Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer

 , and
, and
Abstract
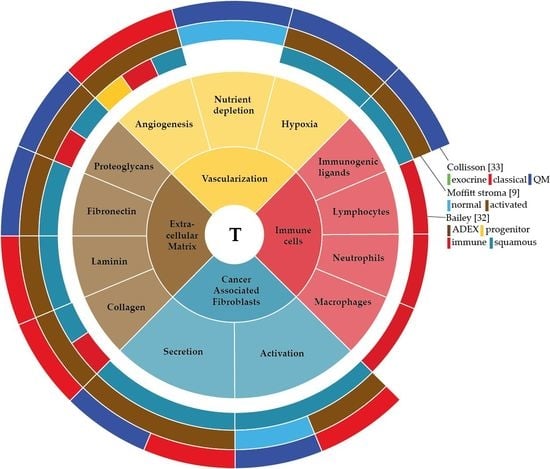
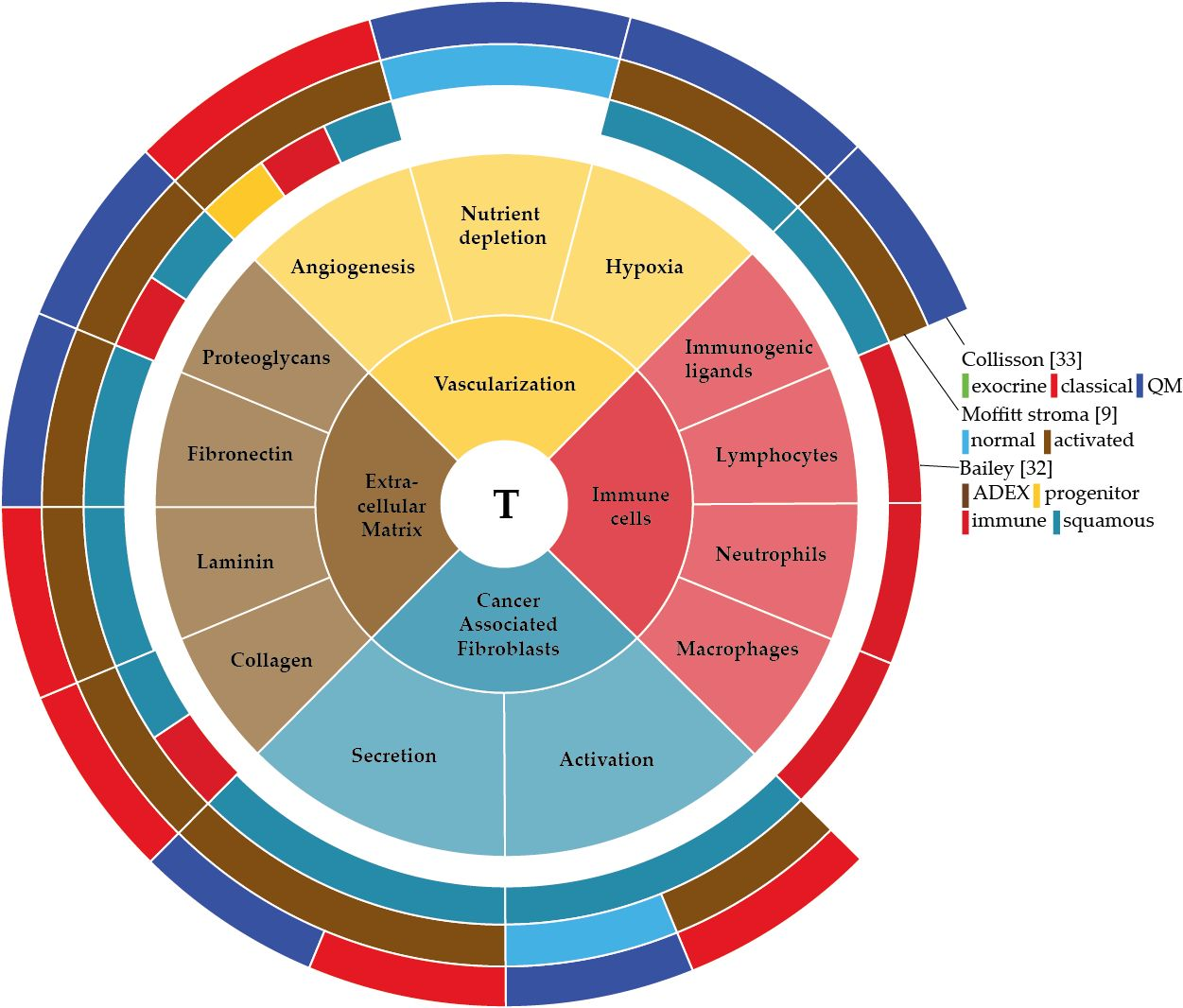
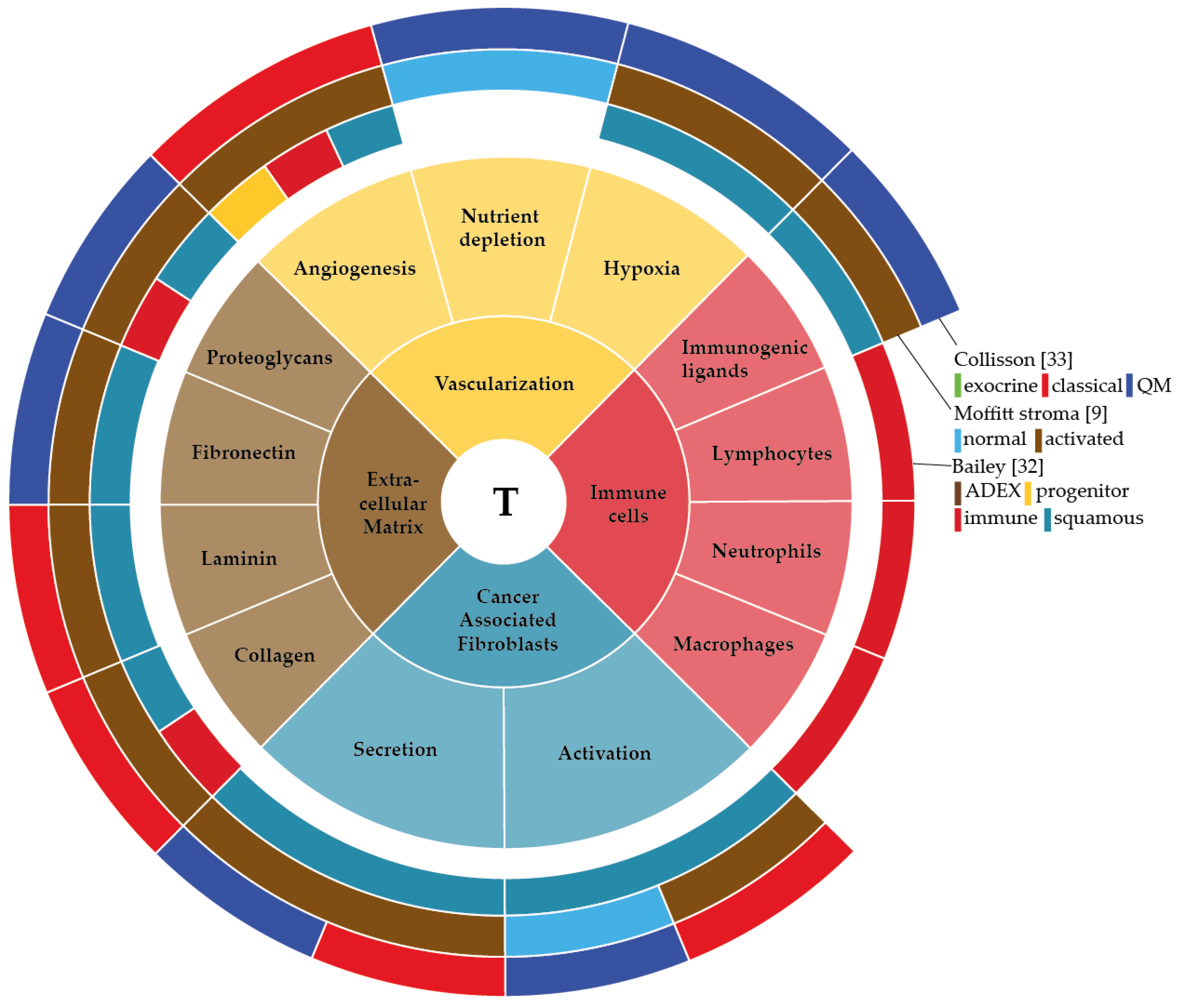
:
1. Introduction
2. Development of PDAC
2.1. Mutational Status of PDAC
2.2. Signalling Pathways in PDAC
2.3. Tumour Heterogeneity
3. Stromal Components: The Extracellular Matrix
3.1. Collagens
3.2. Laminin and Fibronectin
3.3. Proteoglycans
3.4. Hyaluronic Acid
3.5. ECM-Targeting
4. Stromal Component: The Vasculature
4.1. Angiogenesis
4.2. Nutrient Depletion
4.3. Hypoxia
5. Stromal Component: Cancer-Associated Fibroblasts
5.1. Pancreatic Stellate Cells
5.2. PSC-Induced Chemoresistance
6. Stromal Component: Immune Cells
6.1. Macrophages
6.2. Neutrophils
6.3. Lymphocytes
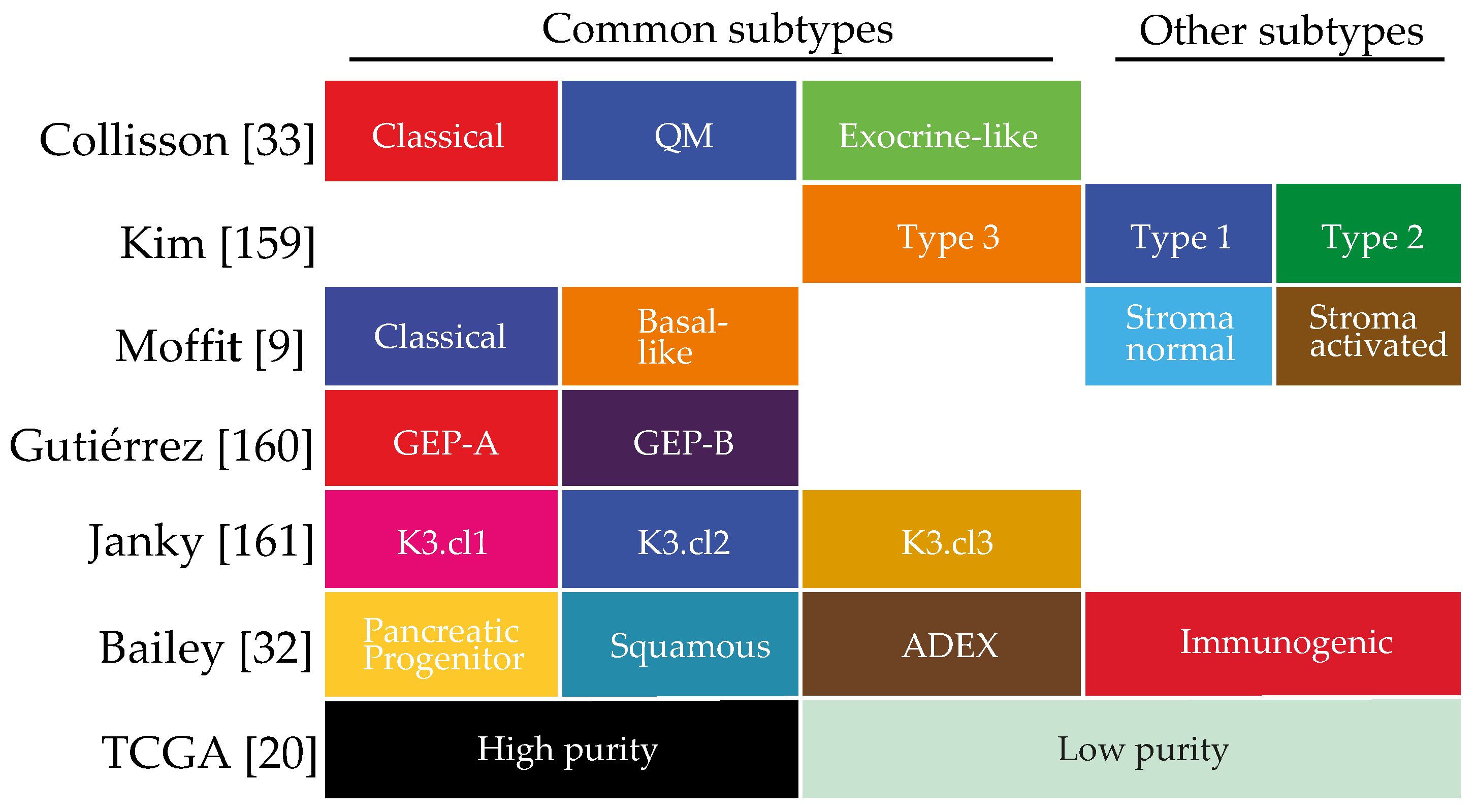
7. Molecular Subtypes in PDAC
7.1. Collisson 2011
7.2. Kim 2013
7.3. Moffitt 2015
7.4. Gutiérrez 2015
7.5. Janky 2016
7.6. Bailey 2016
7.7. The Cancer Genome Atlas (TCGA) Research Network 2017
8. Discussion
9. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic ductal adenocarcinoma |
| PC | Pancreatic cancer |
| CAFs | Cancer associated fibroblasts |
| PSCs | Pancreatic stellate cells |
| ECM | Extracellular matrix |
| MCN | Mucinous cystic neoplasms |
| IPMN | Intraductal papillary mucinous neoplasms |
| PanIN | Pancreatic intraepithelial neoplasia |
| KRAS | KRAS proto-oncogene |
| CDKN2A | Cyclin dependent kinase inhibitor 2A |
| TP53 | Tumor protein 53 |
| SMAD4 | SMAD family member 4 |
| CNV | Copy number variation |
| ERBB2 | Erb-b2 receptor tyrosine kinase 2 |
| MET | MET proto-oncogene, receptor tyrosine kinase |
| CDK6 | Cyclin dependent kinase 6 |
| PIK3CA | Phosphatidylinositol-4,5-biphosphate 3-kinase catalytic subunit alpha |
| PIK3R3 | Phosphoinositide-3-kinase regulatory subunit 3 |
| BRCA | Breast cancer (susceptibility gene) |
| PALB2 | Partner and localizer of BRCA2 |
| Hh | Hedgehog |
| TGF-β | Transcription Growth Factor β |
| SMAD4 | SMAD family member 4 |
| MAPK | Mitogen associated protein kinase |
| TME | Tumor microEnvironment |
| MMPs | Matrix metalloProteases |
| TIMPs | Tissue inhibitor metalloproteases |
| EGF | Epidermal growth factor |
| FAK | Focal adhesion kinase |
| DDR2 | Discoidin domain receptor tyrosine kinase 2 |
| NF-κβ | Nuclear factor kappa beta |
| EMT | Epithelial-to-mesenchymal transition |
| SHH | Sonic Hedgehog |
| SPOCK | Sparc/osteonectin, cwcv and kazal-like domains proteoglycan |
| HA | Hyaluronic acid |
| CD44 | Cluster of differentiation 44 |
| RHAMM | Receptor for HA mediated motility |
| CAM-DR | Cell adhesion mediated drug resistance |
| ROCK | Rho-associated protein kinase |
| ECs | Endothelial cells |
| vSMCs | Vascular smooth muscle cells |
| VEGF | Vascular endothelial growth factor |
| bFGF | basic fibroblast growth factor |
| HIF1α | Hypoxia inducible factor 1α |
| CypA | Cyclophilin A |
| LDH-A | Lactate dehydrogenase A |
| ROS | Reactive oxygen species |
| TCA cycle | Tricarboxylic acid cycle |
| FSP1 | Fibroblast specific protein 1 |
| α-SMA | α-Smooth muscle actin |
| BMDCs | Bone marrow derived cells |
| GFAP | Glial fibrillary acidic protein |
| IL | Interleukin |
| TNFα | Tumor necrosis factor α |
| PDGF | Platelet derived growth factor |
| TAM | Tumor associated macrophage |
| NE | Neutrophil elastase |
| NET | Neutrophil Extracellular Traps |
| NLR | Neutrophil to lymphocyte Ratio |
| TIL | Tumor infiltrating lymphocytes |
| NMF | non-negative matrix factorization |
| QM | Quasi mesenchymal |
| GEP | Gene expression profiles |
| ADEX | Aberrantly differentiated endocrine exocrine |
| TCGA | The Cancer Genome Atlas |
References
- Hernández-muñoz, I.; Skoudy, A.; Real, X. Pancreatic ductal adenocarcinoma: Cellular Origin, signaling pathways and stroma contribution. Pancreatology 2008, 8, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T. Pancreatic ductal adenocarcinoma: Basic and clinical challenges for better prognosis. J. Carcinog. Mutagen. 2013, 9–10. [Google Scholar] [CrossRef]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed]
- Malvezzi, M.; Bertuccio, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year. Ann. Oncol. 2013, 24, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of Pancreatic Ductal Adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44–S47. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.F.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Wilentz, R.E.; Iacobuzio-Donahue, C.A.; Argani, P.; McCarthy, D.M.; Parsons, J.L.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Loss of expression of DPC4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000, 60, 2002–2006. [Google Scholar] [PubMed]
- Hruban, R.H.; Adsay, N.V.; Albores-Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Klöppel, G.; Longnecker, D.S.; et al. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 2001, 25, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Gavin, W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant cK-Ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Smit, V.T.; Boot, A.J.M.; Smits, A.M.M.; Fleuren, G.J.; Cornelisse, C.J.; Bos, J.L. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988, 16, 7773–7782. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.M.; Ying, H.; Juan, J.; Jenkins, N.A.; Copeland, N.G. KRAS-related proteins in pancreatic cancer. Pharmacol. Ther. 2016, 168, 29–42. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Chantrill, L.A.; Nagrial, A.M.; Watson, C.; Johns, A.L.; Martyn-Smith, M.; Simpson, S.; Mead, S.; Jones, M.D.; Samra, J.S.; Gill, A.J.; et al. Precision medicine for advanced pancreas cancer: The individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin. Cancer Res. 2015, 21, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Ringborg, U.; Schilsky, R.L. Strategies to overcome clinical, regulatory, and financial challenges in the implementation of personalized medicine. Am. Soc. Clin. Oncol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Matrisian, L.M.; Berlin, J.D. The past, present, and future of pancreatic cancer clinical trials. Am. Soc. Clin. Oncol. 2016, 35, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Jordan, E.J.; Basturk, O.; Ptashkin, R.N.; Zehir, A.; Berger, M.F.; Leach, T.; Herbst, B.; Askan, G.; Maynard, H.; et al. Real-Time Genomic Profiling of Pancreatic Ductal Adenocarcinoma: Potential Actionability and Correlation with Clinical Phenotype. Clin. Cancer Res. 2017, 23, 6094–6100. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.T.; Hattersley, M.M.; Scarpitti, M.; Yang, B.; Devereaux, E.; Ramachandran, V.; Arumugam, T.; Ji, B.; Logsdon, C.D.; et al. Inhibition of the hedgehog pathway targets the tumor-associated stroma in pancreatic cancer. Mol. Cancer Res. 2012, 10, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [PubMed]
- Damhofer, H.; Medema, J.P.; Veenstra, V.L.; Badea, L.; Popescu, I.; Roelink, H.; Bijlsma, M.F. Assessment of the stromal contribution to Sonic Hedgehog-dependent pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Sadanandam, A.; Tan, P.; Vermeulen, L. Molecular subtypes in cancers of the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 333–342. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.M.F.; Vermeulen, L.; Fessler, E.; Medema, J.P. Cancer heterogeneity—A multifaceted view. EMBO Rep. 2013, 14, 686–695. [Google Scholar]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Giese, T.; Fabio, F.; Wente, M.N.; Esposito, I.; Bachem, M.G.; Giese, N.A.; Bu, M.W.; Friess, H. Pancreatic tumor cells influence the composition of the extracellular matrix. Biochem. Biophys. Res. Commun. 2004, 322, 943–949. [Google Scholar]
- Grzesiak, J.J.; Bouvet, M. The α2β1 integrin mediates the malignant phenotype on type I collagen in pancreatic cancer cell lines. Br. J. Cancer 2006, 94, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen tpe I induces disruption of E-cadherin–mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, Y.; Ko, A.; Gress, T.; Menke, A. Collagen type I-induced Smad-interacting protein 1 expression downregulates E-cadherin in pancreatic cancer. Oncogene 2007, 26, 2381–2385. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Hollingsworth, M.A.; Johnson, K.R.; Wheelock, M.J. Collagen I Promotes Metastasis in Pancreatic Cancer by Activating c-Jun NH 2-Terminal Kinase 1 and Up-regulating N-Cadherin Expression. Cancer Res. 2006, 66, 11745–11753. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.A.; Nawshad, A.; Medici, D. Discoidin domain receptor 2 is a critical regulator of epithelial-mesenchymal transition. Matrix Biol. 2011, 30, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Nawshad, A. Type I collagen promotes epithelial-mesenchymal transition through ILK-dependent activation of NF-κB and LEF-1. Matrix Biol. 2010, 29, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, S.; Grünwald, B.; Krüger, A.; Reithmeier, A.; Hähl, T.; Cheng, T.; Feuchtinger, A.; Born, D.; Erkan, M.; Kleeff, J.; et al. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. 2015, 356, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Halper, J.; Kjaer, M. Basic components of connective tissues and extracellular matrix: Elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv. Exp. Med. Biol. 2014, 802, 31–47. [Google Scholar] [PubMed]
- Vaquero, E.C.; Edderkaoui, M.; Nam, K.J.; Gukovsky, I.; Pandol, S.J.; Gukovskaya, A.S. Extracellular matrix proteins protect pancreatic cancer cells from death via mitochondrial and nonmitochondrial pathways. Gastroenterology 2003, 125, 1188–1202. [Google Scholar] [CrossRef]
- Lowrie, A.G.; Salter, D.M.; Ross, J.A. Latent effects of fibronectin, α5β1 integrin, αVβ5 integrin and the cytoskeleton regulate pancreatic carcinoma cell IL-8 secretion. Br. J. Cancer 2004, 91, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1α co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Ishiwata, T.; Kumbasar, A.; Friess, H.; Buchler, M.W.; Lander, A.D.; Korc, M. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J. Clin. Investig. 1998, 102, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Herreros-Villanueva, M.; Bujanda, L. Glypican-1 in exosomes as biomarker for early detection of pancreatic cancer. Ann. Transl. Med. 2016, 4, 64. [Google Scholar] [PubMed]
- Veenstra, V.L.; Damhofer, H.; Waasdorp, C.; Steins, A.; Kocher, H.M.; Medema, J.P.; van Laarhoven, H.W.; Bijlsma, M.F. Stromal SPOCK1 supports invasive pancreatic cancer growth. Mol. Oncol. 2017, 11, 1050–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kang, Y.; Roife, D.; Lee, Y.; Pratt, M.; Perez, M.R.; Dai, B.; Koay, E.J.; Fleming, J.B. Prolonged exposure to extracellular lumican restrains pancreatic adenocarcinoma growth. Oncogene 2017, 36, 5432–5438. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.R.; Laurent, T.C.; Laurent, U.B. Hyaluronan: Its nature, distribution, functions and turnover. J. Intern. Med. 1997, 242, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.B.; Kohi, S.; Koga, A.; Hirata, K.; Sato, N. Hyaluronan stimulates pancreatic cancer cell motility. Oncotarget 2016, 7, 4829–4840. [Google Scholar] [PubMed]
- Kultti, A.; Zhao, C.; Singha, N.C.; Zimmerman, S.; Osgood, R.J.; Symons, R.; Jiang, P.; Li, X.; Thompson, C.B.; Infante, J.R.; et al. Accumulation of extracellular hyaluronan by hyaluronan synthase 3 promotes tumor growth and modulates the pancreatic cancer microenvironment. Biomed. Res. Int. 2014, 2014, 817613. [Google Scholar] [CrossRef] [PubMed]
- Abetamann, V.; Kern, H.F.; Elsasser, H.P. Differential expression of the hyaluronan receptors CD44 and RHAMM in human pancreatic cancer cells. Clin. Cancer Res. 1996, 2, 1607–1618. [Google Scholar] [PubMed]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-Stroma Interaction of Human Pancreatic Cancer: Acquired Resistance to Anticancer Drugs and Proliferation Regulation Is Dependent on Extracellular Matrix Proteins. Pancres 2004, 28, 38–44. [Google Scholar] [CrossRef]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed]
- Huanwen, W.; Zhiyong, L.; Xiaohua, S.; Xinyu, R.; Kai, W.; Tonghua, L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol. Cancer 2009, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Sebens Muerkoster, S.; Kotteritzsch, J.; Geismann, C.; Gast, D.; Kruse, M.L.; Altevogt, P.; Folsch, U.R.; Schafer, H. α5-integrin is crucial for L1CAM-mediated chemoresistance in pancreatic adenocarcinoma. Int. J. Oncol. 2009, 34, 243–253. [Google Scholar] [PubMed]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.; Gladkov, O.; Holcombe, R.F.; Jiang, P.; et al. Final results of a phase Ib study of gemcitabine plus PEGPH20 in patients with stage IV previously untreated pancreatic cancer. J. Clin. Oncol. 2015, 33, 359. [Google Scholar] [CrossRef]
- Rath, N.; Morton, J.P.; Julian, L.; Helbig, L.; Kadir, S.; McGhee, E.J.; Anderson, K.I.; Kalna, G.; Mullin, M.; Pinho, A.V.; et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol. Med. 2017, 9, 198–218. [Google Scholar] [CrossRef] [PubMed]
- Vennin, C.; Rath, N.; Pajic, M.; Olson, M.F.; Timpson, P. Targeting ROCK activity to disrupt and prime pancreatic cancer for chemotherapy. Small GTPases 2017. [Google Scholar] [CrossRef] [PubMed]
- Keshet, E.; Ben-Sasson, S.A. Anticancer drug targets: Approaching angiogenesis. J. Clin. Investig. 1999, 104, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, L.E.; Hemo, I.; Keshet, E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 125, 1591–1598. [Google Scholar] [PubMed]
- Subarsky, P.; Hill, R.P. The hypoxic tumour microenvironment and metastatic progression. Clin. Exp. Metastasis 2003, 20, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Jurisica, I.; Do, T.; Hedley, D.W. Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Tumor Stem Cell Biol. 2011, 71, 3110–3120. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A Key Regulatory Factor in Tumor Growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.; Brunetti, O.; Gnoni, A.; Cascinu, S.; Gasparini, G.; Lorusso, V.; Ribatti, D.; Silvestris, N. Angiogenesis in pancreatic ductal adenocarcinoma: A controversial issue. Oncotarget 2016, 7, 58649. [Google Scholar] [CrossRef] [PubMed]
- Di Maggio, F.; Arumugam, P.; Delvecchio, F.R.; Batista, S.; Lechertier, T.; Hodivala-Dilke, K.; Kocher, H.M. Pancreatic stellate cells regulate blood vessel density in the stroma of pancreatic ductal adenocarcinoma. Pancreatology 2016, 16, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Bausch, D.; Pausch, T.; Krauss, T.; Hopt, U.T.; Fernandez-Del-Castillo, C.; Warshaw, A.L.; Thayer, S.P.; Keck, T. Neutrophil granulocyte derived MMP-9 is a VEGF independent functional component of the angiogenic switch in pancreatic ductal adenocarcinoma. Angiogenesis 2011, 14, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Heiden, M.G.V.; Miller, G.; Drebin, J.A.; Bar-sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezene, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kimmelman, A.C. A critical role for autophagy in pancreatic cancer. Autophagy 2011, 7, 912–913. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Saiyin, H.; Ardito-abraham, C.M.; Wu, Y.; Wei, Y.; Fang, Y.; Han, X.; Li, J.; Zhou, P.; Yi, Q.; Maitra, A.; et al. Identification of novel vascular projections with cellular trafficking abilities on the microvasculature of pancreatic ductal adenocarcinoma. J. Pathol. 2015, 236, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Rainero, E.; Howe, J.D.; Caswell, P.T.; Jamieson, N.B.; Anderson, K.; Critchley, D.R.; Machesky, L.; Norman, J.C. Ligand-occupied integrin internalization links nutrient signaling to invasive migration. Cell Rep. 2015, 10, 398–413. [Google Scholar] [CrossRef] [PubMed]
- Couvelard, A.; O’Toole, D.; Leek, R.; Turley, H.; Sauvanet, A.; Degott, C.; Ruszniewski, P.; Belghiti, J.; Harris, A.L.; Gatter, K.; et al. Expression of hypoxia-inducible factors is correlated with the presence of a fibrotic focus and angiogenesis in pancreatic ductal adenocarcinomas. Histopathology 2005, 46, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Oong, A.C.; Ehta, V.K.; Uynh, Q.L.; Isher, G.A.; Erris, D.J.; Rown, J.A.; Astidas, A.J.; Ierra, M.R.V. Pancreatic tumors show high levels of hypoxia. J. Radiat. Oncol. 2000, 48, 919–922. [Google Scholar]
- Zhang, Q.; Lou, Y.; Zhang, J.; Fu, Q.; Wei, T.; Sun, X.; Chen, Q.; Yang, J.; Bai, X.; Liang, T. Hypoxia-inducible factor-2α promotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. Mol. Cancer 2017, 16, 119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Liu, F.; Gao, C.; Wang, X.; Zhao, T.; Liu, J. CypA, a gene downstream of HIF-1α, promotes the development of PDAC. PLoS ONE 2014, 9, e92824. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Gao, S.; Ren, H.; Sun, W.; Zhang, H.; Sun, J.; Yang, S.; Hao, J. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014, 74, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Maftouh, M.; Avan, A.; Sciarrillo, R.; Granchi, C.; Leon, L.G.; Rani, R.; Funel, N.; Smid, K.; Honeywell, R.; Boggi, U.; et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer 2014, 110, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Morifuji, Y.; Kai, M.; Suyama, K.; Iwasaki, H.; Katano, M. Hedgehog inhibitor decreases chemosensitivity to 5-fluorouracil and gemcitabine under hypoxic conditions in pancreatic cancer. Cancer Sci. 2012, 103, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.X.; Wang, D.W.; Liu, T.; Liu, W.X.; Xia, W.B.; Xu, J.; Zhang, Y.H.; Qu, Y.K.; Guo, L.Q.; Ding, L.; et al. Effects of the HIF-1α and NF-κB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol. Rep. 2014, 31, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, J.; Wei, W.; Shi, M.; Xin, B.; Zhang, T.; Shen, X. Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1α and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. Ther. 2016, 17, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Dauer, P.; Gupta, V.; McGinn, O.; Arora, N.; Majumdar, K.; Uhlrich, C., 3rd; Dalluge, J.; Dudeja, V.; Saluja, A.; et al. Microenvironment mediated alterations to metabolic pathways confer increased chemo-resistance in CD133+ tumor initiating cells. Oncotarget 2016, 7, 56324–56337. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Groot, A.P.; Oduro, J.P.; Franken, R.J.; Schoenmakers, S.H.; Peppelenbosch, M.P.; Spek, C.A. Hypoxia induces a hedgehog response mediated by HIF-1α. J. Cell. Mol. Med. 2009, 13, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.L.; Lam, J.S.; Ailles, L.E.; Wong, M.Z.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Liao, Q.; Niu, Z.; Zhou, L.; Zhao, Y. Cancer-associated fi broblasts in pancreatic adenocarcinoma. Future Oncol. 2015, 11, 2603–2610. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Shek, F.; Benyon, R.; Walker, F.; McCrudden, P.; Pender, S.; Williams, E.; Johnson, P.; Johnson, C.; Bateman, A.; Fine, D.; et al. Expression of transforming growth factor-β1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 2002, 160, 1787–1798. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Günter, K.; Detlefsen, S.; Feyerabend, B. Fibrosis of the pancreas: The initial tissue damage and the resulting pattern. Virchows Arch. 2004, 445, 1–8. [Google Scholar]
- Wilson, J.S.; Pirola, R.C.; Apte, M.V. Stars and stripes in pancreatic cancer: Role of stellate cells and stroma in cancer progression. Front. Physiol. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Bachem, M.G.; Schünemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Tjomsland, V.; Aasrum, M.; Christoffersen, T.; Gladhaug, I.P. Functional heterogeneity in tumor-derived human pancreatic stellate cells: Differential expression of HGF and implications for mitogenic signaling and migration in pancreatic cancer cells. Oncotarget 2017, 8, 71672–71684. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, H.; Huang, C.; Zhao, T.; Zhang, H.; Zheng, C.; Ren, H.; Hao, J. Wnt2 protein plays a role in the progression of pancreatic cancer promoted by pancreatic stellate cells. Med. Oncol. 2015, 32, 97. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Liu, X.D.; Chen, Z.Y.; Zhu, J.; Xiong, Y.; Li, K.; Dong, J.H.; Li, X. Interaction between cancer cells and stromal fibroblasts is required for activation of the uPAR-uPA-MMP-2 cascade in pancreatic cancer metastasis. Clin. Cancer Res. 2007, 13, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Bo, H.; Zhang, S.; Gao, L.; Chen, Y.; Zhang, J.; Chang, X.; Zhu, M. Upregulation of Wnt5a promotes epithelial-to-mesenchymal transition and metastasis of pancreatic cancer cells. BMC Cancer 2013, 13, 496. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Laarhoven, H.W.M.V. The conflicting roles of tumor stroma in pancreatic cancer and their contribution to the failure of clinical trials: A systematic review and critical appraisal. Cancer Metastasis Rev. 2015, 34, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Hesler, R.A.; Huang, J.J.; Starr, M.D.; Treboschi, V.M.; Bernanke, A.G.; Nixon, A.B.; McCall, S.J.; White, R.R.; Blobe, G.C. TGF-β-induced stromal CYR61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hENT1 and hCNT3. Carcinogenesis 2016, 37, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Esposito, I.; Menicagli, M.; Funel, N.; Bergmann, F.; Boggi, U.; Mosca, F.; Bevilacqua, G.; Campani, D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J. Clin. Pathol. 2004, 57, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Sica, A.; Lewis, C.E. Plasticity of macrophage function during tumor progression: Regulation by distinct molecular mechanisms. J. Immunol. 2008, 180, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Gardian, K.; Janczewska, S.; Olszewski, W.L.; Durlik, M. Analysis of pancreatic cancer microenvironment: Role of macrophage in-filtrates and growth factors expression. J. Cancer 2012, 3, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3K?? drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Caro, G.D.; Cortese, N.; Castino, G.F.; Grizzi, F.; Gavazzi, F.; Ridol, C.; Capretti, G.; Mineri, R.; Todoric, J.; Zerbi, A.; et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Pancreas 2015, 1–11. [Google Scholar]
- Kumar, V.; Sharma, A. Neutrophils: Cinderella of innate immune system. Int. Immunopharmacol. 2010, 10, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, J.; Leschner, S.; Westphal, K.; Lienenklaus, S.; Weiss, S. Neutrophils responsive to endogenous IFN-β regulate tumor angiogenesis and growth in a mouse tumor model. J. Clin. Investig. 2010, 120, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Mishalian, I.; Bayuh, R.; Levy, L.; Zolotarov, L.; Michaeli, J.; Fridlender, Z.G. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol. Immunother. 2013, 62, 1745–1756. [Google Scholar] [CrossRef] [PubMed]
- Gaida, M.M.; Steffen, T.G.; Günther, F.; Tschaharganeh, D.F.; Felix, K.; Bergmann, F.; Schirmacher, P.; Hänsch, G.M. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur. J. Immunol. 2012, 42, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhang, L.; Peng, Y.; Tao, M.; Li, L.; Xiu, D. Neutrophils assist the metastasis of circulating tumor cells in pancreatic ductal adenocarcinoma. Medicine 2016, 95, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Orlichenko, L.; Schapiro, N.E.; Loughran, P.; Gianfrate, G.C.; Ellis, J.T.; Singhi, A.D.; Kang, R.; Tang, D.; Lotze, M.T.; et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015, 22, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Arima, K.; Okabe, H.; Hashimoto, D.; Chikamoto, A.; Tsuji, A.; Yamamura, K.; Kitano, Y.; Inoue, R.; Kaida, T.; Higashi, T.; et al. The diagnostic role of the neutrophil-to-lymphocyte ratio in predicting pancreatic ductal adenocarcinoma in patients with pancreatic diseases. Int. J. Clin. Oncol. 2016, 2, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, R.; Bertolotto, M.; Barisione, G.; Astigiano, S.; Mandruzzato, S.; Ottonello, L.; Dallegri, F.; Bronte, V.; Ferrini, S.; Barbieri, O. Exocytosis of azurophil and arginase 1-containing granules by activated polymorphonuclear neutrophils is required to inhibit T lymphocyte proliferation. J. Leukoc. Biol. 2011, 89, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef] [PubMed]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Vizio, B.; Novarino, A.; Giacobino, A.; Cristiano, C.; Prati, A.; Ciuffreda, L.; Montrucchio, G.; Bellone, G. Potential plasticity of T regulatory cells in pancreatic carcinoma in relation to disease progression and outcome. Exp. Ther. Med. 2012, 4, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The Combined Effects of Trypptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor -Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zhang, B.B.; Hua, H.; Li, B.; Zhang, B.; Yu, Q.; Li, X.Y.; Liu, Y.; Pan, W.; Liu, X.Y.; et al. The dynamics of Treg/Th17 and the imbalance of Treg/Th17 in Clonorchis sinensis-infected mice. PLoS ONE 2015, 10, e0143217. [Google Scholar] [CrossRef] [PubMed]
- Gnerlich, J.L.; Mitchem, J.B.; Weir, J.S.; Sankpal, N.V.; Kashiwagi, H.; Belt, B.A.; Porembka, M.R.; Herndon, J.M.; Eberlein, T.J.; Goedegebuure, P.; et al. Induction of Th17 cells in the tumor microenvironment improves survival in a murine model of pancreatic cancer. J. Immunol. 2010, 185, 4063–4071. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and clinical significance of Th17 cells in the tumor microenvironment and peripheral blood of pancreatic cancer patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef] [PubMed]
- Protti, M.P.; Monte, L.D. Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Front. Physiol. 2013, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Donahue, T.R.; Tran, L.M.; Hill, R.; Li, Y.; Kovochich, A.; Calvopina, J.H.; Patel, S.G.; Wu, N.; Hindoyan, A.; Farrell, J.J.; et al. Integrative survival-based molecular profiling of human pancreatic cancer. Clin. Cancer Res. 2012, 18, 1352–1363. [Google Scholar] [CrossRef] [PubMed]
- Stratford, J.K.; Bentrem, D.J.; Anderson, J.M.; Fan, C.; Volmar, K.A.; Marron, J.S.; Routh, E.D.; Caskey, L.S.; Samuel, J.C.; Der, C.J.; et al. A six-gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med. 2010, 7, e1000307. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Wang, J.; Nagano, A.; Desai, A.; Arumugam, P.; Dumartin, L.; Fitzgibbon, J.; Hagemann, T.; Marshall, J.F.; Kocher, H.M.; et al. A multi-gene signature predicts outcome in patients with pancreatic ductal adenocarcinoma. Genome Med. 2014, 6, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Markowetz, F.; De Sousa, E.M.F.; Medema, J.P.; Vermeulen, L. Dissecting cancer heterogeneity—An unsupervised classification approach. Int. J. Biochem. Cell Biol. 2013, 45, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kang, M.J.; Lee, S.; Bae, S.; Han, S.; Jang, J.-Y.; Park, T. Identifying molecular subtypes related to clinicopathologic factors in pancreatic cancer. Biomed. Eng. Online 2014, 13, S5. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.L.; Corchete, L.; Teodosio, C.; Sarasquete, M.E.; del Mar Abad, M.; Iglesias, M.; Esteban, C.; Sayagues, J.M.; Orfao, A.; Munoz-Bellvis, L. Identification and characterization of the gene expression profiles for protein coding and non-coding RNAs of pancreatic ductal adenocarcinomas. Oncotarget 2015, 6, 19070–19086. [Google Scholar] [CrossRef] [PubMed]
- Janky, R.; Binda, M.M.; Allemeersch, J.; Van den Broeck, A.; Govaere, O.; Swinnen, J.V.; Roskams, T.; Aerts, S.; Topal, B. Prognostic relevance of molecular subtypes and master regulators in pancreatic ductal adenocarcinoma. BMC Cancer 2016, 16, 632. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Start. | Trial | No Pts | Study Design | Treatment | TME Targeting Agent | PI |
|---|---|---|---|---|---|---|
| 2017 | NCT03307148 | 34 | Single group assignment | ATRA with Gemcitabine and Nab-paclitaxel | ATRA: all trans retinoic acid, involved in the inactivation of PSCs | David Propper |
| 2017 | NCT03098160 | 69 | Sequential assignment | Evofosfamide and ipilimumab | Evofosfamide: hypoxia-directed cytotoxic agent Ipilimumab: activating cytotoxic T-cells | David Hong |
| 2017 | NCT03168139 | 20 | Single group assignment | Olaptesed pegol w/wo Pembrolizumab | Olaptesed: targeting CXCL12, involved in vascular homeostasis Pembrolizumab: inhibition of PD-1 receptor, immune checkpoint inhibitor | Unknown |
| 2017 | NCT03277209 | 28 | Single group assignment | Dose escalation plerixafor | Plerixafor: targeting CXCR4, receptor for stroma derived factor-1, involved modulation of the immune micro-environment | Elizabeta Popa |
| 2016 | NCT02777710 | 58 | Single group assignment | Durvalumab and Pexidartinib | Durvalumab: inhibits PD-L1, immune checkpoint inhibitor Pexidartinib: inhibits CSF1R, depleting the TME from M2-like TAMs | Philippe Cassier |
| 2106 | NCT02921022 | 56 | Non-randomized parallel assignment | Prophylactic/therapeutic rivaroxaban with Gemcitabine, Nab-paclitaxel and PEGPH20 | PEGPH20: hyaluronidase enzyme, targeting HA | Kenneth Yu |
| 2016 | NCT02726854 | 30 | Single group assignment | Apatinib | Apatinib: VEGFR-2 inhibitor, inhibiting angiogenesis | Enxiao Li |
| 2015 | NCT02451982 | 50 | Randomized parallel assignment | Cyclophosphamide and GVAX Pancreatic cancer w/wo Nivolumab | GVAX: vaccine secreting granulocyte-macrophage colony stimulating factor, stimulates immune response Nivolumab: inhibits PD-L1, immune checkpoint inhibitor | Lei Zheng |
| 2015 | NCT02546531 | 50 | Non-randomized parallel assignment | Dose escalation/expansion of defactinib, pembrolizumab and gemcitabine | Defactinib: FAK inhibitor, involved in the inhibition of fibrosis and inflammation and anti-tumor effects Pembrolizumab: inhibition of PD-1 receptor, immune checkpoint inhibitor | Andrea Wang-Gillam |
| 2015 | NCT02179970 | 28 | Single group assignment | dose escalation plerixafor | Plerixafor: targeting CXCR4, receptor for stroma derived factor-1, involved modulation of the immune microenvironment | Duncan Jodrell |
| 2014 | NCT02030860 | 15 | Randomized single group assignment | Gemcitabine, Nab-paclitaxel w/wo Paricalcitol | Paricalcitol: active vitamin D2 analog, targeting fibrosis in the TME | Peter O’Dwyer |
| 2014 | NCT02159989 | 69 | Single group assignment | Sapanisertib and Ziv-Aflibercept | Ziv-Aflibercept: VEGF inhibitor, involved in angiogenesis inhibition | Aung Naing |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veenstra, V.L.; Garcia-Garijo, A.; Van Laarhoven, H.W.; Bijlsma, M.F. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers 2018, 10, 34. https://doi.org/10.3390/cancers10020034
Veenstra VL, Garcia-Garijo A, Van Laarhoven HW, Bijlsma MF. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers. 2018; 10(2):34. https://doi.org/10.3390/cancers10020034
Chicago/Turabian StyleVeenstra, Veronique L., Andrea Garcia-Garijo, Hanneke W. Van Laarhoven, and Maarten F. Bijlsma. 2018. "Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer" Cancers 10, no. 2: 34. https://doi.org/10.3390/cancers10020034
APA StyleVeenstra, V. L., Garcia-Garijo, A., Van Laarhoven, H. W., & Bijlsma, M. F. (2018). Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers, 10(2), 34. https://doi.org/10.3390/cancers10020034