Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viruses
2.2. 3D Skin Model
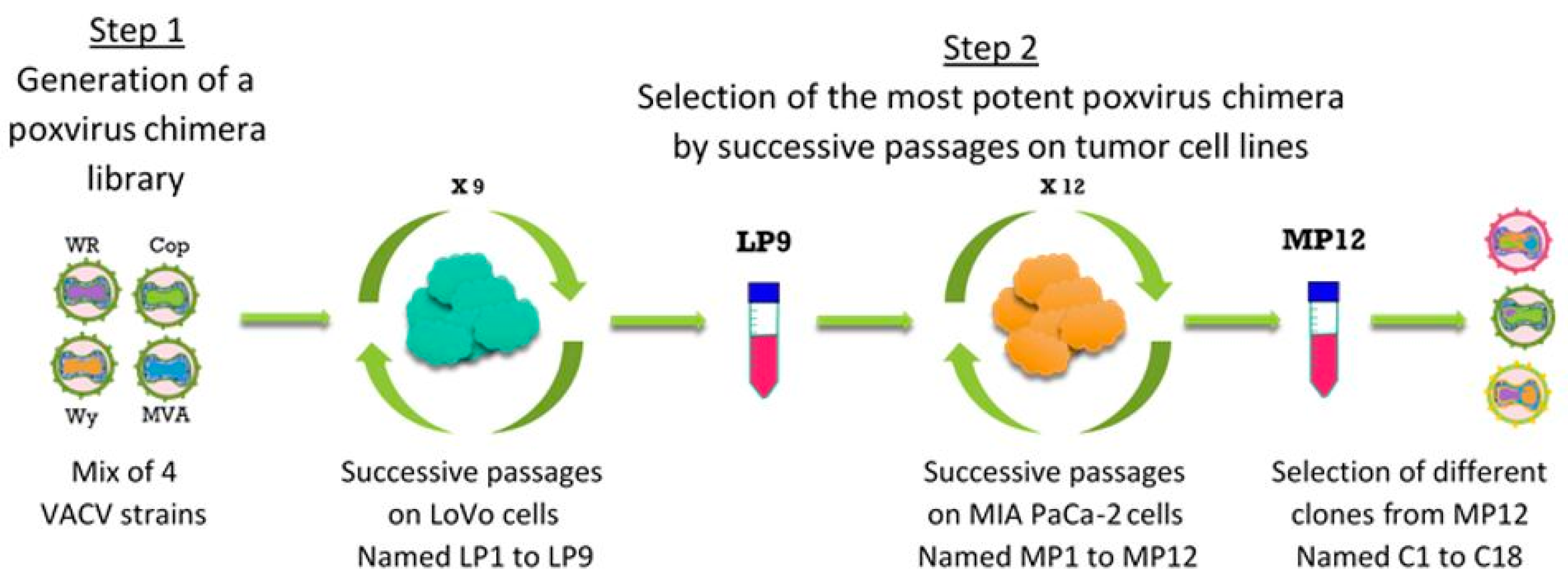
2.3. Directed Evolution for Selection of Chimeric Vaccinia Virus deVV5
2.4. Generation of deVV5-fcu1
2.5. In Vitro Cytotoxicity Assay
2.6. In Vitro Virus Yield Assay
2.7. In Vitro Cell Sensitivity to 5-FC
2.8. Cytosine Deaminase Enzymatic Assay
2.9. DNA Sequencing
2.10. Statistical Analyses
3. Results
3.1. Oncolytic Activity of the Four VACV Parental Strains
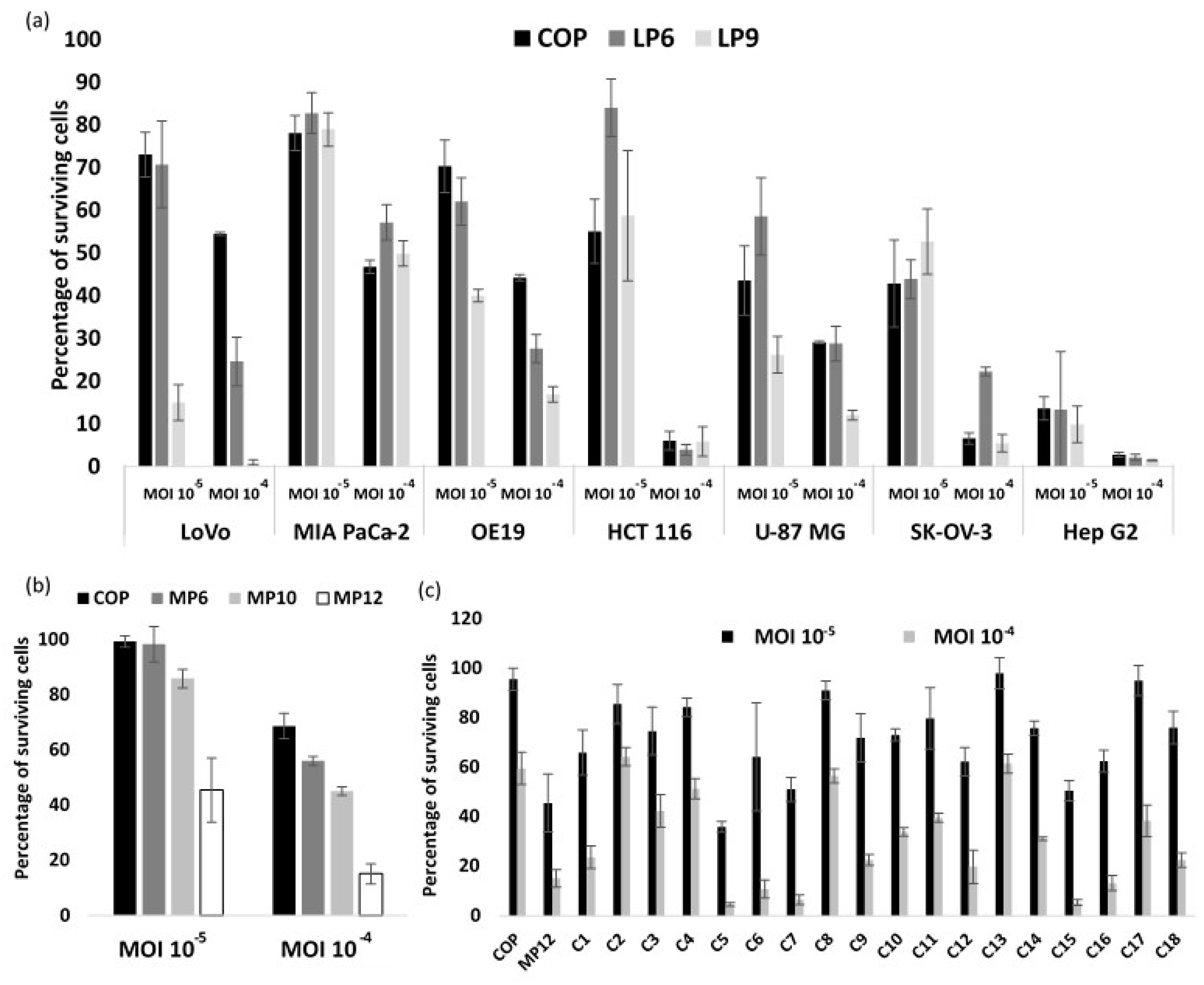
3.2. VACV Shuffling: deVV5 Is a Chimeric Virus with Enhanced Oncolytic Potency In Vitro
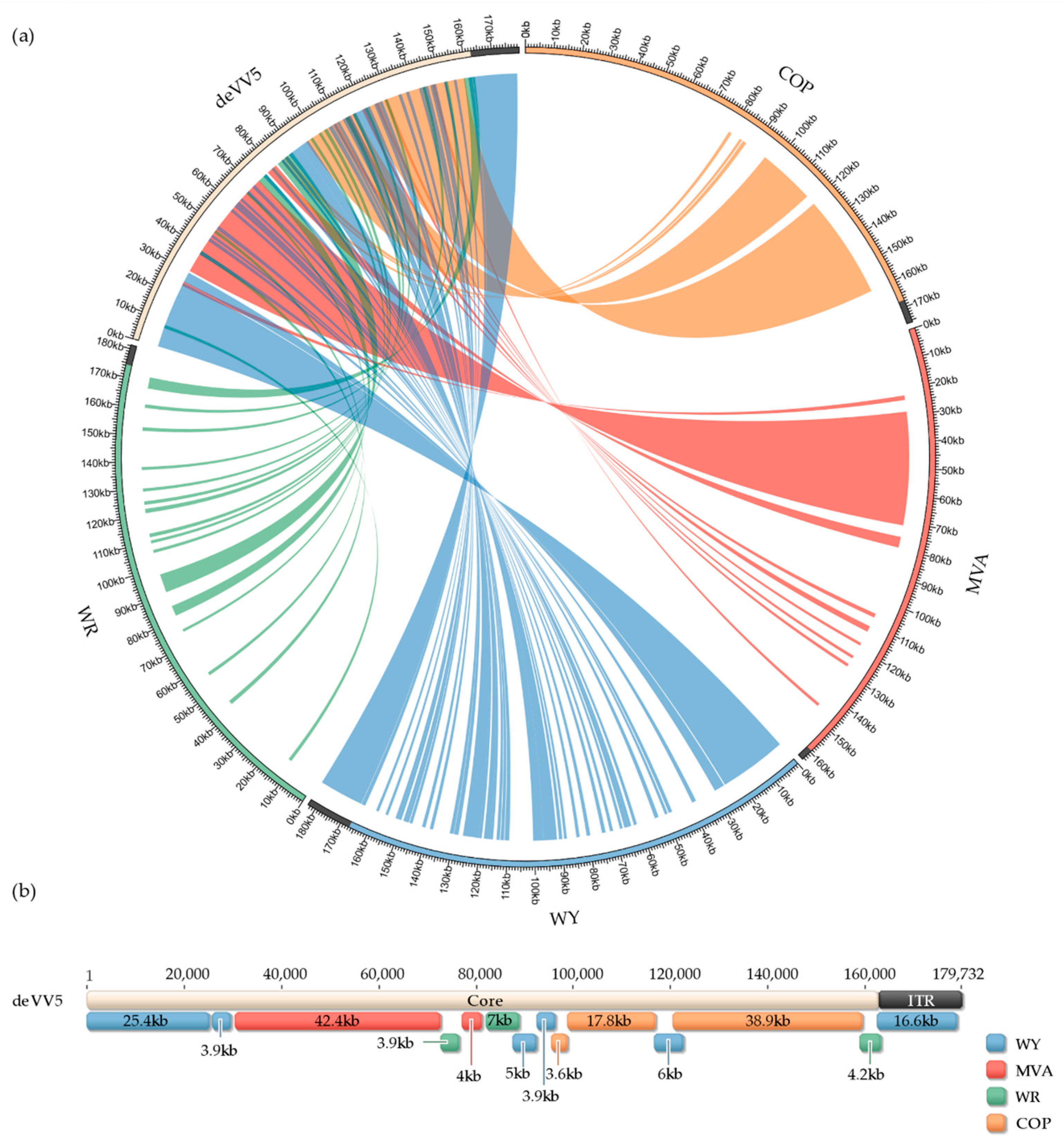
3.3. Genome Analysis
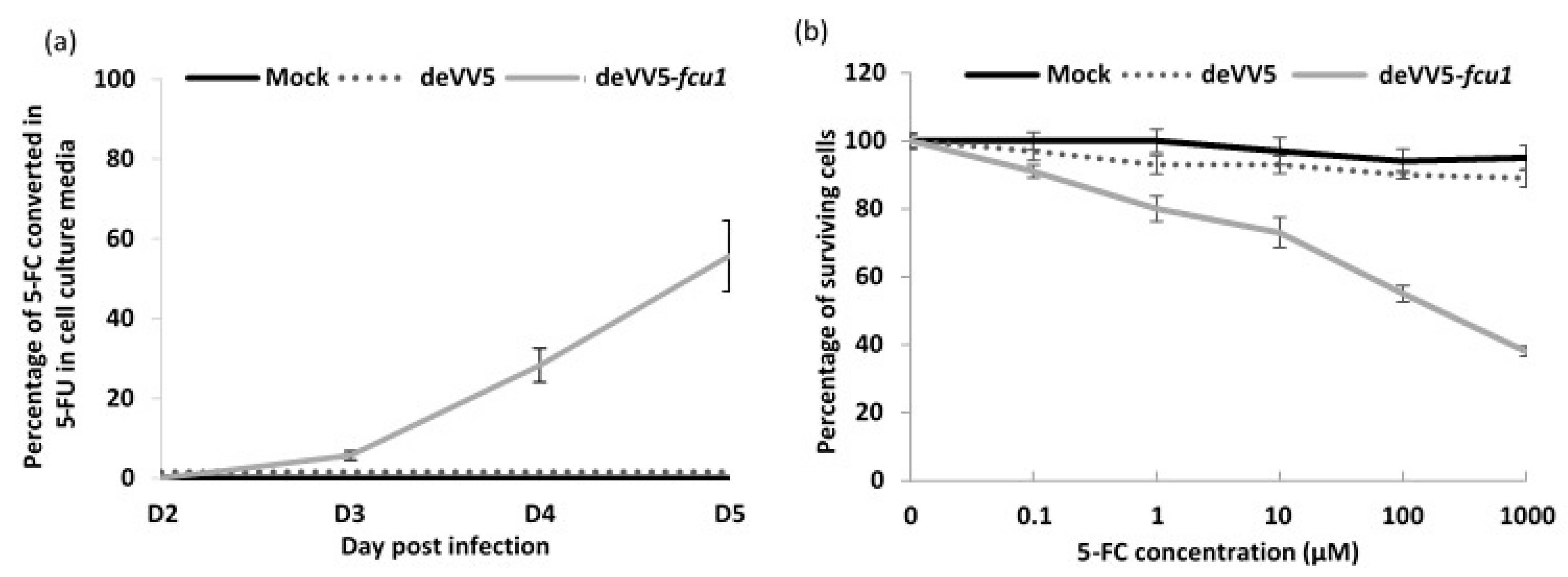
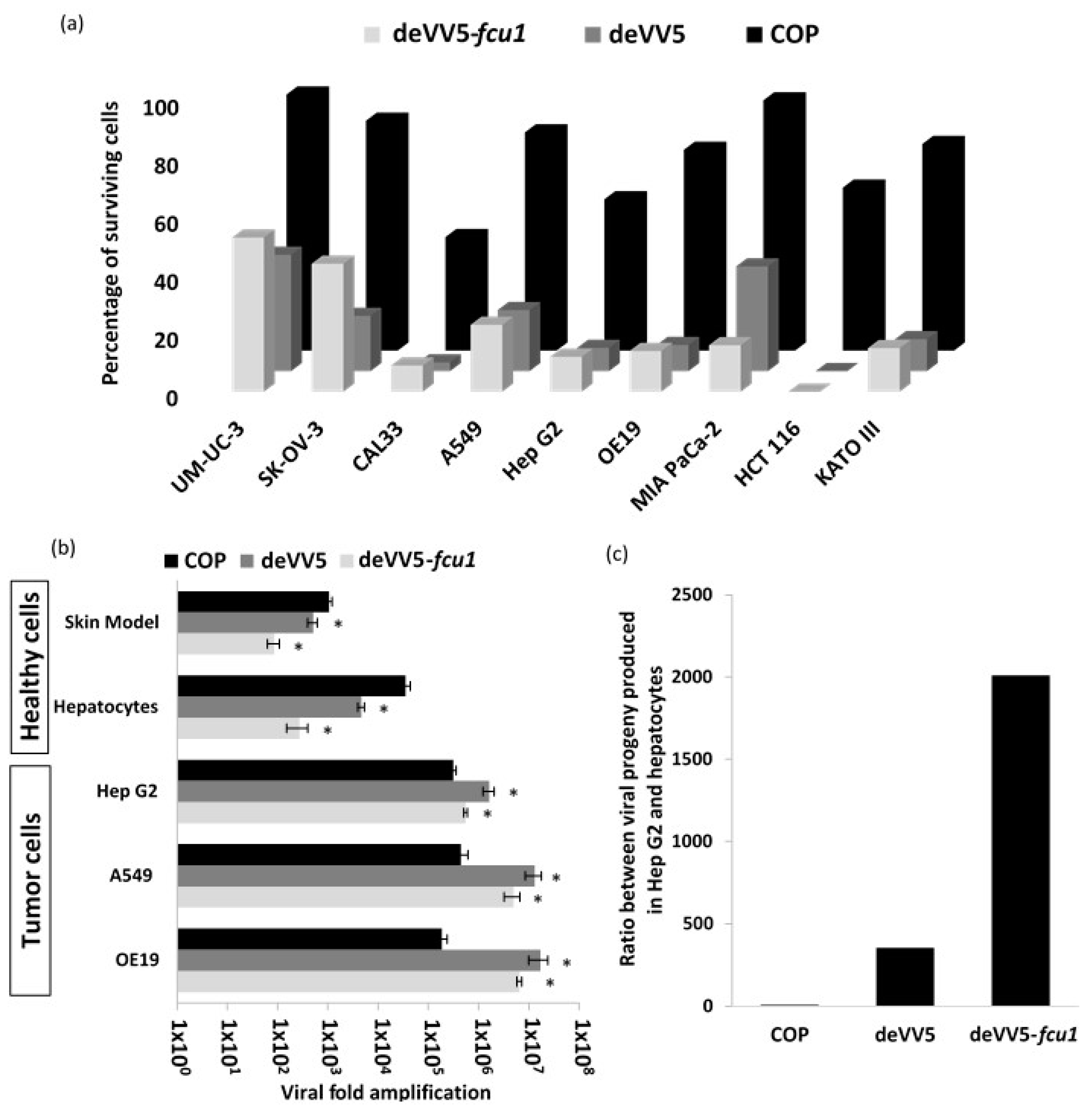
3.4. Arming of deVV5 Leads to Increased Potency
3.5. Increased Oncolytic and Replicative Efficiency of deVV5 and deVV5-fcu1
3.6. Replication of deVV5 and deVV5-fcu1 on Human Primary Cells
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Heinrich, B.; Klein, J.; Delic, M.; Goepfert, K.; Engel, V.; Geberzahn, L.; Lusky, M.; Erbs, P.; Preville, X.; Moehler, M. Immunogenicity of oncolytic vaccinia viruses JX-GFP and TG6002 in a human melanoma in vitro model: Studying immunogenic cell death, dendritic cell maturation and interaction with cytotoxic T lymphocytes. OncoTargets Ther. 2017, 10, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Fend, L.; Yamazaki, T.; Remy, C.; Fahrner, C.; Gantzer, M.; Nourtier, V.; Préville, X.; Quemeneur, E.; Kepp, O.; Adam, J.; et al. Immune Checkpoint Blockade, Immunogenic Chemotherapy or IFN-α Blockade Boost the Local and Abscopal Effects of Oncolytic Virotherapy. Cancer Res. 2017, 77, 4146–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Dey, M. Immune System, Friend or Foe of Oncolytic Virotherapy? Front. Oncol. 2017, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, J.F.; de Vor, L.; Fouchier, R.A.; van den Hoogen, B.G. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D. Genetically Engineered Vaccinia Viruses as Agents for Cancer Treatment, Imaging, and Transgene Delivery. Front. Oncol. 2017, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G. Poxvirus tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeh, H.J.; Downs-Canner, S.; McCart, J.A.; Guo, Z.S.; Rao, U.N.; Ramalingam, L.; Thorne, S.H.; Jones, H.L.; Kalinski, P.; Wieckowski, E.; et al. First-in-man study of western reserve strain oncolytic vaccinia virus: Safety, systemic spread, and antitumor activity. Mol. Ther. 2015, 23, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, J.; Kintz, J.; Futin, N.; Findeli, A.; Cordier, P.; Schlesinger, Y.; Hoffmann, C.; Tosch, C.; Balloul, J.M.; Erbs, P. Targeted delivery of a suicide gene to human colorectal tumors by a conditionally replicating vaccinia virus. Gene Ther. 2008, 15, 1361–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mell, L.K.; Brumund, K.T.; Daniels, G.A.; Advani, S.J.; Zakeri, K.; Wright, M.E.; Onyeama, S.J.; Weisman, R.A.; Sanghvi, P.R.; Martin, P.J.; et al. Phase I Trial of Intravenous Oncolytic Vaccinia Virus (GL-ONC1) with Cisplatin and Radiotherapy in Patients with Locoregionally Advanced Head and Neck Carcinoma. Clin. Cancer Res. 2017, 23, 5696–5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.Y.; Jeong, S.N.; Kang, D.H.; Heo, J. Evolutionary cancer-favoring engineered vaccinia virus for metastatic hepatocellular carcinoma. Oncotarget 2017, 8, 71489–71499. [Google Scholar] [CrossRef] [PubMed]
- Hruby, D.E. Vaccinia virus vectors: New strategies for producing recombinant vaccines. Clin. Microbiol. Rev. 1990, 3, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F. Genetic studies with mammalian poxviruses. II. Recombination between two strains of vaccinia virus in single HeLa cells. Virology 1959, 8, 499–507. [Google Scholar] [CrossRef]
- Woodroofe, G.M.; Fenner, F. Genetic studies with mammalian poxviruses. IV. Hybridization between several different poxviruses. Virology 1960, 12, 272–282. [Google Scholar] [CrossRef]
- Paszkowski, P.; Noyce, R.S.; Evans, D.H. Live-Cell Imaging of Vaccinia Virus Recombination. PLoS Pathog. 2016, 12, e1005824. [Google Scholar] [CrossRef] [PubMed]
- Koerber, J.T.; Maheshri, N.; Kaspar, B.K.; Schaffer, D.V. Construction of diverse adeno-associated viral libraries for directed evolution of enhanced gene delivery vehicles. Nat. Protoc. 2006, 1, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Harden, P.; Bauzon, M.; Chartier, C.; Nye, J.; Thorne, S.; Reid, T.; Ni, S.; Lieber, A.; Fisher, K.; et al. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS ONE 2008, 3, e2409. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Bauzon, M.; Green, N.; Seymour, L.; Fisher, K.; Hermiston, T. OvAd1, a Novel, Potent, and Selective Chimeric Oncolytic Virus Developed for Ovarian Cancer by 3D-Directed Evolution. Mol. Ther. Oncolytics 2016, 4, 55–66. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.P.; Choi, A.H.; Kim, S.I.; Chaurasiya, S.; Lu, J.; Park, A.K.; Woo, Y.; Warner, S.G.; Fong, Y.; Chen, N.G. Novel oncolytic chimeric orthopoxvirus causes regression of pancreatic cancer xenografts and exhibits abscopal effect at a single low dose. J. Transl. Med. 2018, 16, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Sampedro, L.; Perdiguero, B.; Mejías-Pérez, E.; García-Arriaza, J.; Di Pilato, M.; Esteban, M. The evolution of poxvirus vaccines. Viruses 2015, 7, 1726–1803. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Stickl, H.; Müller, H.K.; Danner, K.; Singer, H. The smallpox vaccination strain MVA: Marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism. Zentralbl. Bakteriol. B 1978, 167, 375–390. [Google Scholar] [PubMed]
- Erbs, P.; Findeli, A.; Kintz, J.; Cordier, P.; Hoffmann, C.; Geist, M.; Balloul, J.M. Modified vaccinia virus Ankara as a vector for suicide gene therapy. Cancer Gene Ther. 2008, 15, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Husseini, F.; Delord, J.P.; Fournel-Federico, C.; Guitton, J.; Erbs, P.; Homerin, M.; Halluard, C.; Jemming, C.; Orange, C.; Limacher, J.M.; et al. Vectorized gene therapy of liver tumors: Proof-of-concept of TG4023 (MVA-FCU1) in combination with flucytosine. Ann. Oncol. 2017, 28, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Quoix, E.; Lena, H.; Losonczy, G.; Forget, F.; Chouaid, C.; Papai, Z.; Gervais, R.; Ottensmeier, C.; Szczesna, A.; Kazarnowicz, A.; et al. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): Results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol. 2016, 17, 212–223. [Google Scholar] [CrossRef]
- Erbs, P.; Regulier, E.; Kintz, J.; Leroy, P.; Poitevin, Y.; Exinger, F.; Jund, R.; Mehtali, M. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 2000, 60, 3813–3822. [Google Scholar] [PubMed]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricordel, M.; Foloppe, J.; Pichon, C.; Sfrontato, N.; Antoine, D.; Tosch, C.; Cochin, S.; Cordier, P.; Quemeneur, E.; Camus-Bouclainville, C.; et al. Cowpox Virus: A New and Armed Oncolytic Poxvirus. Mol. Ther. Oncolytics 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.R.; Horvath, A.; Annels, N.E.; Pencavel, T.; Metcalf, S.; Seth, R.; Peschard, P.; Price, T.; Coffin, R.S.; Mostafid, H.; et al. Combination of a fusogenic glycoprotein, pro-drug activation and oncolytic HSV as an intravesical therapy for superficial bladder cancer. Br. J. Cancer 2012, 106, 496–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, J.D.; Liikanen, I.; Guse, K.; Foloppe, J.; Sloniecka, M.; Diaconu, I.; Rantanen, V.; Eriksson, M.; Hakkarainen, T.; Lusky, M.; et al. Targeted chemotherapy for head and neck cancer with a chimeric oncolytic adenovirus coding for bifunctional suicide protein FCU1. Clin. Cancer Res. 2010, 16, 2540–2549. [Google Scholar] [CrossRef] [PubMed]
- Quirin, C.; Rohmer, S.; Fernández-Ulibarri, I.; Behr, M.; Hesse, A.; Engelhardt, S.; Erbs, P.; Enk, A.H.; Nettelbeck, D.M. Selectivity and efficiency of late transgene expression by transcriptionally targeted oncolytic adenoviruses are dependent on the transgene insertion strategy. Hum. Gene Ther. 2011, 22, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, J.K.; Bossow, S.; Grossardt, C.; Sawall, S.; Kupsch, J.; Erbs, P.; Hassel, J.C.; von Kalle, C.; Enk, A.H.; Nettelbeck, D.M.; et al. Chemovirotherapy of malignant melanoma with a targeted and armed oncolytic measles virus. J. Investig. Dermatol. 2013, 133, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Hammer, K.; Kazcorowski, A.; Liu, L.; Behr, M.; Schemmer, P.; Herr, I.; Nettelbeck, D.M. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int. J. Cancer 2015, 137, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | MVA | WY | WR | COP |
|---|---|---|---|---|
| A549 | 94.5 ± 3.6 | 90.2 ± 3.2 | 58.6 ± 3.7 | 47.8 ± 4.8 |
| LoVo | 98.6 ± 2.7 | 90.4 ± 5.9 | 70.8 ± 6.2 | 73.1 ± 5.2 |
| MIA PaCa-2 | 103.2 ± 4.2 | 92.8 ± 2.9 | 89.4 ± 2.4 | 78.1 ± 1.5 |
| U-87 MG | 102.8 ± 4.8 | 64.6 ± 1.6 | 41.0 ± 0.8 | 43.6 ± 0.3 |
| Hep G2 | 97.3 ± 3.9 | 66.8 ± 4.9 | 70.9 ± 9.4 | 13.6 ± 2.7 |
| HCT 116 | 99.2 ± 5.4 | 91.5 ± 3.4 | 94.6 ± 3.4 | 55.1 ± 7.5 |
| OE19 | 105.2 ± 6.2 | 78.9 ± 2.2 | 77.6 ± 3.3 | 70.3 ± 0.7 |
| SK-OV-3 | 95.4 ± 7.3 | 59.8 ± 1.8 | 99.7 ± 1.5 | 42.9 ± 1.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricordel, M.; Foloppe, J.; Antoine, D.; Findeli, A.; Kempf, J.; Cordier, P.; Gerbaud, A.; Grellier, B.; Lusky, M.; Quemeneur, E.; et al. Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency. Cancers 2018, 10, 231. https://doi.org/10.3390/cancers10070231
Ricordel M, Foloppe J, Antoine D, Findeli A, Kempf J, Cordier P, Gerbaud A, Grellier B, Lusky M, Quemeneur E, et al. Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency. Cancers. 2018; 10(7):231. https://doi.org/10.3390/cancers10070231
Chicago/Turabian StyleRicordel, Marine, Johann Foloppe, Delphine Antoine, Annie Findeli, Juliette Kempf, Pascale Cordier, Aude Gerbaud, Benoit Grellier, Monika Lusky, Eric Quemeneur, and et al. 2018. "Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency" Cancers 10, no. 7: 231. https://doi.org/10.3390/cancers10070231
APA StyleRicordel, M., Foloppe, J., Antoine, D., Findeli, A., Kempf, J., Cordier, P., Gerbaud, A., Grellier, B., Lusky, M., Quemeneur, E., & Erbs, P. (2018). Vaccinia Virus Shuffling: deVV5, a Novel Chimeric Poxvirus with Improved Oncolytic Potency. Cancers, 10(7), 231. https://doi.org/10.3390/cancers10070231