Mitochondrial RNA Expression and Single Nucleotide Variants in Association with Clinical Parameters in Primary Breast Cancers

Abstract
:1. Introduction
2. Results
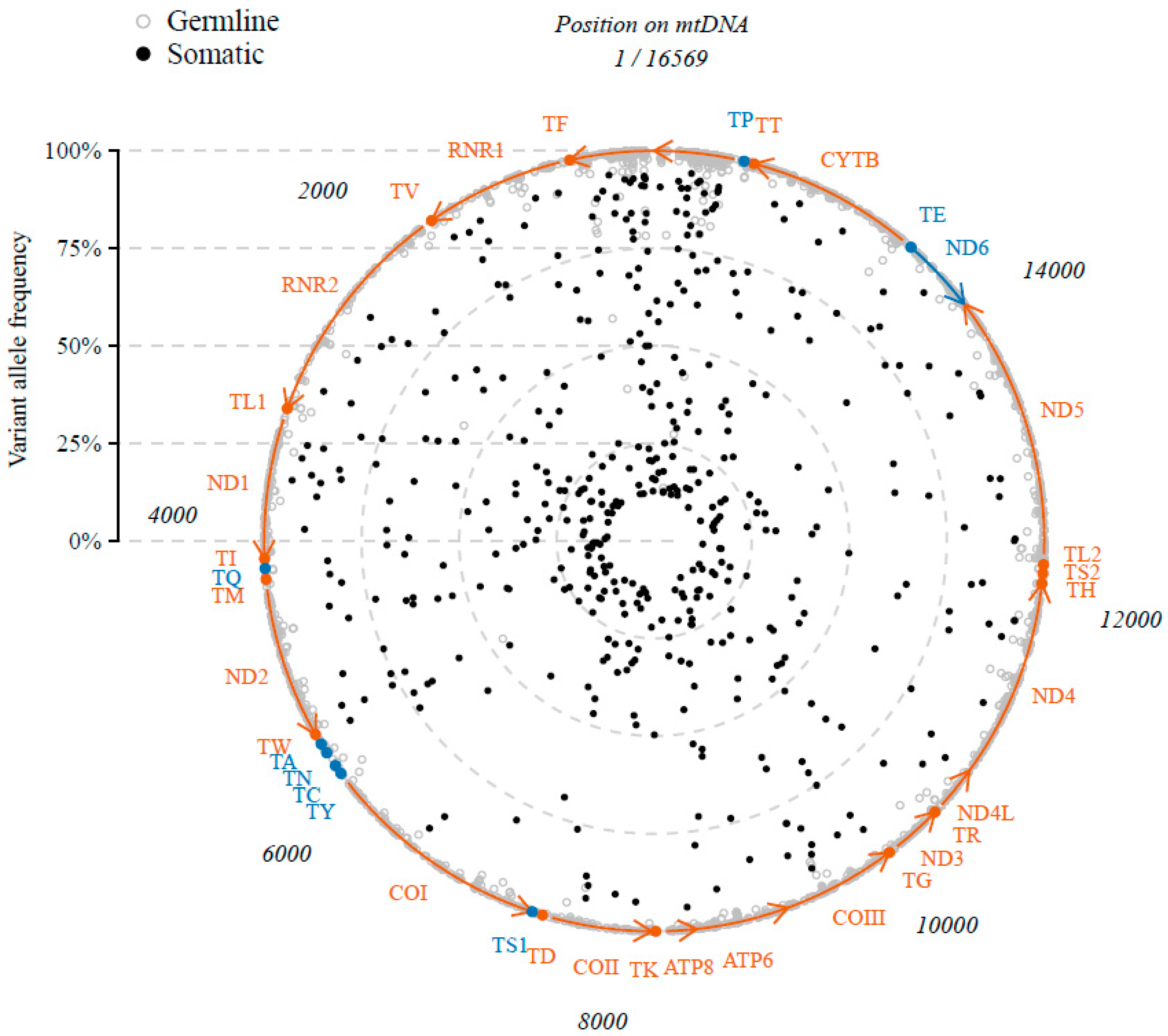
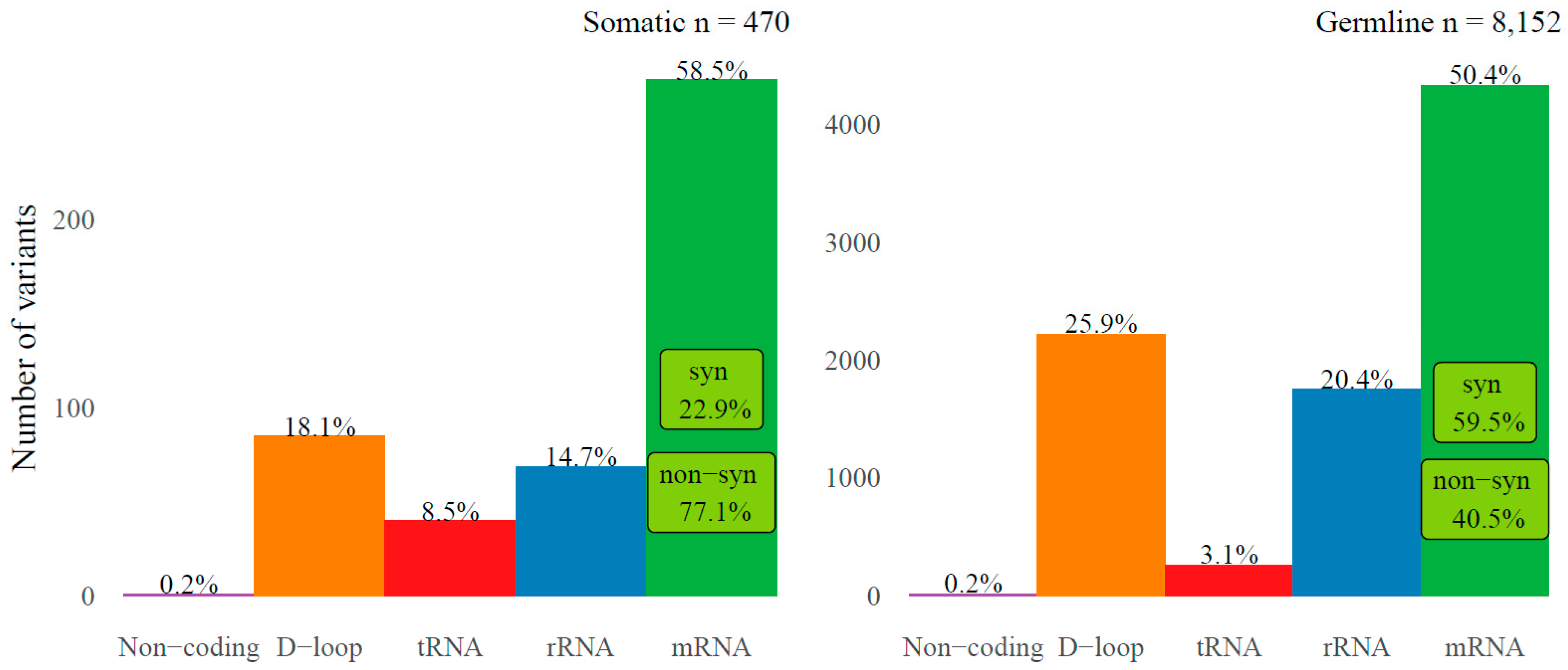
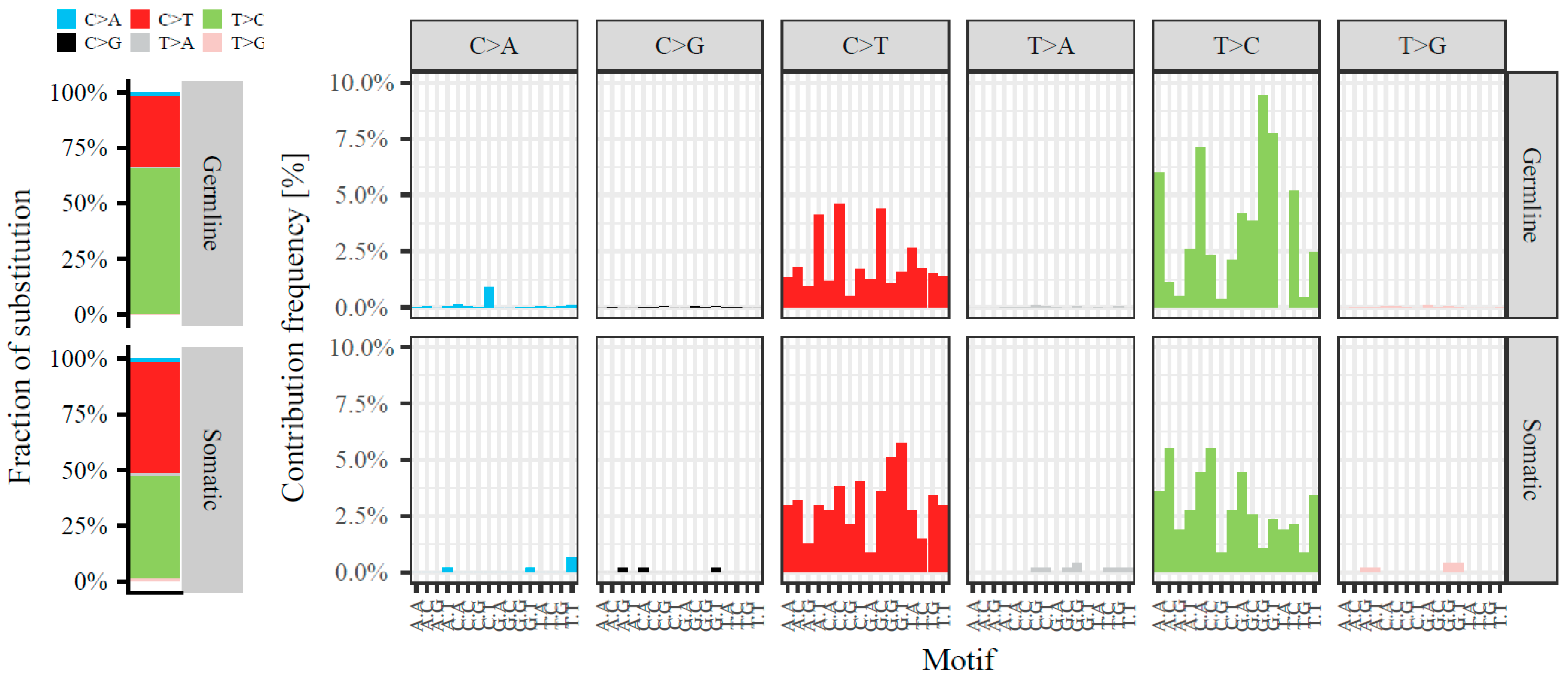
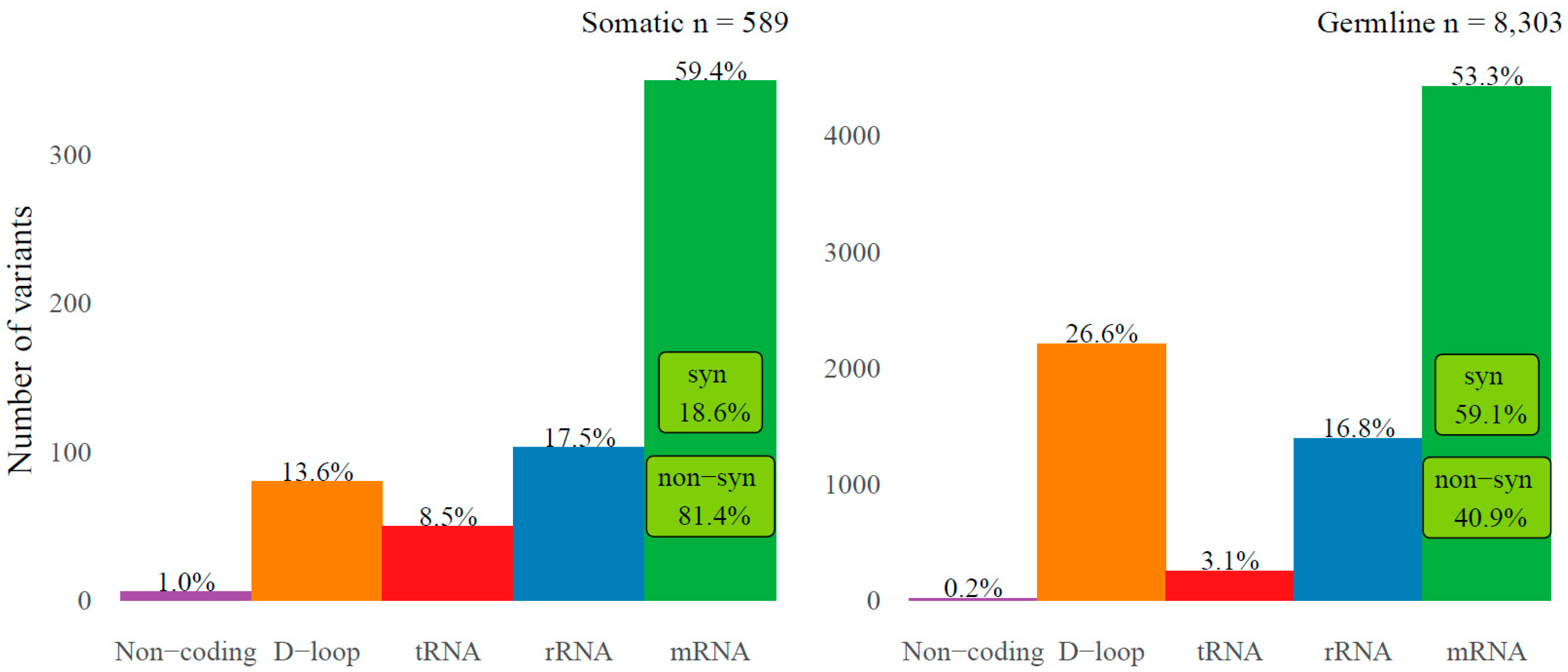
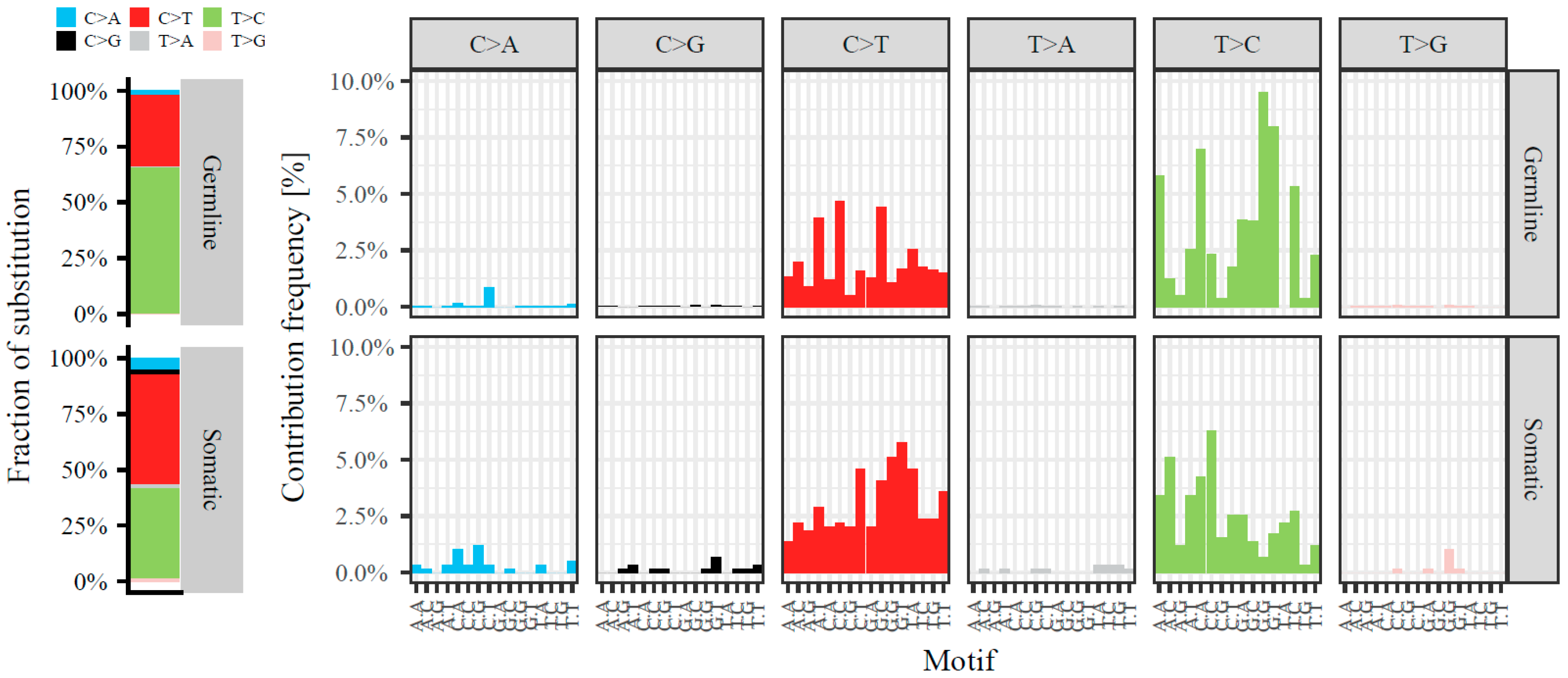
2.1. Somatic Variants in mtRNA
2.2. Somatic Mitochondrial Variants in Relation to Somatic Variants in the Nuclear Genome
2.3. Mitochondrial Gene Expression
2.4. Association with Clinicopathological Parameters
3. Discussion
4. Materials and Methods
4.1. Data
4.2. Bioinformatics
4.3. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
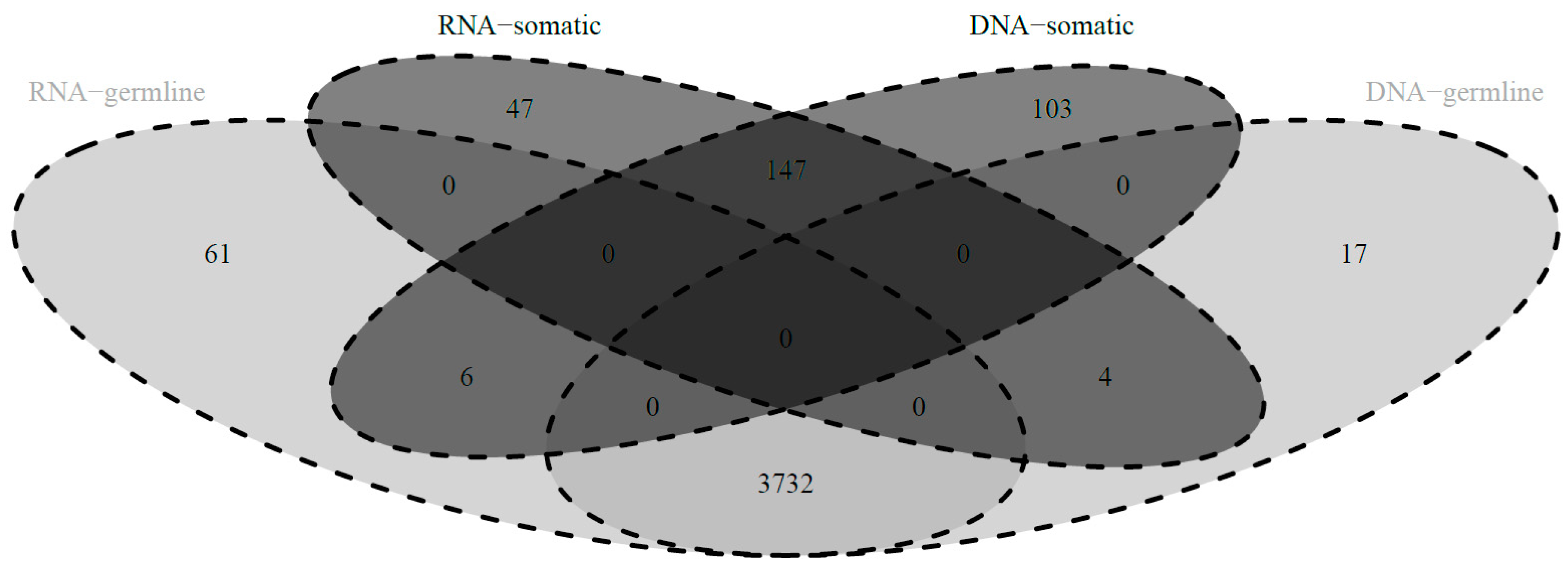
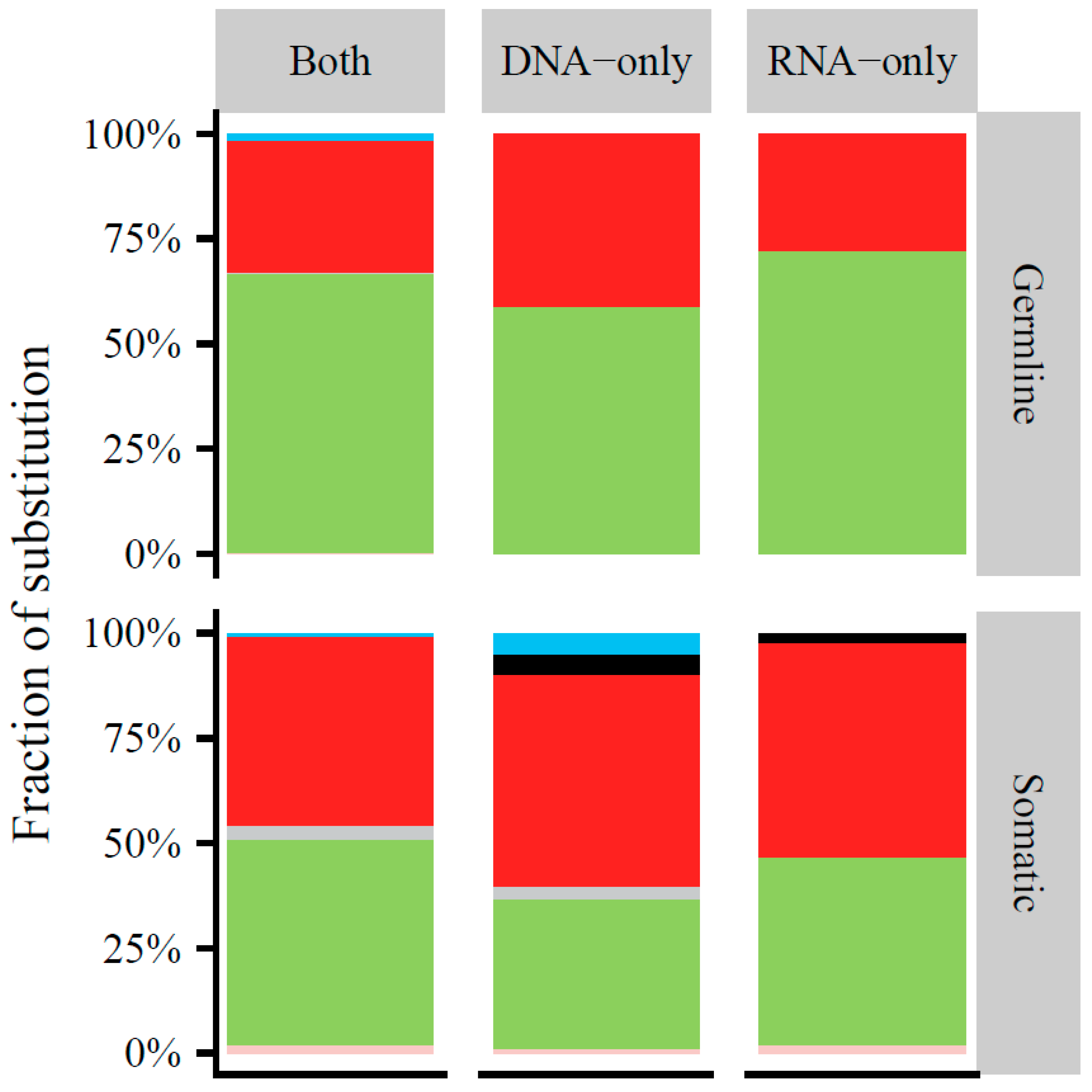
Appendix A.1. Comparison of Mitochondrial Variants at the RNA and DNA Level




Appendix A.2. Direct Comparison of Mitochondrial Variants at the RNA and DNA Level of Overlapping Cases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Variant | Depth RNA Tumor | VAF RNA Tumor | Class RNA | Variant | Depth DNA Tumor | VAF DNA Tumor | Depth DNA Normal | VAF DNA Normal | Class DNA |
|---|---|---|---|---|---|---|---|---|---|---|
| P_6042a | r.199u>c | 1953 | 99.3% | Germline | g.199T>C | 6585 | 67.1% | 208 | 0.5% | Somatic |
| P_9571a | r.1010a>c | 1655 | 97.1% | Germline | g.1010A>C | 21,840 | 94.9% | 763 | 0.0% | Somatic |
| P_6409a | r.4344u>c | 146 | 97.9% | Germline | g.4344T>C | 16,068 | 68.9% | 1707 | 0.1% | Somatic |
| P_6043a | r.5353g>a | 1099 | 97.6% | Germline | g.5353G>A | 20,514 | 88.0% | 544 | 0.0% | Somatic |
| P_5956a | r.11453g>a | 1655 | 96.6% | Germline | g.11453G>A | 21,214 | 96.3% | 4230 | 0.0% | Somatic |
| P_9568a | r.14841a>g | 1877 | 97.4% | Germline | g.14841A>G | 12,060 | 86.5% | 806 | 0.1% | Somatic |
| P_4982a | r.94g>a | 698 | 91.1% | Somatic | g.94G>A | 9643 | 99.8% | 1449 | 99.5% | Germline |
| P_4982a | r.152u>c | 614 | 93.0% | Somatic | g.152T>C | 11,738 | 99.8% | 2090 | 99.0% | Germline |
| P_8622a | r.497c>u | 39 | 84.6% | Somatic | g.497C>T | 11,020 | 99.8% | 1975 | 99.5% | Germline |
| P_4963a | r.16302a>g | 708 | 94.7% | Somatic | g.16302A>G | 7042 | 97.3% | 7860 | 86.6% | Germline |
| Sample | Variant | Gene | Depth DNA Tumor | VAF DNA Tumor | Depth DNA Normal | VAF DNA Normal | Depth RNA Tumor | VAF RNA Tumor | Concordant? |
|---|---|---|---|---|---|---|---|---|---|
| P_6719a | g.2A>T | Control-Region | 81 | 4.94 | 59 | 0 | 15 | 0.00 | na |
| P_6409a | g.66G>T | Control-Region | 1712 | 3.27 | 465 | 0 | 644 | 0.16 | na |
| P_9569a | g.73A>G | Control-Region | 6118 | 11.43 | 146 | 0 | 1010 | 11.58 | Yes |
| P_6719a | g.185G>A | Control-Region | 3193 | 99.72 | 3421 | 99.18 | 10 | 100.00 | Yes |
| P_9592a | g.195T>C | Control-Region | 7012 | 4.04 | 2672 | 0.04 | 1076 | 3.35 | Yes |
| P_4977a | g.263A>G | Control-Region | 10,036 | 100 | 2142 | 99.91 | 9 | 100.00 | Yes |
| P_9597a | g.293T>C | Control-Region | 2568 | 12.27 | 810 | 0 | 1503 | 7.25 | Yes |
| P_4847a | g.307C>A | Control-Region | 1275 | 22.98 | 2124 | 0.09 | 228 | 0.00 | No |
| P_4958a | g.316G>A | Control-Region | 3513 | 10.65 | 698 | 2.72 | 376 | 3.99 | Yes |
| P_5947a | g.319T>C | Control-Region | 2942 | 4.11 | 1185 | 0.25 | 1015 | 26.21 | Yes |
| P_5947a | g.321T>C | Control-Region | 3184 | 4.74 | 1227 | 0.08 | 1017 | 26.16 | Yes |
| P_4847a | g.346T>C | Control-Region | 1557 | 31.34 | 2300 | 0.04 | 304 | 37.17 | Yes |
| P_4847a | g.347G>A | Control-Region | 1492 | 31.37 | 2030 | 0.05 | 305 | 35.08 | Yes |
| P_11340a | g.456C>T | Control-Region | 8351 | 99.8 | 462 | 99.78 | 7 | 100.00 | Yes |
| P_9571a | g.462C>T | Control-Region | 17,653 | 99.93 | 584 | 99.83 | 0 | - | Yes |
| P_6719a | g.462C>T | Control-Region | 5300 | 99.87 | 5718 | 99.88 | 4 | 100.00 | Yes |
| P_4069a | g.462C>T | Control-Region | 9108 | 99.89 | 1760 | 100 | 21 | 95.24 | Yes |
| P_9571a | g.489T>C | Control-Region | 18,133 | 99.98 | 619 | 100 | 2 | 100.00 | Yes |
| P_6719a | g.489T>C | Control-Region | 5798 | 99.91 | 5977 | 99.97 | 4 | 100.00 | Yes |
| P_4069a | g.489T>C | Control-Region | 9408 | 99.97 | 1766 | 100 | 18 | 100.00 | Yes |
| P_6422a | g.549C>T | Control-Region | 15,224 | 99.76 | 9454 | 99.27 | 15 | 93.33 | Yes |
| P_5928a | g.730A>T | MT-RNR1 | 12,334 | 12.93 | 395 | 0 | 5056 | 4.94 | Yes |
| P_11389a | g.903T>C | MT-RNR1 | 11,568 | 4.76 | 1006 | 0 | 5053 | 0.26 | Yes |
| P_6413a | g.1284T>C | MT-RNR1 | 8416 | 25.5 | 2589 | 0.04 | 5030 | 1.83 | Yes |
| P_11399a | g.1320G>A | MT-RNR1 | 6173 | 4.47 | 1995 | 0 | 5039 | 0.02 | No* |
| P_4845a | g.1464G>A | MT-RNR1 | 6496 | 12.96 | 7902 | 0.19 | 5018 | 6.50 | Yes |
| P_6719a | g.1748G>A | MT-RNR2 | 10,396 | 25.31 | 9000 | 0.06 | 4674 | 10.68 | Yes |
| P_9754a | g.1758T>C | MT-RNR2 | 10,021 | 3.42 | 2541 | 0.04 | 5035 | 1.81 | Yes |
| P_8618a | g.1906G>C | MT-RNR2 | 8094 | 3.43 | 5953 | 0.03 | 5038 | 0.02 | No* |
| P_11384a | g.1913G>A | MT-RNR2 | 8494 | 4.07 | 2090 | 0.05 | 5031 | 0.58 | Yes |
| P_9592a | g.1939G>A | MT-RNR2 | 16,324 | 3.59 | 6432 | 0.06 | 5022 | 2.59 | Yes |
| P_6413a | g.1987G>A | MT-RNR2 | 8817 | 11.47 | 2498 | 0.04 | 4972 | 1.57 | Yes |
| P_11380a | g.2024C>T | MT-RNR2 | 12,996 | 14.06 | 647 | 0 | 5024 | 2.31 | Yes |
| P_11377a | g.2343G>A | MT-RNR2 | 7852 | 3.48 | 1118 | 0 | 5049 | 0.04 | No* |
| P_9572a | g.2492G>A | MT-RNR2 | 13,639 | 3.43 | 671 | 0.15 | 5049 | 5.27 | Yes |
| P_7221a | g.2571G>A | MT-RNR2 | 8410 | 3.22 | 2488 | 0.08 | 5007 | 3.40 | Yes |
| P_11374a | g.2695G>A | MT-RNR2 | 4771 | 3.98 | 1416 | 0 | 5045 | 0.71 | Yes |
| P_4976a | g.2716G>A | MT-RNR2 | 21,489 | 7.3 | 18,243 | 0.06 | 4996 | 4.06 | Yes |
| P_5950a | g.3065T>C | MT-RNR2 | 7371 | 5.24 | 1548 | 0 | 4870 | 3.72 | Yes |
| P_8980a | g.3068G>A | MT-RNR2 | 15,125 | 12.65 | 3374 | 0.12 | 4997 | 4.16 | Yes |
| P_9573a | g.3097T>C | MT-RNR2 | 10,249 | 9.23 | 592 | 0.17 | 5036 | 5.90 | Yes |
| P_7215a | g.3617T>C | MT-ND1 | 12,614 | 3.24 | 2196 | 0 | 1400 | 4.07 | Yes |
| P_11375a | g.3715G>C | MT-ND1 | 9098 | 3.7 | 339 | 0.59 | 4889 | 1.82 | Yes |
| P_4080a | g.4153G>A | MT-ND1 | 14,450 | 7.7 | 2259 | 0.04 | 53 | 3.77 | Yes |
| P_6411a | g.4308G>A | MT-TI | 13,780 | 3.72 | 1097 | 0 | 738 | 6.91 | Yes |
| P_7218a | g.4336T>C | MT-TQ | 11,432 | 99.9 | 2181 | 100 | 3 | 66.67 | Yes |
| P_8979a | g.4399T>C | MT-TQ | 7338 | 20.37 | 1572 | 0 | 29 | 20.69 | Yes |
| P_4833a | g.4412G>A | MT-TM | 10,532 | 5.86 | 4078 | 0.02 | 1039 | 87.20 | Yes |
| P_4072a | g.4429G>A | MT-TM | 16,360 | 55.94 | 2245 | 0.04 | 548 | 85.22 | Yes |
| P_9777a | g.4582T>C | MT-ND2 | 3336 | 17.99 | 2439 | 0 | 19 | 0.00 | na |
| P_11819a | g.4924G>A | MT-ND2 | 7357 | 9.11 | 4036 | 0.05 | 3760 | 6.97 | Yes |
| P_4080a | g.5581A>G | Non-Coding | 17,806 | 99.66 | 2669 | 99.96 | 0 | - | Yes |
| P_6728a | g.5582A>G | Non-Coding | 13,659 | 99.34 | 11,657 | 99.91 | 9 | 100.00 | Yes |
| P_4982a | g.5703G>A | MT-TN | 13,401 | 91.4 | 2276 | 0.04 | 1963 | 89.61 | Yes |
| P_4971a | g.5920G>A | MT-CO1 | 6202 | 3.58 | 1517 | 0.07 | 4376 | 7.24 | Yes |
| P_11340a | g.6255G>A | MT-CO1 | 11,906 | 6.16 | 824 | 0 | 1638 | 9.77 | Yes |
| P_6422a | g.6673T>C | MT-CO1 | 20,090 | 4.39 | 12,278 | 0.02 | 1080 | 5.28 | Yes |
| P_9574a | g.6724T>C | MT-CO1 | 10,549 | 6.03 | 871 | 0 | 5018 | 6.10 | Yes |
| P_9574a | g.7191T>C | MT-CO1 | 11,204 | 6.12 | 932 | 0.11 | 5024 | 5.77 | Yes |
| P_11377a | g.7207G>A | MT-CO1 | 8421 | 5.85 | 1093 | 0 | 5036 | 2.76 | Yes |
| P_7214a | g.7219G>A | MT-CO1 | 11,168 | 3.36 | 2565 | 0 | 4983 | 3.45 | Yes |
| P_4080a | g.7595G>A | MT-CO2 | 17,880 | 20.37 | 2789 | 0.07 | 3 | 0.00 | na |
| P_7219a | g.7652T>C | MT-CO2 | 9000 | 7.7 | 2654 | 0.04 | 4787 | 7.65 | Yes |
| P_11336a | g.7935T>C | MT-CO2 | 9586 | 5.42 | 594 | 0.34 | 4985 | 7.36 | Yes |
| P_9592a | g.8213G>A | MT-CO2 | 15,889 | 3.63 | 6009 | 0.02 | 4964 | 3.28 | Yes |
| P_4967a | g.8249G>A | MT-CO2 | 13,941 | 3.21 | 7740 | 0.03 | 5031 | 0.85 | Yes |
| P_9572a | g.8269G>C | MT-CO2 | 6539 | 3.17 | 404 | 1.24 | 4858 | 0.00 | No |
| P_11372a | g.8270C>T | Non-Coding | 1447 | 98.48 | 184 | 96.2 | 4755 | 99.75 | Yes |
| P_9002a | g.8278C>G | Non-Coding | 2694 | 15.55 | 75 | 6.67 | 3092 | 0.00 | No |
| P_9572a | g.8290G>C | Non-Coding | 6059 | 5.73 | 370 | 2.43 | 96 | 1.04 | na |
| P_9572a | g.8291A>C | Non-Coding | 6217 | 4.68 | 382 | 2.36 | 93 | 5.38 | Yes |
| P_3989a | g.8448T>C | MT-ATP8 | 11,980 | 8.68 | 5300 | 0.02 | 5042 | 1.23 | Yes |
| P_7214a | g.8547T>C | MT-ATP8/6 | 7924 | 3.14 | 1835 | 0.05 | 3949 | 0.20 | Yes |
| P_4977a | g.8860A>G | MT-ATP6 | 13,075 | 99.99 | 3626 | 99.97 | 46 | 97.83 | Yes |
| P_9754a | g.9053G>A | MT-ATP6 | 10,819 | 4.92 | 2532 | 0 | 5011 | 3.47 | Yes |
| P_9001a | g.9078T>C | MT-ATP6 | 14,729 | 5.89 | 497 | 0.2 | 2045 | 3.03 | Yes |
| P_4958a | g.9181A>G | MT-ATP6 | 9010 | 39.35 | 2221 | 0 | 4870 | 49.96 | Yes |
| P_5936a | g.9285A>T | MT-CO3 | 6732 | 11.51 | 3408 | 0.03 | 4916 | 6.96 | Yes |
| P_6413a | g.9286T>C | MT-CO3 | 6771 | 11.79 | 1989 | 0.2 | 4904 | 5.69 | Yes |
| P_11337a | g.9429G>A | MT-CO3 | 16,236 | 3.47 | 404 | 0 | 4975 | 3.50 | Yes |
| P_5960a | g.9497T>C | MT-CO3 | 10,947 | 3.27 | 2603 | 0 | 4961 | 2.80 | Yes |
| P_11336a | g.9594C>T | MT-CO3 | 10,659 | 5.65 | 623 | 0.16 | 4845 | 5.49 | Yes |
| P_9567a | g.9645G>A | MT-CO3 | 11,647 | 6.36 | 700 | 0.14 | 5021 | 3.94 | Yes |
| P_9847a | g.10177G>A | MT-ND3 | 22,790 | 3.05 | 18,685 | 0.02 | 3403 | 7.35 | Yes |
| P_7426a | g.10463T>C | MT-TR | 9817 | 99.98 | 1300 | 99.92 | 7 | 100.00 | Yes |
| P_9757a | g.10747T>C | MT-ND4L | 6998 | 7.82 | 2229 | 0.04 | 4963 | 10.50 | Yes |
| P_11336a | g.10838A>G | MT-ND4 | 10,950 | 9.52 | 617 | 0 | 5006 | 5.81 | Yes |
| P_5936a | g.11195G>A | MT-ND4 | 7126 | 8.73 | 3615 | 0.03 | 4923 | 7.39 | Yes |
| P_9582a | g.11477G>A | MT-ND4 | 11,726 | 3.77 | 12,886 | 0.02 | 4963 | 5.00 | Yes |
| P_7206a | g.11825G>A | MT-ND4 | 12,293 | 3.54 | 1094 | 0.09 | 4999 | 5.82 | Yes |
| P_4266a | g.11984T>C | MT-ND4 | 14,006 | 13.17 | 8778 | 0.03 | 4951 | 9.41 | Yes |
| P_9755a | g.12154C>T | MT-TH | 8116 | 3.7 | 1603 | 0.06 | 263 | 8.75 | Yes |
| P_7221a | g.12618G>A | MT-ND5 | 9056 | 6.1 | 2555 | 0.04 | 2853 | 8.24 | Yes |
| P_8981a | g.12769G>A | MT-ND5 | 10,621 | 7.06 | 3108 | 0.03 | 4976 | 8.16 | Yes |
| P_5930a | g.12771G>A | MT-ND5 | 10,265 | 11.83 | 3150 | 0 | 4972 | 9.61 | Yes |
| P_6413a | g.12977T>C | MT-ND5 | 7340 | 34.1 | 2123 | 0.09 | 4942 | 2.47 | Yes |
| P_4847a | g.13099G>A | MT-ND5 | 13,805 | 5.16 | 4972 | 0.02 | 4931 | 1.60 | Yes |
| P_7409a | g.13156C>T | MT-ND5 | 8790 | 4.55 | 2599 | 0.04 | 4891 | 7.56 | Yes |
| P_5946a | g.13178G>A | MT-ND5 | 5740 | 8.89 | 2846 | 0.07 | 3483 | 11.08 | Yes |
| P_8979a | g.13198G>A | MT-ND5 | 11,789 | 8.59 | 2741 | 0 | 2877 | 7.92 | Yes |
| P_11336a | g.13272C>A | MT-ND5 | 9660 | 5.48 | 579 | 0 | 4603 | 2.00 | Yes |
| P_8979a | g.13496C>A | MT-ND5 | 10,985 | 5.5 | 2332 | 0.04 | 3916 | 5.80 | Yes |
| P_11374a | g.13531G>A | MT-ND5 | 4731 | 5.14 | 1252 | 0 | 5007 | 3.87 | Yes |
| P_11391a | g.13567A>G | MT-ND5 | 1322 | 23.83 | 856 | 0 | 5009 | 0.86 | Yes |
| P_11372a | g.14112C>A | MT-ND5 | 8553 | 10.1 | 1251 | 0.08 | 4959 | 10.71 | Yes |
| P_8611a | g.14197T>C | MT-ND6 | 10,577 | 5.01 | 4713 | 0.04 | 5005 | 5.97 | Yes |
| P_7215a | g.14447T>C | MT-ND6 | 13,776 | 3.47 | 2242 | 0 | 2321 | 2.76 | Yes |
| P_11374a | g.14760G>A | MT-CYB | 5961 | 19.28 | 1421 | 0.07 | 921 | 0.00 | No |
| P_4959a | g.14788T>C | MT-CYB | 13,250 | 10.17 | 8208 | 0.12 | 4608 | 6.45 | Yes |
| P_9570a | g.14888G>A | MT-CYB | 15,466 | 3.84 | 517 | 0 | 3649 | 5.10 | Yes |
| P_5954a | g.14939T>C | MT-CYB | 8033 | 3.39 | 2633 | 0.04 | 2959 | 6.69 | Yes |
| P_4982a | g.15012T>C | MT-CYB | 22,666 | 8.39 | 4348 | 0.05 | 5016 | 7.93 | Yes |
| P_5950a | g.15093G>A | MT-CYB | 8082 | 5.26 | 1601 | 0 | 5037 | 7.72 | Yes |
| P_9582a | g.15170G>A | MT-CYB | 11,759 | 3.46 | 13,082 | 0.02 | 3899 | 9.69 | Yes |
| P_11342a | g.15242G>A | MT-CYB | 11,745 | 3.32 | 373 | 0 | 4339 | 6.04 | Yes |
| P_9582a | g.15854T>C | MT-CYB | 10,545 | 6.89 | 12,711 | 0.06 | 3895 | 7.01 | Yes |
| P_4977a | g.15970T>C | MT-TP | 19,783 | 99.96 | 5232 | 100 | 3 | 100.00 | Yes |
| P_4847a | g.16033G>A | Control-Region | 14,332 | 7.59 | 5556 | 0.02 | 2732 | 0.48 | Yes |
| P_7433a | g.16147C>T | Control-Region | 15,174 | 11 | 3874 | 0.03 | 2873 | 1.98 | Yes |
| P_9539a | g.16293A>G | Control-Region | 2683 | 9.58 | 470 | 0.21 | 3315 | 9.20 | Yes |
| Sample | Variant | Gene | Class | Depth RNA Tumor | VAF RNA Tumor | Comment |
|---|---|---|---|---|---|---|
| P_4982a | r.72u>c | Control-Region | Somatic | 318 | 20.44 | True variant (mutually exclusive 73G, 94T) |
| P_6406a | r.72u>c | Control-Region | Germline | 1906 | 99.79 | True variant |
| P_9589a | r.73a>g | Control-Region | Germline | 592 | 99.83 | True variant |
| P_5959a | r.73a>g | Control-Region | Germline | 954 | 100.00 | True variant |
| P_11394a | r.73a>g | Control-Region | Germline | 923 | 100.00 | True variant |
| P_11389a | r.146u>c | Control-Region | Germline | 694 | 99.71 | True variant |
| P_8978a | r.146u>c | Control-Region | Germline | 371 | 99.73 | True variant (phased with 185A and 204C) |
| P_8611a | r.146u>c | Control-Region | Germline | 1599 | 99.75 | True variant (phased with 195C) |
| P_8981a | r.146u>c | Control-Region | Germline | 308 | 100.00 | True variant |
| P_8609a | r.152u>c | Control-Region | Somatic | 1448 | 69.96 | True variant (phased with 195C) |
| P_10014a | r.152u>c | Control-Region | Germline | 2272 | 95.38 | True variant |
| P_4606a | r.152u>c | Control-Region | Germline | 98 | 98.98 | True variant |
| P_4266a | r.152u>c | Control-Region | Germline | 956 | 99.37 | True variant |
| P_8618a | r.152u>c | Control-Region | Germline | 437 | 99.77 | True variant |
| P_8979a | r.152u>c | Control-Region | Germline | 371 | 100.00 | True variant |
| P_4261a | r.182c>u | Control-Region | Somatic | 1220 | 77.54 | True variant |
| P_11383a | r.185g>a | Control-Region | Germline | 301 | 99.67 | True variant (phased with 150T and 228A) |
| P_5928a | r.185g>a | Control-Region | Germline | 723 | 100.00 | True variant (phased with 188G and 228A) |
| P_9592a | r.188a>g | Control-Region | Germline | 814 | 99.63 | True variant (phased with 185A and 228A) |
| P_5928a | r.188a>g | Control-Region | Germline | 716 | 100.00 | True variant (phased with 185A and 228A) |
| P_5956a | r.188a>g | Control-Region | Germline | 1450 | 100.00 | True variant (phased with 185A and 228A) |
| P_9571a | r.188a>g | Control-Region | Germline | 37 | 100.00 | True variant (phased with 185A, 222T and 228A) |
| P_4069a | r.189a>g | Control-Region | Germline | 997 | 100.00 | True variant |
| P_11372a | r.195u>c | Control-Region | Germline | 715 | 99.72 | True variant (phased with 152C and 263G) |
| P_8978a | r.228g>a | Control-Region | Somatic | 573 | 82.72 | True variant (phased with 185A, 204C, 263G) |
| P_11383a | r.228g>a | Control-Region | Germline | 585 | 99.83 | True variant (phased with 185A, 263G, 295T) |
| P_9597a | r.263a>g | Control-Region | Germline | 2341 | 99.83 | True variant (phased with 228A and 295T) |
| P_10010a | r.263a>g | Control-Region | Germline | 1331 | 99.85 | True variant |
| P_7238a | r.263a>g | Control-Region | Germline | 1388 | 99.86 | True variant (phased with 295T) |
| P_8611a | r.263a>g | Control-Region | Germline | 1948 | 99.95 | True variant (phased with 195C) |
| P_8830a | r.263a>g | Control-Region | Germline | 892 | 100.00 | True variant (phased with 207A and 234G) |
| P_7238a | r.295c>u | Control-Region | Germline | 766 | 99.48 | True variant (phased with 263G) |
| P_5956a | r.295c>u | Control-Region | Germline | 1121 | 99.91 | True variant (phased with 263G) |
| P_6732a | r.295c>u | Control-Region | Germline | 545 | 100.00 | True variant |
| P_9597a | r.295c>u | Control-Region | Germline | 1272 | 100.00 | True variant (phased with 228A and 263G) |
| P_7316a | r.456c>u | Control-Region | Germline | 88 | 95.45 | True variant |
| P_9758a | r.1604g>a | MT-TV | Somatic | 159 | 23.27 | True variant |
| P_11399a | r.1669g>a | MT-TV | Somatic | 534 | 41.20 | True variant |
| P_6730a | r.1973g>a | MT-RNR2 | Somatic | 1171 | 57.05 | True variant |
| P_4977a | r.2166c>u | MT-RNR2 | Somatic | 1433 | 18.42 | Potential artefact; at start of read ACCxATA context |
| P_10010a | r.2300g>a | MT-RNR2 | Somatic | 2066 | 13.84 | True variant (in DNA! 2300G>A, 9106|5497|37.64%) |
| P_7431a | r.2416u>c | MT-RNR2 | Somatic | 855 | 84.80 | True variant |
| P_6406a | r.3109u>c | MT-RNR2 | Somatic | 1407 | 38.38 | True variant |
| P_4963a | r.3283g>a | MT-TL1 | Somatic | 908 | 29.52 | True variant |
| P_4606a | r.3535u>c | MT-ND1 | Somatic | 2203 | 92.74 | True variant |
| P_4080a | r.3705g>a | MT-ND1 | Somatic | 51 | 94.12 | True variant |
| P_5942a | r.3796a>g | MT-ND1 | Germline | 2032 | 98.43 | True variant |
| P_9754a | r.3913g>a | MT-ND1 | Somatic | 1685 | 12.11 | True variant |
| P_7219a | r.4282g>a | MT-TI | Somatic | 76 | 35.53 | True variant |
| P_6733a | r.4360g>a | MT-TQ | Somatic | 54 | 40.74 | True variant |
| P_6728a | r.4408g>a | MT-TM | Somatic | 199 | 17.09 | True variant |
| P_6043a | r.4986a>g | MT-ND2 | Somatic | 2073 | 39.22 | True variant |
| P_4847a | r.5479u>c | MT-ND2 | Somatic | 890 | 13.03 | Potential artefact; at end of read TCCxACC context |
| P_9567a | r.6569c>u | MT-CO1 | Somatic | 1773 | 88.72 | True variant |
| P_4970a | r.7045u>c | MT-CO1 | Somatic | 761 | 12.22 | True variant |
| P_9847a | r.7146a>g | MT-CO1 | Germline | 757 | 98.15 | True variant |
| P_9757a | r.7579u>c | MT-TD | Somatic | 109 | 16.51 | True variant |
| P_11383a | r.7698u>c | MT-CO2 | Somatic | 2277 | 12.60 | True variant |
| P_9541a | r.7765a>g | MT-CO2 | Somatic | 797 | 17.69 | True variant |
| P_4976a | r.7895u>c | MT-CO2 | Somatic | 2368 | 28.08 | True variant |
| P_11819a | r.8149a>g | MT-CO2 | Somatic | 660 | 92.58 | True variant |
| P_7216a | r.8286u>c | Non-Coding | Somatic | 364 | 49.73 | True variant |
| P_8978a | r.8408c>u | MT-ATP8 | Germline | 2239 | 99.60 | True variant |
| P_4604a | r.9989u>c | MT-CO3 | Germline | 302 | 98.68 | True variant |
| P_4833a | r.10306a>c | MT-ND3 | Somatic | 1004 | 35.26 | True variant |
| P_4976a | r.11718g>a | MT-ND4 | Somatic | 1304 | 92.33 | True variant |
| P_6722a | r.11899u>c | MT-ND4 | Germline | 691 | 95.37 | True variant (phased with 11914A) |
| P_8978a | r.12763g>a | MT-ND5 | Somatic | 1602 | 89.20 | True variant |
| P_9754a | r.12876c>u | MT-ND5 | Germline | 1478 | 97.29 | True variant |
| P_6411a | r.13528a>g | MT-ND5 | Somatic | 1701 | 91.59 | True variant |
| P_4983a | r.13552g>a | MT-ND5 | Somatic | 1569 | 91.65 | True variant |
| P_9002a | r.14389c>u | MT-ND6 | Somatic | 1718 | 93.95 | True variant |
| P_5930a | r.14721g>c | MT-TE | Somatic | 302 | 27.81 | True variant (phased with 14766T) |
| P_8978a | r.15495u>c | MT-CYB | Somatic | 1812 | 94.09 | True variant |
| P_11337a | r.15607a>g | MT-CYB | Germline | 959 | 99.06 | True variant |
| P_4982a | r.15904c>u | MT-TT | Somatic | 344 | 34.01 | True variant (overlapping 15927A) |
| P_4982a | r.15927g>a | MT-TT | Somatic | 344 | 36.92 | True variant (overlapping with 15904) |
| P_4847a | r.16092u>c | Control-Region | Germline | 1838 | 99.78 | True variant |
| P_11389a | r.16093u>c | Control-Region | Somatic | 745 | 12.62 | True variant |
| P_9582a | r.16093u>c | Control-Region | Somatic | 753 | 12.88 | True variant (phased with 16126C) |
| P_4072a | r.16093u>c | Control-Region | Germline | 1041 | 27.76 | True variant |
| P_9567a | r.16093u>c | Control-Region | Somatic | 2459 | 92.19 | True variant |
| P_4955a | r.16093u>c | Control-Region | Germline | 1298 | 98.69 | True variant |
| P_4970a | r.16104c>u | Control-Region | Germline | 800 | 81.88 | True variant |
| P_8979a | r.16184c>u | Control-Region | Germline | 317 | 100.00 | True variant |
| P_4606a | r.16186c>u | Control-Region | Germline | 181 | 96.69 | True variant |
| P_8830a | r.16209u>c | Control-Region | Germline | 863 | 100.00 | True variant (phased with 16171A, 16183C, 16188C, 16233T, 16258T) |
| P_8830a | r.16223c>u | Control-Region | Germline | 1197 | 100.00 | True variant (phased with 16171A, 16183C, 16188C, 1609C, 16258T) |
| P_9002a | r.16235a>g | Control-Region | Germline | 1409 | 82.82 | True variant (phased with 16183C, 16184A, 16189C, 16217C) |
| P_11338a | r.16235a>g | Control-Region | Somatic | 2188 | 93.01 | True variant (phased with 16293G and 16304C) |
| P_6730a | r.16267c>u | Control-Region | Somatic | 2167 | 34.98 | True variant |
| P_6732a | r.16278c>u | Control-Region | Somatic | 2661 | 47.35 | True variant |
| P_9575a | r.16290c>u | Control-Region | Somatic | 1530 | 25.16 | True variant (phased with 16265C, 16291T, 16335G) |
| P_11341a | r.16293a>g | Control-Region | Germline | 1205 | 100.00 | True variant (phased with 16331C, 16354T) |
| P_9582a | r.16294c>u | Control-Region | Germline | 870 | 99.89 | True variant (phased with 16304C) |
| P_9847a | r.16294c>u | Control-Region | Germline | 731 | 100.00 | True variant (phased with 16278T, 16293G, 16311C, 16360T) |
| P_9582a | r.16304u>c | Control-Region | Germline | 663 | 99.85 | True variant (phased with 16294T) |
| P_9596a | r.16311u>c | Control-Region | Germline | 802 | 98.63 | True variant |
| P_5956a | r.16311u>c | Control-Region | Germline | 757 | 100.00 | True variant |
| P_9761a | r.16336g>a | Control-Region | Germline | 1571 | 100.00 | True variant |
| P_9541a | r.16342u>c | Control-Region | Germline | 806 | 99.75 | True variant |
| P_11381a | r.16356u>c | Control-Region | Germline | 1466 | 99.86 | True variant |
| P_9568a | r.16362u>c | Control-Region | Germline | 1232 | 99.84 | True variant (phased with 16304C) |
| P_5946a | r.16362u>c | Control-Region | Germline | 327 | 100.00 | True variant |
| P_9599a | r.16362u>c | Control-Region | Germline | 376 | 100.00 | True variant (phased with 16325C) |
| P_11819a | r.16362u>c | Control-Region | Germline | 373 | 100.00 | True variant |
| P_10010a | r.16519u>c | Control-Region | Somatic | 732 | 56.15 | True variant |
| P_6730a | r.16540c>u | Control-Region | Somatic | 1142 | 61.56 | True variant (phased with 16519C) |
References
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. Elife 2016, 5. [Google Scholar] [CrossRef]
- Mambo, E.; Chatterjee, A.; Xing, M.; Tallini, G.; Haugen, B.R.; Yeung, S.C.; Sukumar, S.; Sidransky, D. Tumor-specific changes in mtDNA content in human cancer. Int. J. Cancer 2005, 116, 920–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Zhou, Y.; Shi, Y.; Ning, L.; Yang, Y.; Wei, X.; Zhang, N.; Hao, X.; Niu, R. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 2007, 59, 450–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, L.M.; Yin, P.H.; Chi, C.W.; Hsu, C.Y.; Wu, C.W.; Lee, L.M.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef]
- Fan, A.X.; Radpour, R.; Haghighi, M.M.; Kohler, C.; Xia, P.; Hahn, S.; Holzgreve, W.; Zhong, X.Y. Mitochondrial DNA content in paired normal and cancerous breast tissue samples from patients with breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 983–989. [Google Scholar] [CrossRef]
- Barekati, Z.; Radpour, R.; Kohler, C.; Zhang, B.; Toniolo, P.; Lenner, P.; Lv, Q.; Zheng, H.; Zhong, X.Y. Methylation profile of TP53 regulatory pathway and mtDNA alterations in breast cancer patients lacking TP53 mutations. Hum. Mol. Genet. 2010, 19, 2936–2946. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.; LaFramboise, T. Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 2014, 35, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.K.; Chang, J.; Yeh, K.T.; Lou, M.A.; Lu, J.F.; Tan, D.J.; Liu, H.; Wong, L.J. Mitochondrial DNA content varies with pathological characteristics of breast cancer. J. Oncol. 2011, 2011, 496189. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Yin, P.H.; Lee, H.C.; Chi, C.W.; Tseng, L.M. Mitochondrial DNA content as a potential marker to predict response to anthracycline in breast cancer patients. Breast J. 2010, 16, 264–270. [Google Scholar] [CrossRef]
- Reznik, E.; Wang, Q.G.; La, K.; Schultz, N.; Sander, C. Mitochondrial respiratory gene expression is suppressed in many cancers. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Weerts, M.J.; Sieuwerts, A.M.; Smid, M.; Look, M.P.; Foekens, J.A.; Sleijfer, S.; Martens, J.W. Mitochondrial DNA content in breast cancer: Impact on in vitro and in vivo phenotype and patient prognosis. Oncotarget 2016, 7, 29166–29176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weerts, M.J.A.; Hollestelle, A.; Sieuwerts, A.M.; Foekens, J.A.; Sleijfer, S.; Martens, J.W.M. Low tumor mitochondrial DNA content is associated with better outcome in breast cancer patients receiving anthracycline-based chemotherapy. Clin. Cancer Res. 2017, 23, 4735–4743. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.B.; Alaei-Mahabadi, B.; Sabarinathan, R.; Samuelsson, T.; Gorodkin, J.; Gustafsson, C.M.; Larsson, E. Simultaneous DNA and RNA mapping of somatic mitochondrial mutations across diverse human cancers. PLoS Genet. 2015, 11, e1005333. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Bar-Yaacov, D.; Avital, G.; Levin, L.; Richards, A.L.; Hachen, N.; Rebolledo Jaramillo, B.; Nekrutenko, A.; Zarivach, R.; Mishmar, D. RNA-DNA differences in human mitochondria restore ancestral form of 16S ribosomal RNA. Genome Res. 2013, 23, 1789–1796. [Google Scholar] [CrossRef]
- Hodgkinson, A.; Idaghdour, Y.; Gbeha, E.; Grenier, J.C.; Hip-Ki, E.; Bruat, V.; Goulet, J.P.; de Malliard, T.; Awadalla, P. High-resolution genomic analysis of human mitochondrial RNA sequence variation. Science 2014, 344, 413–415. [Google Scholar] [CrossRef]
- Nielsen, M.M.; Tehler, D.; Vang, S.; Sudzina, F.; Hedegaard, J.; Nordentoft, I.; Orntoft, T.F.; Lund, A.H.; Pedersen, J.S. Identification of expressed and conserved human noncoding RNAs. RNA 2014, 20, 236–251. [Google Scholar] [CrossRef]
- Grandhi, S.; Bosworth, C.; Maddox, W.; Sensiba, C.; Akhavanfard, S.; Ni, Y.; LaFramboise, T. Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue-specific signals of relaxed and positive selection. Hum. Mol. Genet. 2017, 3798753. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Q.; Delannoy, M.; Cooke, C.; Yager, J.D. Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1011–E1022. [Google Scholar] [CrossRef] [PubMed]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, 434. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.L.; MacDonald, L.R.; Rogers, J.V.; Aravkin, A.; Haseley, D.R.; Beatty, J.D. Positron emission mammography: Correlation of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 status and 18F-FDG. AJR Am. J. Roentgenol. 2011, 197, W247–W255. [Google Scholar] [CrossRef]
- Yoon, H.J.; Kang, K.W.; Chun, I.K.; Cho, N.; Im, S.A.; Jeong, S.; Lee, S.; Jung, K.C.; Lee, Y.S.; Jeong, J.M.; et al. Correlation of breast cancer subtypes, based on estrogen receptor, progesterone receptor, and HER2, with functional imaging parameters from (6)(8)Ga-RGD PET/CT and (1)(8)F-FDG PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1534–1543. [Google Scholar] [CrossRef]
- Gil-Rendo, A.; Martinez-Regueira, F.; Zornoza, G.; Garcia-Velloso, M.J.; Beorlegui, C.; Rodriguez-Spiteri, N. Association between [18F]fluorodeoxyglucose uptake and prognostic parameters in breast cancer. Br. J. Surg. 2009, 96, 166–170. [Google Scholar] [CrossRef]
- Ikenaga, N.; Otomo, N.; Toyofuku, A.; Ueda, Y.; Toyoda, K.; Hayashi, T.; Nishikawa, K.; Tanaka, M. Standardized uptake values for breast carcinomas assessed by fluorodeoxyglucose-positron emission tomography correlate with prognostic factors. Am. Surg. 2007, 73, 1151–1157. [Google Scholar] [PubMed]
- Mavi, A.; Cermik, T.F.; Urhan, M.; Puskulcu, H.; Basu, S.; Yu, J.Q.; Zhuang, H.; Czerniecki, B.; Alavi, A. The effects of estrogen, progesterone, and C-erbB-2 receptor states on 18F-FDG uptake of primary breast cancer lesions. J. Nucl. Med. 2007, 48, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Nakajo, M.; Kajiya, Y.; Kaneko, T.; Kaneko, Y.; Takasaki, T.; Tani, A.; Ueno, M.; Koriyama, C.; Nakajo, M. FDG PET/CT and diffusion-weighted imaging for breast cancer: Prognostic value of maximum standardized uptake values and apparent diffusion coefficient values of the primary lesion. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.R.; Port, E.; Gonen, M.; Doane, A.; Yeung, H.; Gerald, W.; Cook, J.B.; Larson, S. 18F-FDG PET of locally invasive breast cancer and association of estrogen receptor status with standardized uptake value: Microarray and immunohistochemical analysis. J. Nucl. Med. 2010, 51, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.M.; Yin, P.H.; Yang, C.W.; Tsai, Y.F.; Hsu, C.Y.; Chi, C.W.; Lee, H.C. Somatic mutations of the mitochondrial genome in human breast cancers. Genes Chromosomes Cancer 2011, 50, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.J.; Bai, R.K.; Wong, L.J. Comprehensive scanning of somatic mitochondrial DNA mutations in breast cancer. Cancer Res. 2002, 62, 972–976. [Google Scholar] [PubMed]
- Coller, H.A.; Khrapko, K.; Bodyak, N.D.; Nekhaeva, E.; Herrero-Jimenez, P.; Thilly, W.G. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat. Genet. 2001, 28, 147–150. [Google Scholar] [CrossRef]
- Samuels, D.C.; Li, C.; Li, B.; Song, Z.; Torstenson, E.; Boyd Clay, H.; Rokas, A.; Thornton-Wells, T.A.; Moore, J.H.; Hughes, T.M.; et al. Recurrent tissue-specific mtDNA mutations are common in humans. PLoS Genet. 2013, 9, e1003929. [Google Scholar] [CrossRef]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [Green Version]
- Li, M.K.; Schroder, R.; Ni, S.Y.; Madea, B.; Stoneking, M. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 2491–2496. [Google Scholar] [CrossRef] [Green Version]
- Calloway, C.D.; Reynolds, R.L.; Herrin, G.L., Jr.; Anderson, W.W. The frequency of heteroplasmy in the HVII region of mtDNA differs across tissue types and increases with age. Am. J. Hum. Genet. 2000, 66, 1384–1397. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Johnson, K.A. Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 2001, 276, 38097–38107. [Google Scholar] [CrossRef] [PubMed]
- Collura, R.V.; Auerbach, M.R.; Stewart, C.B. A quick, direct method that can differentiate expressed mitochondrial genes from their nuclear pseudogenes. Curr. Biol. 1996, 6, 1337–1339. [Google Scholar] [CrossRef]
- Ramos, A.; Barbena, E.; Mateiu, L.; del Mar Gonzalez, M.; Mairal, Q.; Lima, M.; Montiel, R.; Aluja, M.P.; Santos, C. Nuclear insertions of mitochondrial origin: Database updating and usefulness in cancer studies. Mitochondrion 2011, 11, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.L.; Maki, J.; Reguly, B.; Dakubo, G.D.; Aguirre, A.; Wittock, R.; Robinson, K.; Jakupciak, J.P.; Thayer, R.E. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genomics 2006, 7, 185. [Google Scholar] [CrossRef] [PubMed]
- Parfait, B.; Rustin, P.; Munnich, A.; Rotig, A. Co-amplification of nuclear pseudogenes and assessment of heteroplasmy of mitochondrial DNA mutations. Biochem. Biophys. Res. Commun. 1998, 247, 57–59. [Google Scholar] [CrossRef]
- Albayrak, L.; Khanipov, K.; Pimenova, M.; Golovko, G.; Rojas, M.; Pavlidis, I.; Chumakov, S.; Aguilar, G.; Chavez, A.; Widger, W.R.; et al. The ability of human nuclear DNA to cause false positive low-abundance heteroplasmy calls varies across the mitochondrial genome. BMC Genomics 2016, 17, 1017. [Google Scholar] [CrossRef]
- Hazkani-Covo, E.; Zeller, R.M.; Martin, W. Molecular poltergeists: Mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010, 6, e1000834. [Google Scholar] [CrossRef]
- Knoop, V. When you can’t trust the DNA: RNA editing changes transcript sequences. Cell Mol. Life Sci. 2011, 68, 567–586. [Google Scholar] [CrossRef]
- Smid, M.; Rodriguez-Gonzalez, F.G.; Sieuwerts, A.M.; Salgado, R.; Prager-Van der Smissen, W.J.; Vlugt-Daane, M.V.; van Galen, A.; Nik-Zainal, S.; Staaf, J.; Brinkman, A.B.; et al. Breast cancer genome and transcriptome integration implicates specific mutational signatures with immune cell infiltration. Nat. Commun. 2016, 7, 12910. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics 2015, 51, 11.14.1–11.14.19. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Idaghdour, Y.; Hodgkinson, A. Integrated genomic analysis of mitochondrial RNA processing in human cancers. Genome Med. 2017, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA variation and analysis using Mitomap and Mitomaster. Curr. Protoc. Bioinformatics 2013, 44, 1.23.1–1.23.26. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstatter, A.; Forer, L.; Specht, G.; Bandelt, H.J.; Kronenberg, F.; Salas, A.; Schonherr, S. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef]
- Gehring, J.S.; Fischer, B.; Lawrence, M.; Huber, W. SomaticSignatures: Inferring mutational signatures from single-nucleotide variants. Bioinformatics 2015, 31, 3673–3675. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]





| Variable | No. of Cases | mtRNA Somatic Variants | p | mtRNA Expression | p | ||
|---|---|---|---|---|---|---|---|
| 0 Variants | 1 Variant | >1 Variants | Median (IQR) TPM | ||||
| Age | 0.022 a | 0.049 d | |||||
| 56 (28–85) | 291 (100%) | 53 (17) | 55 (23) | 61 (24) | 0.11 c | ||
| unknown | 53 | ||||||
| Tumor size | 0.07 b | 0.051 a | |||||
| T1 ≤ 2 cm | 76 (35.2%) | 33.8% | 25.0% | 44.4% | 19.9202 (0.0043) | ||
| T2 > 2–5 cm | 109 (50.5%) | 47.9% | 64.1% | 42.0% | 19.9207 (0.0045) | ||
| T3 > 5 cm | 31 (14.4%) | 18.3% | 10.9% | 13.6% | 19.9223 (0.0047) | ||
| unknown | 128 | ||||||
| Grade | 0.4 b | 0.1 a | |||||
| I | 24 (8.5%) | 9.9% | 12.2% | 5.1% | 19.9202 (0.0037) | ||
| II | 111 (39.4%) | 40.7% | 35.1% | 41.0% | 19.9216 (0.0044) | ||
| III | 147 (52.1%) | 49.5% | 52.7% | 53.8% | 19.9209 (0.0049) | ||
| unknown | 62 | ||||||
| ER | 0.3 b | <0.001 a | |||||
| Negative | 81 (27.8%) | 21.7% | 31.2% | 30.3% | 19.9196 (0.0050) | ||
| Positive | 210 (72.2%) | 78.3% | 68.8% | 69.7% | 19.9216 (0.0041) | ||
| unknown | 53 | ||||||
| PR | 0.5 b | 0.006 a | |||||
| Negative | 102 (35.4%) | 31.5% | 40.5% | 35.0% | 19.9204 (0.0048) | ||
| Positive | 186 (64.6%) | 68.5% | 59.5% | 65.0% | 19.9215 (0.0042) | ||
| unknown | 56 | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weerts, M.J.A.; Smid, M.; Foekens, J.A.; Sleijfer, S.; Martens, J.W.M. Mitochondrial RNA Expression and Single Nucleotide Variants in Association with Clinical Parameters in Primary Breast Cancers. Cancers 2018, 10, 500. https://doi.org/10.3390/cancers10120500
Weerts MJA, Smid M, Foekens JA, Sleijfer S, Martens JWM. Mitochondrial RNA Expression and Single Nucleotide Variants in Association with Clinical Parameters in Primary Breast Cancers. Cancers. 2018; 10(12):500. https://doi.org/10.3390/cancers10120500
Chicago/Turabian StyleWeerts, Marjolein J. A., Marcel Smid, John A. Foekens, Stefan Sleijfer, and John W. M. Martens. 2018. "Mitochondrial RNA Expression and Single Nucleotide Variants in Association with Clinical Parameters in Primary Breast Cancers" Cancers 10, no. 12: 500. https://doi.org/10.3390/cancers10120500
APA StyleWeerts, M. J. A., Smid, M., Foekens, J. A., Sleijfer, S., & Martens, J. W. M. (2018). Mitochondrial RNA Expression and Single Nucleotide Variants in Association with Clinical Parameters in Primary Breast Cancers. Cancers, 10(12), 500. https://doi.org/10.3390/cancers10120500