Interplay between Epigenetics, Expression of Estrogen Receptor- α, HER2/ERBB2 and Sensitivity of Triple Negative Breast Cancer Cells to Hormonal Therapy

 and
and
Abstract
:1. Introduction
2. Results
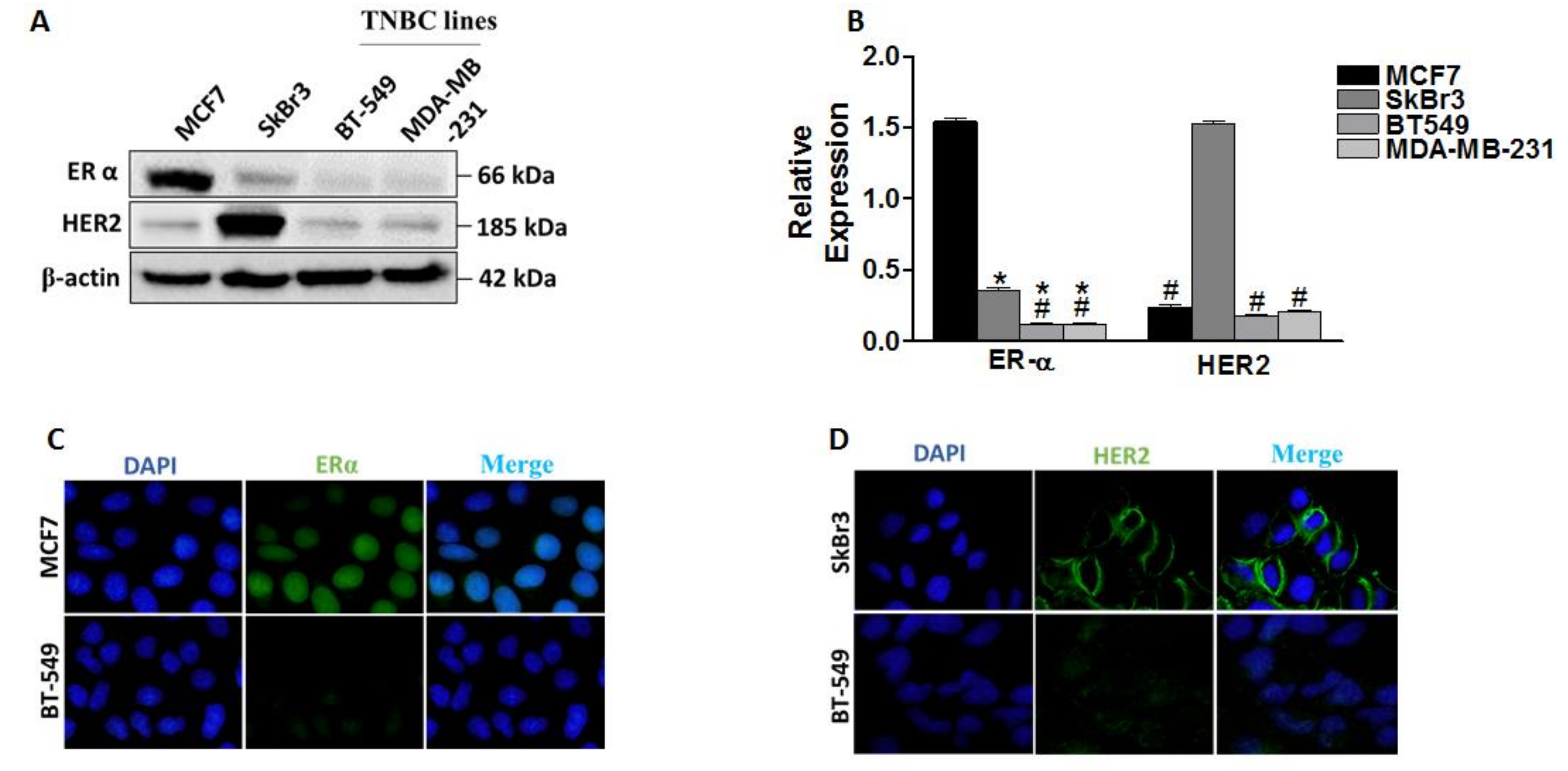
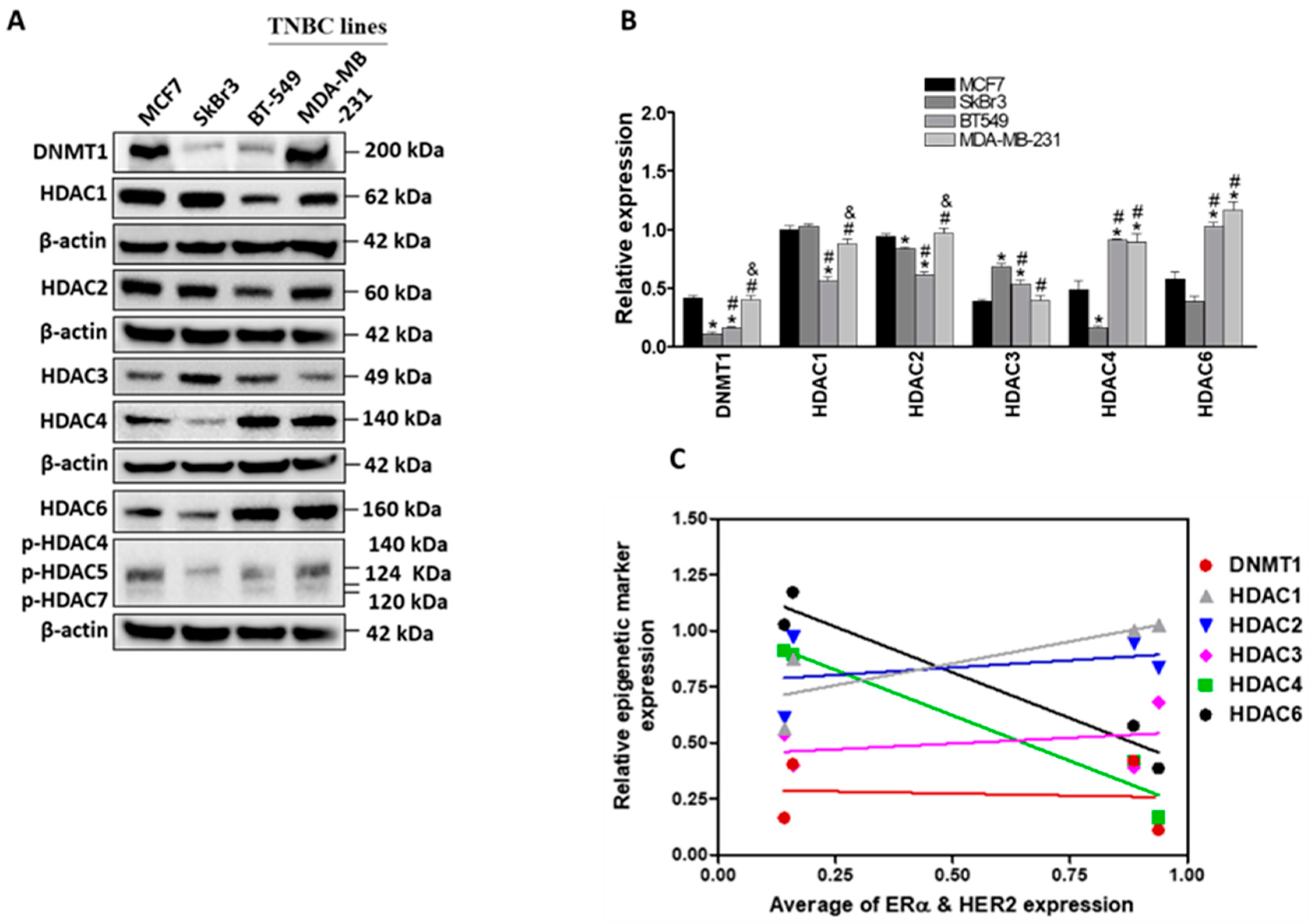
2.1. Differential Expression of ERα, HER2/ERBB2, DNMT1 and HDACs in Breast Cancer Cells
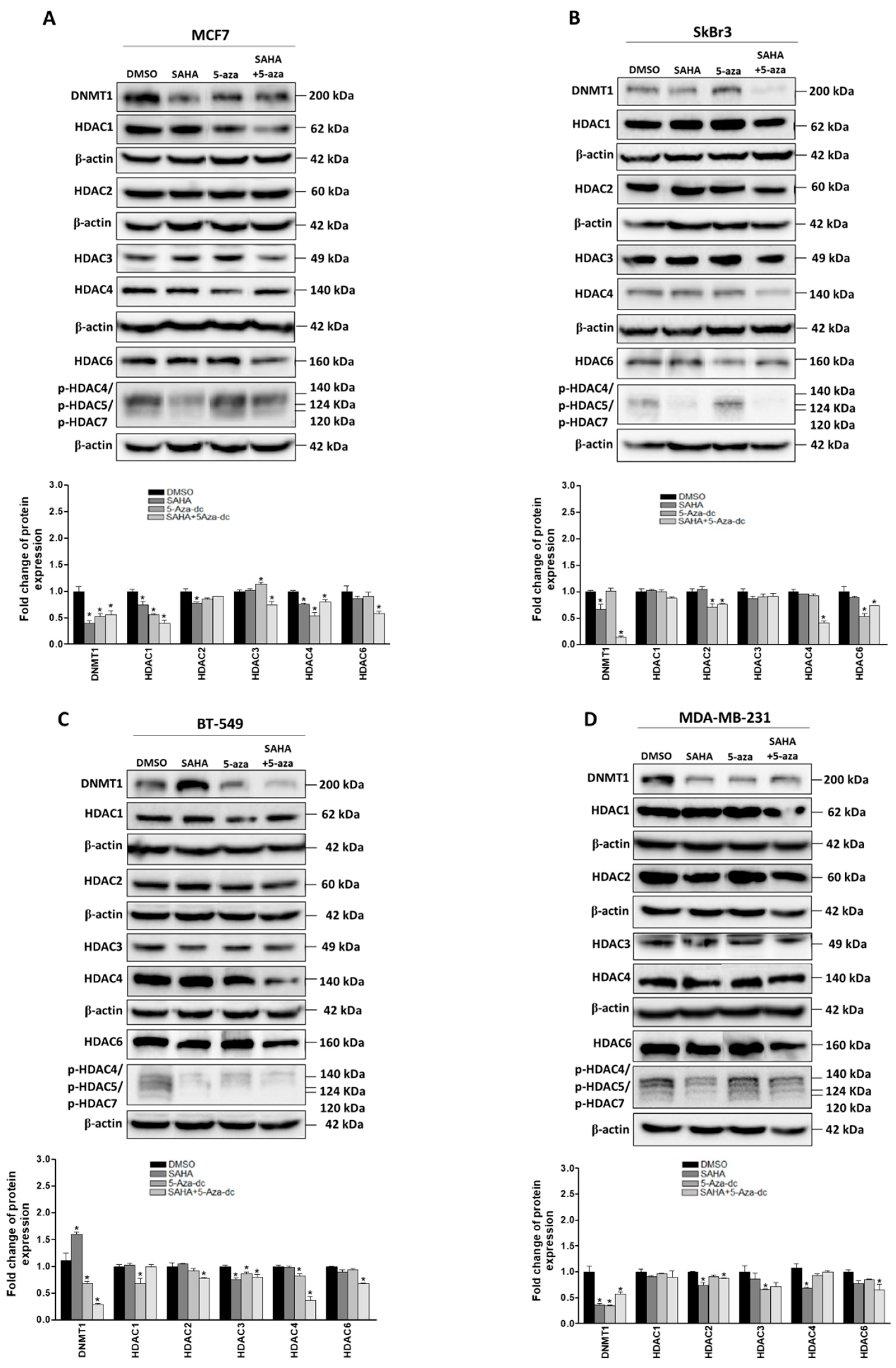
2.2. Effect of SAHA and/or 5-aza-dc on the Expression of Epigenetic Modulators
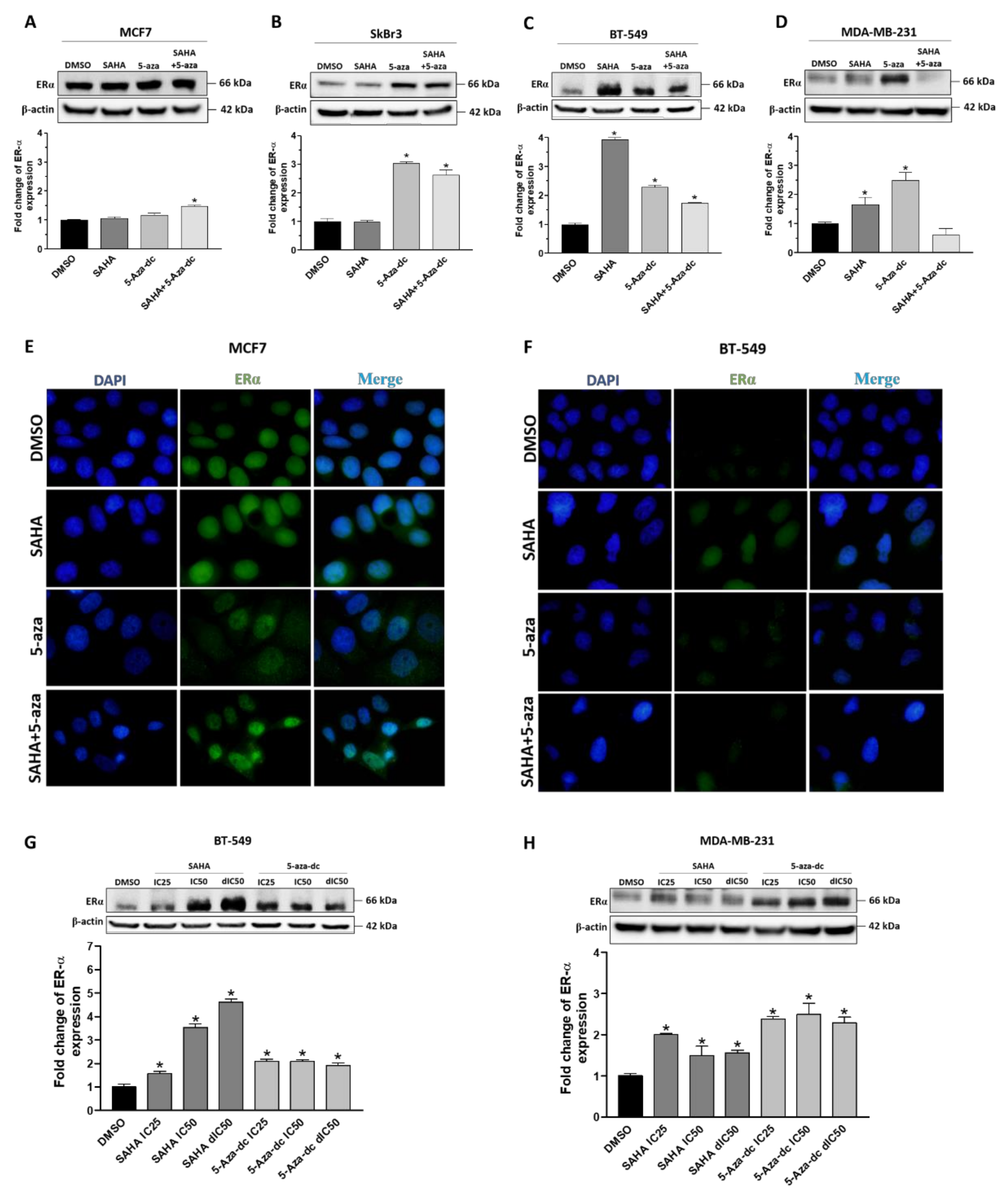
2.3. Effect of SAHA and/or 5-aza-dc on the Expression Level of ERα
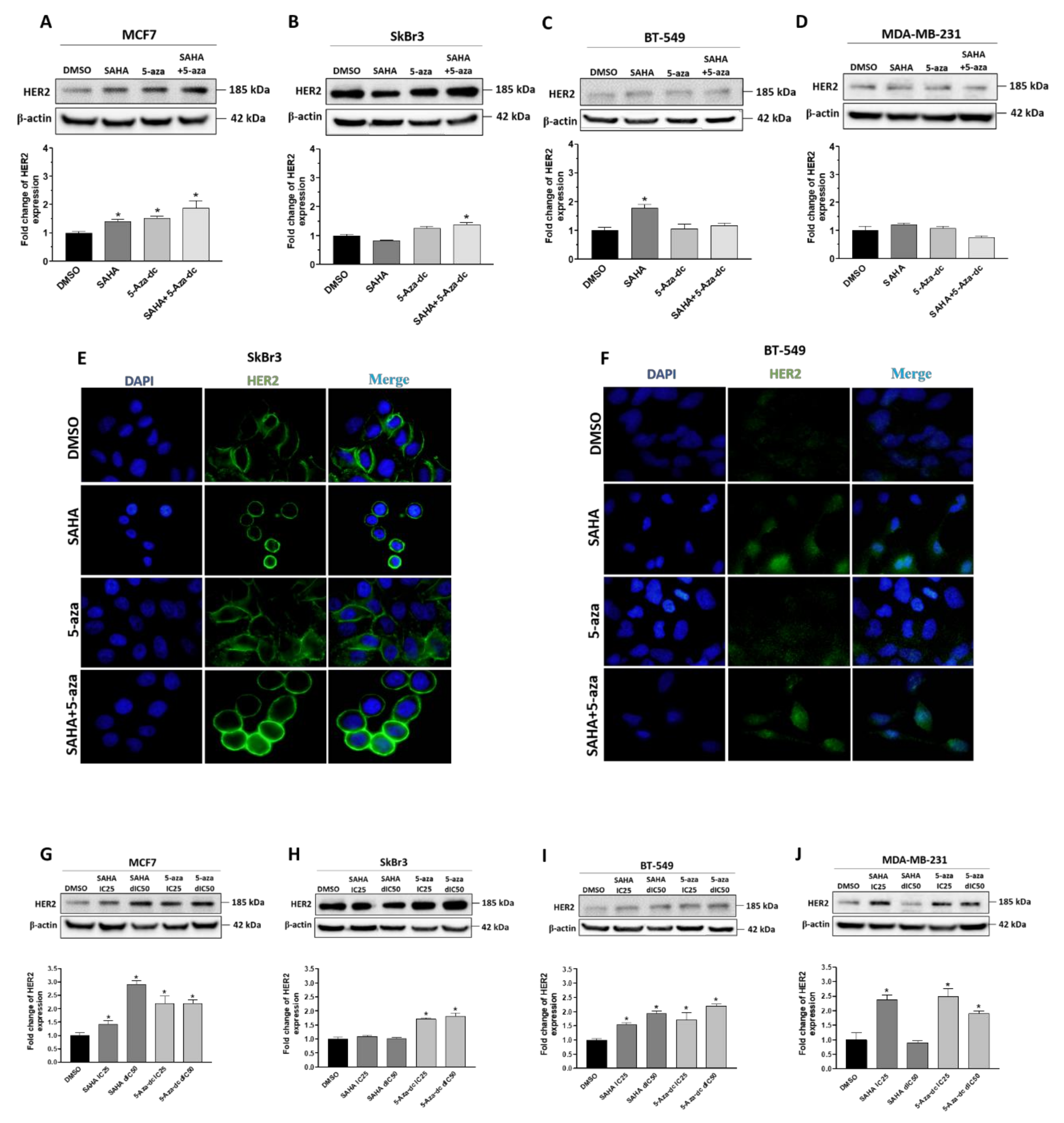
2.4. Effect of SAHA and/or 5-aza-dc on the Expression Level of HER2/ERBB2
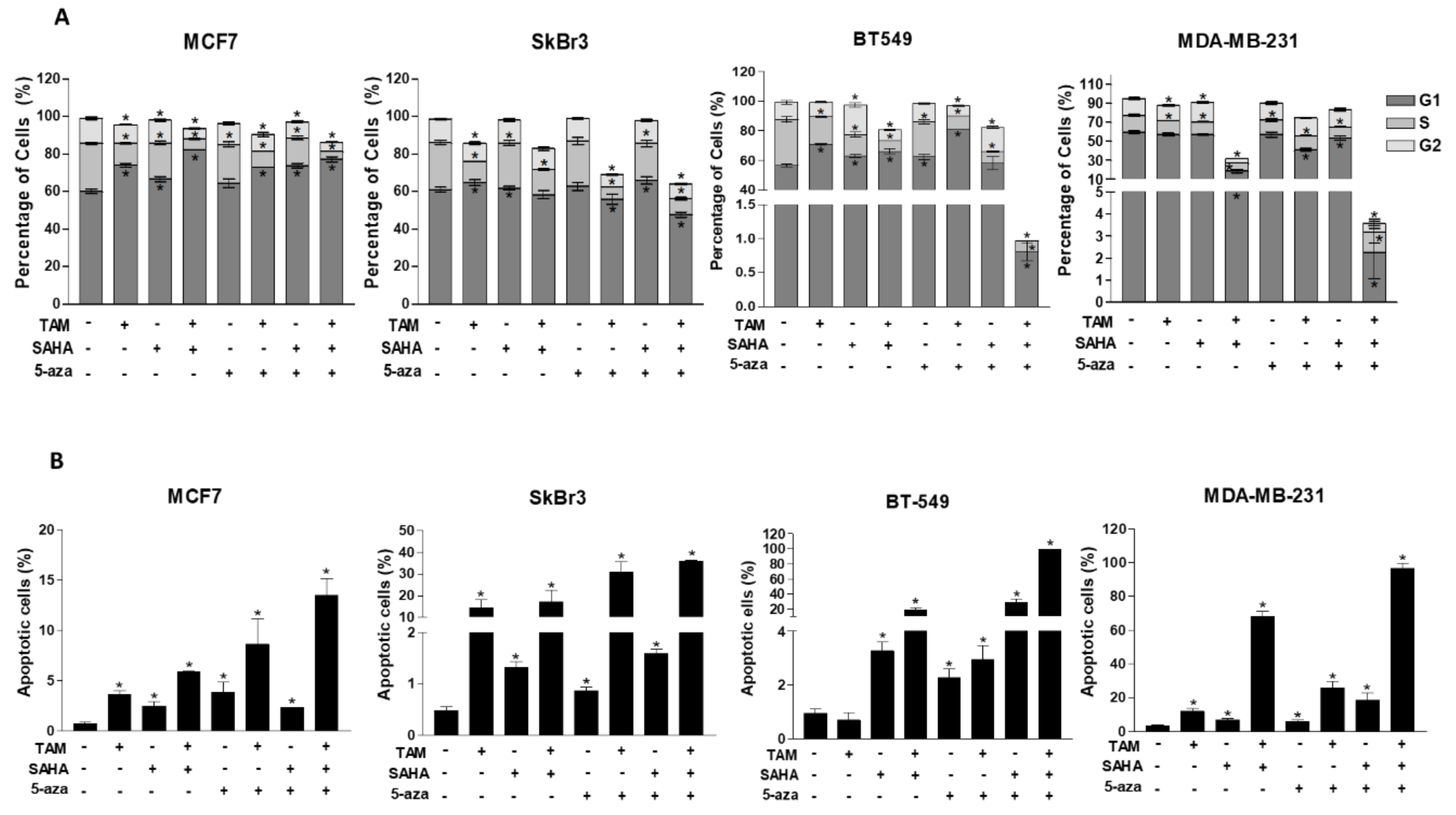
2.5. Effect of SAHA and/or 5-aza-dc on the Response of Breast Cancer Cells to Hormonal Therapy
2.6. Effect of SAHA and/or 5-aza-dc on Cell Cycle Distribution and Apoptosis in Breast Cancer Cells
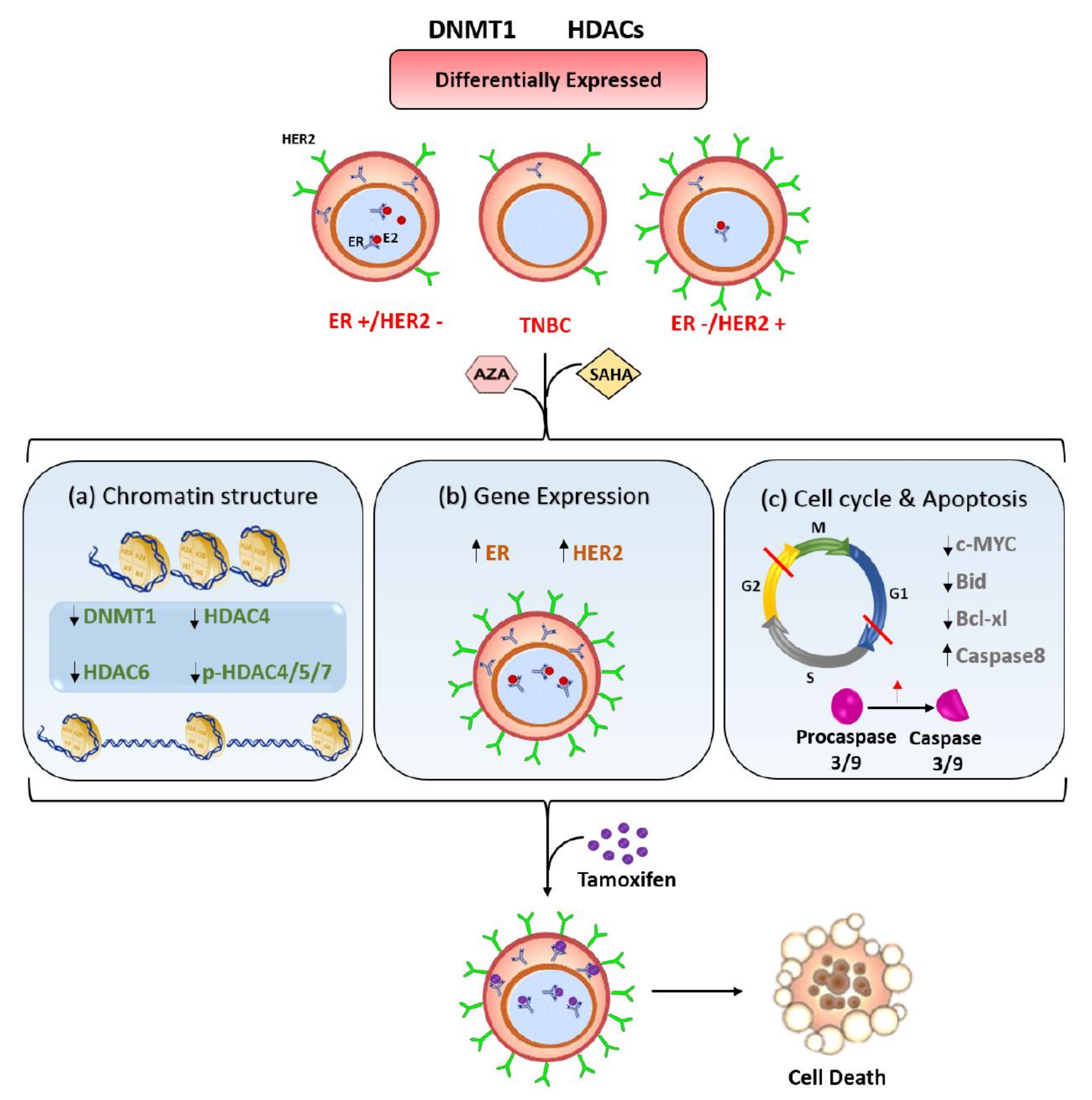
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Colony Formation Assay
4.3. Sulforhodamine B (SRB) Assay
4.4. Western Blot
4.5. Immunofluorescence
4.6. Cell Cycle Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Russnes, H.G.; Lingjaerde, O.C.; Borresen-Dale, A.L.; Caldas, C. Breast Cancer Molecular Stratification: From Intrinsic Subtypes to Integrative Clusters. Am. J. Pathol. 2017, 187, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Podo, F.; Buydens, L.M.; Degani, H.; Hilhorst, R.; Klipp, E.; Gribbestad, I.S.; Van Huffel, S.; van Laarhoven, H.W.; Luts, J.; Monleon, D.; et al. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 2010, 4, 209–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peddi, P.F.; Ellis, M.J.; Ma, C. Molecular basis of triple negative breast cancer and implications for therapy. Int. J. Breast Cancer 2012, 2012, 217185. [Google Scholar] [CrossRef] [PubMed]
- Diana, A.; Franzese, E.; Centonze, S.; Carlino, F.; Della Corte, C.M.; Ventriglia, J.; Petrillo, A.; De Vita, F.; Alfano, R.; Ciardiello, F.; et al. Triple-Negative Breast Cancers: Systematic Review of the Literature on Molecular and Clinical Features with a Focus on Treatment with Innovative Drugs. Curr. Oncol. Rep. 2018, 20, 76. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jin, J.; Ji, W.; Guan, X. Therapeutic landscape in mutational triple negative breast cancer. Mol. Cancer 2018, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef] [PubMed]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, S142–S150. [Google Scholar] [CrossRef]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Strauss, J.; Figg, W.D. Using Epigenetic Therapy to Overcome Chemotherapy Resistance. Anticancer Res. 2016, 36, 1–4. [Google Scholar] [PubMed]
- Connolly, R.; Stearns, V. Epigenetics as a therapeutic target in breast cancer. J. Mammary Gland Biol. Neoplasia 2012, 17, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.S. Targeting epigenetics using synthetic lethality in precision medicine. Cell. Mol. Life Sci. 2018, 75, 3381–3392. [Google Scholar] [CrossRef] [PubMed]
- Howell, P.M.; Liu, Z.; Khong, H.T. Demethylating Agents in the Treatment of Cancer. Pharmaceuticals 2010, 3, 2022–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzbecker, U.; Middeke, J.M.; Sockel, K.; Herbst, R.; Wolf, D.; Baldus, C.D.; Oelschlagel, U.; Mutherig, A.; Fransecky, L.; Noppeney, R.; et al. Measurable residual disease-guided treatment with azacitidine to prevent haematological relapse in patients with myelodysplastic syndrome and acute myeloid leukaemia (RELAZA2): An open-label, multicentre, phase 2 trial. Lancet Oncol. 2018, 19, 1668–1679. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Grandjenette, C.; Ghelfi, J.; Karius, T.; Foliguet, B.; Dicato, M.; Diederich, M. Sustained exposure to the DNA demethylating agent, 2′-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem. Pharmacol. 2011, 81, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, C.; Chatterji, B.P. HDAC Inhibitors as Novel Anti-Cancer Therapeutics. Recent Pat. Anticancer Drug Discov. 2015, 10, 145–162. [Google Scholar] [CrossRef]
- Ghayad, S.E.; Rammal, G.; Sarkis, O.; Basma, H.; Ghamloush, F.; Fahs, A.; Karam, M.; Harajli, M.; Rabeh, W.; Mouawad, J.E.; et al. The histone deacetylase inhibitor Suberoylanilide Hydroxamic Acid (SAHA) as a therapeutic agent in rhabdomyosarcoma. Cancer Biol. Ther. 2018, 1–12. [Google Scholar] [CrossRef]
- Min, A.; Im, S.A.; Kim, D.K.; Song, S.H.; Kim, H.J.; Lee, K.H.; Kim, T.Y.; Han, S.W.; Oh, D.Y.; Kim, T.Y.; et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015, 17, 33. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Y.; Kiani, M.F.; Wang, B. Classification, Treatment Strategy, and Associated Drug Resistance in Breast Cancer. Clin. Breast Cancer 2016, 16, 335–343. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Elledge, R. Molecular markers for predicting response to tamoxifen in breast cancer patients. Endocrine 2000, 13, 1–10. [Google Scholar] [CrossRef]
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 2008, 29, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, O.A.; Esteller, M. Epigenetic modifications in breast cancer and their role in personalized medicine. Am. J. Pathol. 2013, 183, 1052–1063. [Google Scholar] [CrossRef]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epigenetics in breast and prostate cancer. Methods Mol. Biol. 2015, 1238, 425–466. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.J.; Dietel, M.; Weichert, W.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer—Overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Sunami, E.; Shinozaki, M.; Sim, M.S.; Nguyen, S.L.; Vu, A.T.; Giuliano, A.E.; Hoon, D.S. Estrogen receptor and HER2/neu status affect epigenetic differences of tumor-related genes in primary breast tumors. Breast Cancer Res. 2008, 10, R46. [Google Scholar] [CrossRef]
- Roll, J.D.; Rivenbark, A.G.; Jones, W.D.; Coleman, W.B. DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol. Cancer 2008, 7, 15. [Google Scholar] [CrossRef]
- Saji, S.; Kawakami, M.; Hayashi, S.; Yoshida, N.; Hirose, M.; Horiguchi, S.; Itoh, A.; Funata, N.; Schreiber, S.L.; Yoshida, M.; et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene 2005, 24, 4531–4539. [Google Scholar] [CrossRef] [Green Version]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Richon, V. Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer 2006, 95, S2. [Google Scholar] [CrossRef]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Med. J. 2009, 50, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Broche, J.; Bashtrykov, P. Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes. Genes 2018, 9, 566. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Blum, J.; Yang, X.; Beaulieu, N.; Macleod, A.R.; Davidson, N.E. Release of methyl CpG binding proteins and histone deacetylase 1 from the Estrogen receptor alpha (ER) promoter upon reactivation in ER-negative human breast cancer cells. Mol. Endocrinol. 2005, 19, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Pryzbylkowski, P.; Obajimi, O.; Keen, J.C. Trichostatin A and 5 Aza-2′ deoxycytidine decrease estrogen receptor mRNA stability in ER positive MCF7 cells through modulation of HuR. Breast Cancer Res. Treat. 2008, 111, 15–25. [Google Scholar] [CrossRef]
- Billam, M.; Sobolewski, M.D.; Davidson, N.E. Effects of a novel DNA methyltransferase inhibitor zebularine on human breast cancer cells. Breast Cancer Res. Treat. 2010, 120, 581–592. [Google Scholar] [CrossRef]
- Chen, M.Y.; Liao, W.S.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C., Jr. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef] [Green Version]
- Henderson, C.; Mizzau, M.; Paroni, G.; Maestro, R.; Schneider, C.; Brancolini, C. Role of caspases, Bid, and p53 in the apoptotic response triggered by histone deacetylase inhibitors trichostatin-A (TSA) and suberoylanilide hydroxamic acid (SAHA). J. Biol. Chem. 2003, 278, 12579–12589. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Wakimoto, N.; Yin, D.; Gery, S.; Kawamata, N.; Takai, N.; Komatsu, N.; Chumakov, A.; Imai, Y.; Koeffler, H.P. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int. J. Cancer 2007, 121, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Awady, R.A.; Saleh, E.M.; Dahm-Daphi, J. Targeting DNA double-strand break repair: Is it the right way for sensitizing cells to 5-fluorouracil? Anticancer Drugs 2010, 21, 277–287. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, R.A.; Saleh, E.M.; Ezz, M.; Elsayed, A.M. Interaction of celecoxib with different anti-cancer drugs is antagonistic in breast but not in other cancer cells. Toxicol. Appl. Pharmacol. 2011, 255, 271–286. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, R.A.; Semreen, M.H.; Saber-Ayad, M.M.; Cyprian, F.; Menon, V.; Al-Tel, T.H. Modulation of DNA damage response and induction of apoptosis mediates synergism between doxorubicin and a new imidazopyridine derivative in breast and lung cancer cells. DNA Repair 2016, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Saleh, E.M.; El-awady, R.A.; Eissa, N.A.; Abdel-Rahman, W.M. Antagonism between curcumin and the topoisomerase II inhibitor etoposide: A study of DNA damage, cell cycle regulation and death pathways. Cancer Biol. Ther. 2012, 13, 1058–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, E.M.; El-Awady, R.A.; Abdel Alim, M.A.; Abdel Wahab, A.H. Altered expression of proliferation-inducing and proliferation-inhibiting genes might contribute to acquired doxorubicin resistance in breast cancer cells. Cell Biochem. Biophys. 2009, 55, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Wlodkowic, D.; Skommer, J.; Darzynkiewicz, Z. Flow cytometry-based apoptosis detection. Methods Mol. Biol. 2009, 559, 19–32. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | MCF7 | SkBr3 | BT549 | MDA-MB-231 |
|---|---|---|---|---|
| TAM IC50 (µM) | 6.8 ± 2.4 | 12.1 ± 0.89 | 11.9 ± 0.56 | 13.8 ± 0.1 |
| Relative ERα expression | 1.54 ± 0.02 | 0.35 ± 0.02 | 0.11 ± 0.01 | 0.11 ± 0.01 |
| Correlation | r = −0.09654, p = 0.0346 | |||
| (TAM IC50 versus Relative ERα expression) | ||||
| Relative HER2 expression | 0.23 ± 0.03 | 1.53 ± 0.02 | 0.17 ± 0.01 | 0.21 ± 0.005 |
| Correlation | r = −0.1877, p = 0.8123 | |||
| (TAM IC50 versus Relative HER2 expression) | ||||
| Correlation | ||
|---|---|---|
| Epigenetic Markers | r Values | p Values |
| DNMT1 | −0.1003 | ns |
| HDAC1 | 0.8099 | ns |
| HDAC2 | 0.3509 | ns |
| HDAC3 | 0.3238 | ns |
| HDAC4 | −0.9731 | 0.0269 |
| HDAC6 | −0.9711 | 0.0289 |
| IC50 (μM) ± SEM | ||||
|---|---|---|---|---|
| Cell Line | MCF7 | SkBr3 | BT-549 | MDA-MB-231 |
| SAHA | 0.600 ± 0.034 | 0.570 ± 0.042 | 1.500 ± 0.089 | 1.580 ± 0.12 |
| 5-aza-dc | 0.040 ± 0.0028 | 0.080 ± 0.0062 | 1.040 ± 0.074 | 0.026 ± 0.0011 |
| SAHA + 5-aza-dc | 0.107 ± 0.0312 | 0.0284 ± 0.0056 | 0.0097 ± 0.0021 | 0.0082 ± 0.0024 |
| Tamoxifen | 6.4481 ± 0.23 | > 10 | > 10 | >10 |
| Tam + IC25 SAHA | 6.3241 ± 0.31 | > 10 | > 10 | 0.0126 ± 0.00078 |
| Tam + IC50 SAHA | 0.5420 ± 0.014 | > 10 | 0.0022 ± 0.0001 | 0.0025 ± 0.0001 |
| Tam + IC25 5-aza-dc | 0.0117 ± 0.0009 | 0.0059 ± 0.00024 | 0.0022 ± 0.0001 | 0.0030 ± 0.0002 |
| Tam + IC50 5-aza-dc | 0.0031± 0.00013 | 0.0027 ± 0.00008 | 0.0021 ± 0.00023 | 0.0025 ± 0.0001 |
| Tam + IC25 SAHA + IC25 5-aza-dc | 0.0087± 0.00035 | 0.0022 ± 0.0001 | 0.0020 ± 0.00008 | 0.0021 ± 0.00009 |
| Tam + IC50 SAHA + IC50 5-aza-dc | 0.0024 ± 0.00011 | 0.0022 ± 0.00009 | 0.0020 ± 0.00015 | 0.0021 ± 0.00007 |
| Cell Line | Estrogen Receptorα (ERα) | Progesterone Receptor (PR) | HER2/ERBB2 (Overexpression) |
|---|---|---|---|
| MCF7 | Yes | Yes | No |
| SkBr3 | No | No | Yes |
| BT-549 | No | No | No |
| MDA-MB-231 | No | No | No |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramadan, W.S.; Vazhappilly, C.G.; Saleh, E.M.; Menon, V.; AlAzawi, A.M.; El-Serafi, A.T.; Mansour, W.; El-Awady, R. Interplay between Epigenetics, Expression of Estrogen Receptor- α, HER2/ERBB2 and Sensitivity of Triple Negative Breast Cancer Cells to Hormonal Therapy. Cancers 2019, 11, 13. https://doi.org/10.3390/cancers11010013
Ramadan WS, Vazhappilly CG, Saleh EM, Menon V, AlAzawi AM, El-Serafi AT, Mansour W, El-Awady R. Interplay between Epigenetics, Expression of Estrogen Receptor- α, HER2/ERBB2 and Sensitivity of Triple Negative Breast Cancer Cells to Hormonal Therapy. Cancers. 2019; 11(1):13. https://doi.org/10.3390/cancers11010013
Chicago/Turabian StyleRamadan, Wafaa S, Cijo George Vazhappilly, Ekram M Saleh, Varsha Menon, Aya M AlAzawi, Ahmed T El-Serafi, Wael Mansour, and Raafat El-Awady. 2019. "Interplay between Epigenetics, Expression of Estrogen Receptor- α, HER2/ERBB2 and Sensitivity of Triple Negative Breast Cancer Cells to Hormonal Therapy" Cancers 11, no. 1: 13. https://doi.org/10.3390/cancers11010013
APA StyleRamadan, W. S., Vazhappilly, C. G., Saleh, E. M., Menon, V., AlAzawi, A. M., El-Serafi, A. T., Mansour, W., & El-Awady, R. (2019). Interplay between Epigenetics, Expression of Estrogen Receptor- α, HER2/ERBB2 and Sensitivity of Triple Negative Breast Cancer Cells to Hormonal Therapy. Cancers, 11(1), 13. https://doi.org/10.3390/cancers11010013