CsCl (LiCl) in view of the higher (lower) diffusion coefficient of the Cs
+ (Li
+) ion that leads to a higher (lower) fluid velocity [
27]. The behavior predicted by the Standard + BMCSL model is qualitatively similar to that of the standard model but with much greater mobility values (in modulus). Again, this difference originates in the increase of the diffuse double layer thickness with the ionic size,
Figure 1, which increases the fluid flow around the particle.
However, this is not a trivial conclusion as can be seen comparing the results of this work with those obtained considering the standard model with the presence of stagnant layer conductivity [
28]. As can be seen, there is a striking similarity between the dipolar coefficient and the permittivity and conductivity increment spectra when the surface conductivity is enhanced by either an increase of the double layer thickness or the presence of an anomalous surface conductivity. On the contrary, the mobility results are completely different: this magnitude increases with the ionic size but decreases with the anomalous surface conductivity. The reason for this contrasting behavior is that the dielectric response depends on the electric current density while the electrophoretic mobility depends on the fluid flow. Both magnitudes increase with the ionic size while the anomalous surface conductivity only increases the current density since fluid flow is not allowed inside the stagnant layer. Therefore, the current density in the stagnant layer decreases the (1 −
d*) coefficient leading to a decrement of the electrophoretic mobility.
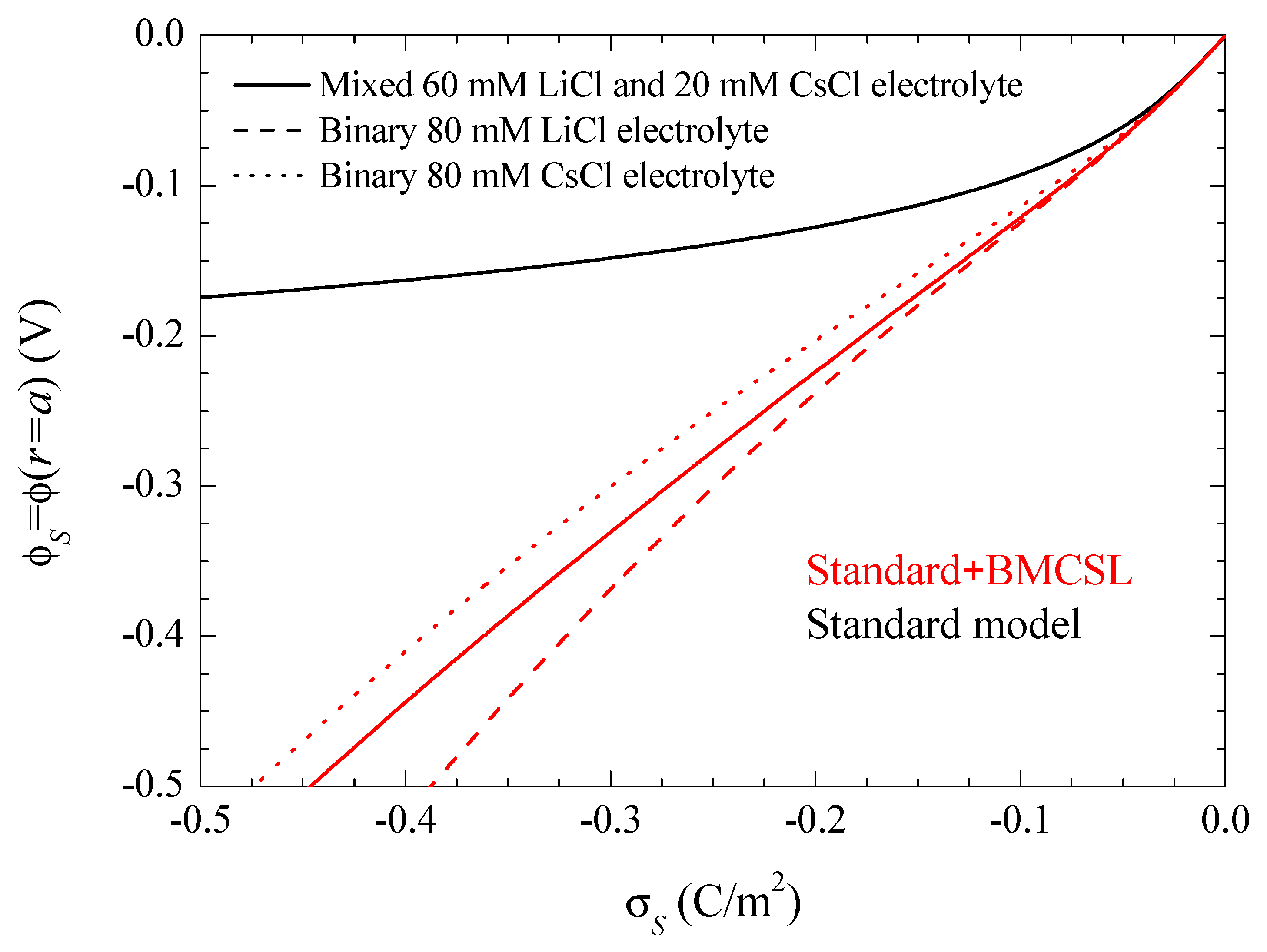
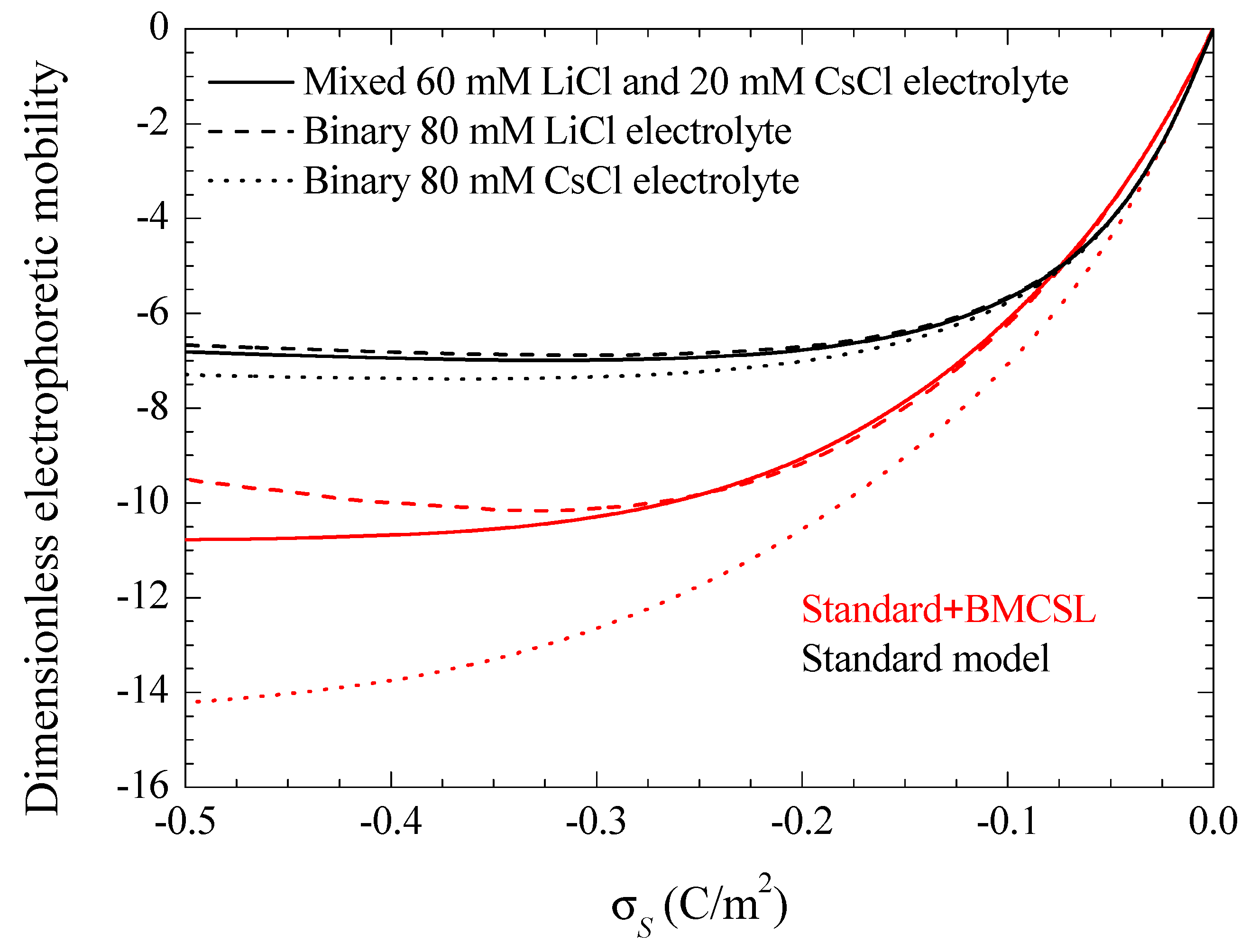
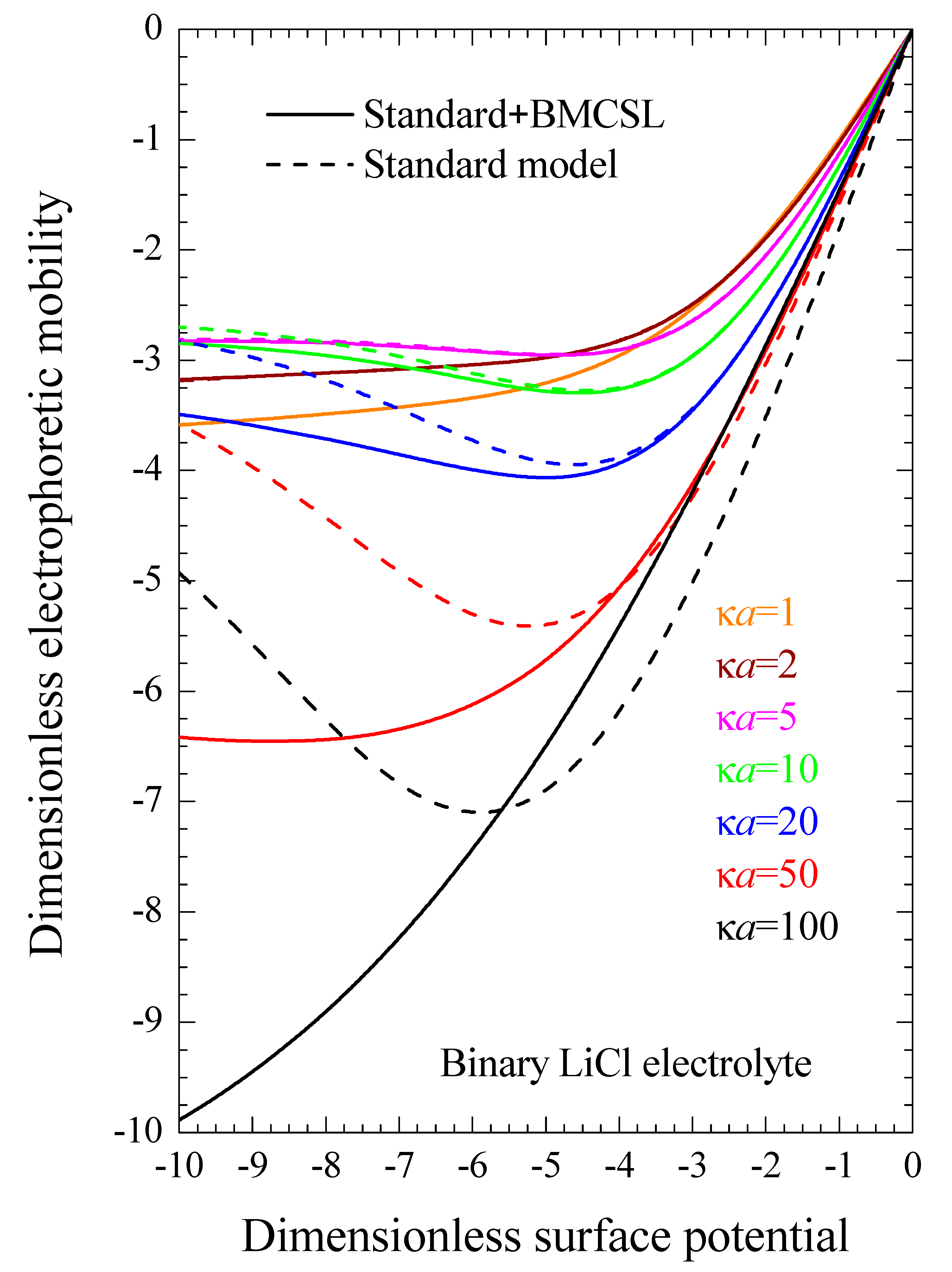
Figure 8 represents the electrophoretic mobility value calculated in the limit
as a function of the surface charge. The standard model behavior corresponds to the well-known results calculated in Reference [
29], except for the figure inversion due to the negative sign of the particle charge and to the use of its surface charge rather than its surface potential as an independent variable. The rather weak differences among the three plotted curves are due to the diffusion coefficient values: the fluid flow in the diffuse double layer increases with the counterion diffusion coefficient so that the electrophoretic mobility is highest (in absolute value) for CsCl, lowest for LiCl, and intermediate for the mixed electrolyte solution. The Standard + BMCSL model leads to a strong increase (in absolute value) of the electrophoretic mobility. Again, the main reason for this behavior is the greater thickness of the diffuse double layer that increases the distance of the charged fluid away from the zero-velocity boundary condition at the particle surface. However, two competing effects are present in this case: the Cs
+ (Li
+) ion has a high (low) diffusion coefficient but a small (large) size that corresponds to a thin (thick) diffuse double layer. For weakly charged particles, the second effect outweighs the first so that the mixed electrolyte closely follows the binary LiCl behavior. On the contrary, for highly-charged particles, the Li
+ ions are practically expelled from the interface neighborhood,
Figure 1, because of which the mixed electrolyte tends to the binary CsCl behavior.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}