Structure-Based Discovery of a Selective KDM5A Inhibitor that Exhibits Anti-Cancer Activity via Inducing Cell Cycle Arrest and Senescence in Breast Cancer Cell Lines




Abstract
:1. Introduction
2. Results
2.1. Compound 1 Identified as a Novel KMD5A Inhibitor via In Silico Screening
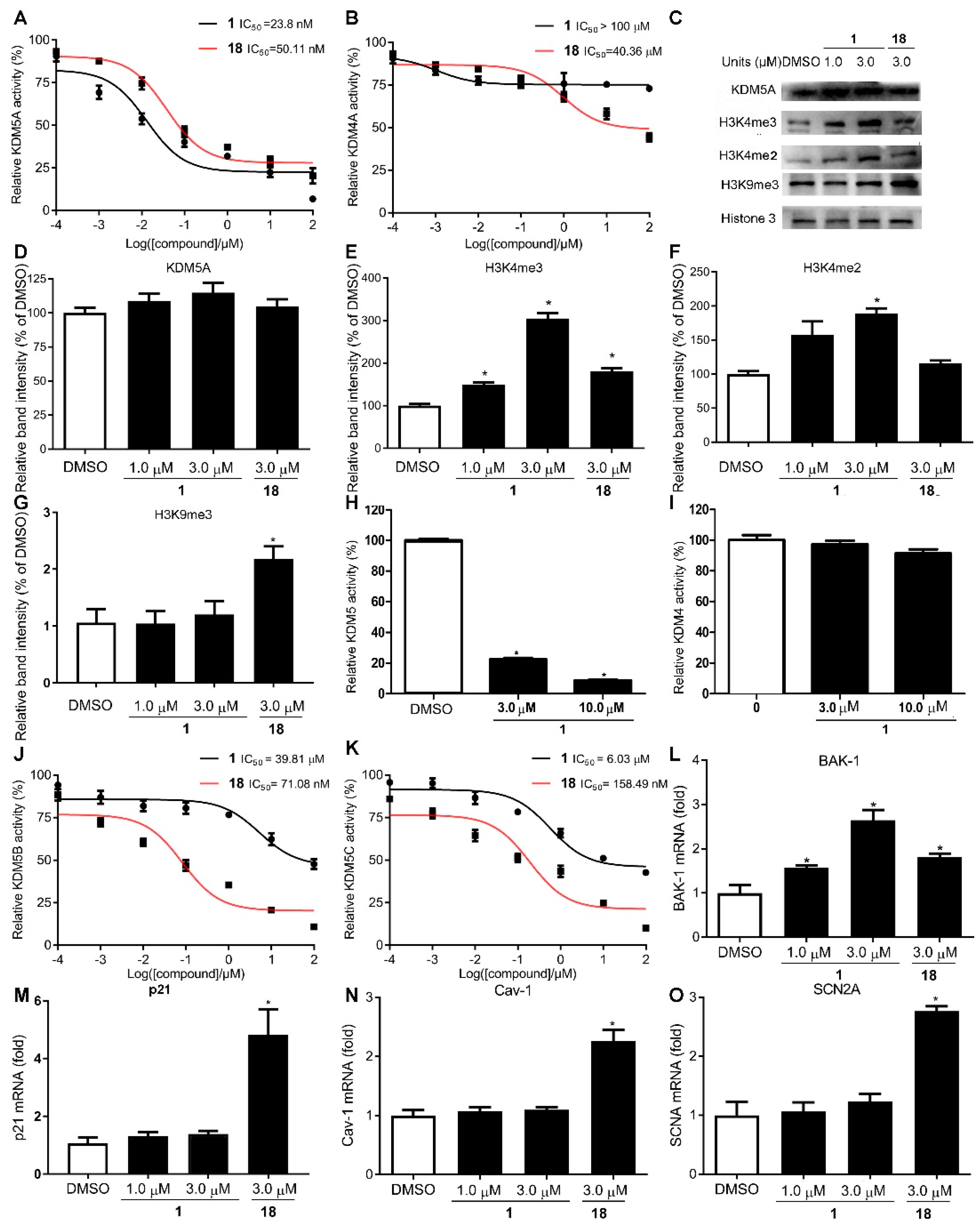
2.2. Compound 1 Is a Potent and Selective KDM5A Inhibitor
2.3. In Silico Identification of Potentially Druggable Surface Sites on the KDM5A-H3K4me3-Binding Domain and Binding Mode of Compound 1
2.4. Compound 1 Exhibits Potent Cytotoxicity Activity Against KDM5A-Overexpressing Breast Cancer Cells
2.5. Effect of Compound 1 on KDM5A-Mediated Transcriptional Activity
2.6. Anti-Proliferative Activity of Compound 1 Results from Inducing Cell Cycle Arrest and Cell Senescence
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Molecular Docking and Virtual Screening
4.3. KDM5A Inhibitor Screening Assay
4.4. KDM4A Activity and Inhibition Assay
4.5. Cellular Thermal Shift Assay
4.6. KDM5A Knockdown Assay
4.7. Cell Cytotoxicity and Proliferation Assay
4.8. Western Blotting
4.9. Cell Cycle Analysis
4.10. Cell Senescence Analysis
4.11. Co-Immunoprecipitation (co-IP) Assay
4.12. Chromatin Immunoprecipitation Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blair, L.P.; Cao, J.; Zou, M.R.; Sayegh, J.; Yan, Q. Epigenetic regulation by lysine demethylase 5 (KDM5) enzymes in cancer. Cancers 2011, 3, 1383–1404. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Walport, L.J.; Hopkinson, R.J.; Schofield, C.J. Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 2012, 16, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.R.; Engstrom, A.; Zoeller, E.L.; Liu, X.; Shanks, J.R.; Zhang, X.; Johns, M.A.; Vertino, P.M.; Fu, H.; Cheng, X. Characterization of a Linked Jumonji Domain of the KDM5/JARID1 Family of Histone H3 Lysine 4 Demethylases. J. Biol. Chem. 2016, 291, 2631–2646. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.H.; Albert, M.; Sroczynska, P.; Cruickshank, V.A.; Guo, Y.; Rossi, D.J.; Helin, K.; Enver, T. The histone demethylase Jarid1b is required for hematopoietic stem cell self-renewal. Blood 2015, 125, 2075–2078. [Google Scholar] [CrossRef]
- Choi, H.; Joo, H.; Won, H.; Min, K.; Kim, H.; Son, T.; Oh, Y.; Lee, J.; Kong, G. Role of RBP2-Induced ER and IGF1R-ErbB Signaling in Tamoxifen Resistance in Breast Cancer. J. Natl. Cancer Inst. 2018, 110, 400–410. [Google Scholar] [CrossRef]
- Banelli, B.; Daga, A.; Forlani, A.; Allemanni, G.; Marubbi, D.; Pistillo, M.P.; Profumo, A.; Romani, M. Small molecules targeting histone demethylase genes (KDMs) inhibit growth of temozolomide-resistant glioblastoma cells. Oncotarget 2017, 8, 34896–34910. [Google Scholar] [CrossRef]
- Cao, J.; Liu, Z.; Cheung, W.; Zhao, M.; Chen, S.; Chan, S.; Booth, C.; Nguyen, D.; Yan, Q. Histone demethylase RBP2 is critical for breast cancer progression and metastasis. Cell Rep. 2014, 6, 868–877. [Google Scholar] [CrossRef]
- Teng, Y.C.; Lee, C.F.; Li, Y.S.; Chen, Y.R.; Hsiao, P.W.; Chan, M.Y.; Lin, F.M.; Huang, H.D.; Chen, Y.T.; Jeng, Y.M.; et al. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013, 73, 4711–4721. [Google Scholar] [CrossRef]
- Zeng, J.; Ge, Z.; Wang, L.; Li, Q.; Wang, N.; Björkholm, M.; Jia, J.; Xu, D. The histone demethylase RBP2 Is overexpressed in gastric cancer and its inhibition triggers senescence of cancer cells. Gastroenterology 2010, 138, 981–992. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Song, P.; Geng, X.; Liang, X.; Zhou, M.; Wang, Y.; Chen, C.; Jia, J.; Zeng, J. Critical role of histone demethylase RBP2 in human gastric cancer angiogenesis. Mol. Cancer 2014, 13, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.; Hengst, L.; Slingerland, J. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhang, B.; Labadie, S.; Ortwine, D.F.; Vinogradova, M.; Kiefer, J.R.; Gehling, V.S.; Harmange, J.C.; Cummings, R.; Lai, T.; et al. Lead optimization of a pyrazolo[1,5-a]pyrimidin-7(4H)-one scaffold to identify potent, selective and orally bioavailable KDM5 inhibitors suitable for in vivo biological studies. Bioorg. Med. Chem. Lett. 2016, 26, 4036–4041. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.R.; Liu, X.; Gale, M.; Wu, L.; Shanks, J.R.; Zhang, X.; Webber, P.J.; Bell, J.S.; Kales, S.C.; Mott, B.T.; et al. Structural Basis for KDM5A Histone Lysine Demethylase Inhibition by Diverse Compounds. Cell Chem. Biol. 2016, 23, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Gale, M.; Sayegh, J.; Cao, J.; Norcia, M.; Gareiss, P.; Hoyer, D.; Merkel, J.S.; Yan, Q. Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget 2016, 7, 39931–39944. [Google Scholar] [CrossRef] [Green Version]
- Bavetsias, V.; Lanigan, R.M.; Ruda, G.F.; Atrash, B.; McLaughlin, M.G.; Tumber, A.; Mok, N.Y.; Le Bihan, Y.-V.; Dempster, S.; Boxall, K.J. 8-Substituted Pyrido [3, 4-d] pyrimidin-4 (3 H)-one Derivatives as Potent, Cell Permeable, KDM4 (JMJD2) and KDM5 (JARID1) Histone Lysine Demethylase Inhibitors. J. Med. Chem. 2016, 59, 1388–1409. [Google Scholar] [CrossRef]
- Johansson, C.; Velupillai, S.; Tumber, A.; Szykowska, A.; Hookway, E.S.; Nowak, R.P.; Strain-Damerell, C.; Gileadi, C.; Philpott, M.; Burgess-Brown, N. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat. Chem. Biol. 2016, 12, 539–545. [Google Scholar] [CrossRef]
- Tumber, A.; Nuzzi, A.; Hookway, E.S.; Hatch, S.B.; Velupillai, S.; Johansson, C.; Kawamura, A.; Savitsky, P.; Yapp, C.; Szykowska, A. Potent and selective kdm5 inhibitor stops cellular demethylation of H3K4me3 at transcription start sites and proliferation of MM1S myeloma cells. Cell Chem. Biol. 2017, 24, 371–380. [Google Scholar] [CrossRef]
- Vazquez-Rodriguez, S.; Wright, M.; Rogers, C.M.; Cribbs, A.; Velupillai, S.; Philpott, M.; Lee, H.; Dunford, J.E.; Huber, K.V.; Robers, M.B. Design, Synthesis and Characterization of Covalent KDM5 Inhibitors. Angew. Chem. 2018. [Google Scholar] [CrossRef]
- Yang, G.J.; Wang, W.; Mok, S.W.F.; Wu, C.; Law, B.Y.K.; Miao, X.M.; Wu, K.J.; Zhong, H.J.; Wong, C.Y.; Wong, V.K.W.; et al. Selective inhibition of lysine-specific demethylase 5A (KDM5A) using a rhodium (III) complex for triple-negative breast cancer therapy. Angew. Chem. Int. Ed. 2018, 57, 13091–13095. [Google Scholar] [CrossRef]
- Zhong, H.-J.; Lee, B.R.; Boyle, J.W.; Wang, W.; Ma, D.-L.; Chan, P.W.H.; Leung, C.-H. Structure-based screening and optimization of cytisine derivatives as inhibitors of the menin–MLL interaction. Chem. Commun. 2016, 52, 5788–5791. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zhu, K.; Zhang, X.; Zhang, S.; Liu, Y.; Ren, J.; Li, C.; Ye, M.; Ling, X. Nonimmobilized Biomaterial Capillary Electrophoresis for Screening Drugs Targeting Human Glucose Transporter 1. Anal. Chem. 2017, 89, 12951–12959. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Gao, J.; Li, G.Q.; Xie, Z. Quantifying four-probe metabolites in a single UPLC–MS/MS run to explore the effects of cooked rhubarb on cytochrome P450 isozymes. Bioanalysis 2012, 4, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.-M.; Gao, J.-W.; Shi, Z.; Huang, P.; Lu, Y.-S.; Yao, M.-C.; Huang, M. Herb–drug pharmacokinetic interaction between radix astragali and pioglitazone in rats. J. Ethnopharmacol. 2012, 144, 300–304. [Google Scholar] [CrossRef]
- Kuang, Y.; Lin, Y.; Li, K.; Song, W.; Ji, S.; Qiao, X.; Zhang, Q.; Ye, M. Screening of hepatoprotective compounds from licorice against carbon tetrachloride and acetaminophen induced HepG2 cells injury. Phytomedicine 2017, 34, 59–66. [Google Scholar] [CrossRef]
- Ji, S.; Tang, S.; Li, K.; Li, Z.; Liang, W.; Qiao, X.; Wang, Q.; Yu, S.; Ye, M. Licoricidin inhibits the growth of SW480 human colorectal adenocarcinoma cells in vitro and in vivo by inducing cycle arrest, apoptosis and autophagy. Toxicol. Appl. Pharmacol. 2017, 326, 25–33. [Google Scholar] [CrossRef]
- Li, P.-K.; Pandit, B.; Sackett, D.L.; Hu, Z.; Zink, J.; Zhi, J.; Freeman, D.; Robey, R.W.; Werbovetz, K.; Lewis, A. A thalidomide analogue with in vitro antiproliferative, antimitotic, and microtubule-stabilizing activities. Mol. Cancer Ther. 2006, 5, 450–456. [Google Scholar] [CrossRef]
- Leung, C.-H.; Grill, S.P.; Lam, W.; Han, Q.-B.; Sun, H.-D.; Cheng, Y.-C. Novel mechanism of inhibition of nuclear factor-κB DNA-binding activity by diterpenoids isolated from Isodon rubescens. Mol. Pharmacol. 2005, 68, 286–297. [Google Scholar]
- Ying, C.; Li, Y.; Leung, C.-H.; Robek, M.D.; Cheng, Y.-C. Unique antiviral mechanism discovered in anti-hepatitis B virus research with a natural product analogue. Proc. Natl. Acad. Sci. USA 2007, 104, 8526–8531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Jin, H.; Liu, Z.; Zhang, L.; Wang, X.S. An unbiased method to build benchmarking sets for ligand-based virtual screening and its application to GPCRs. J. Chem. Inf. Model. 2014, 54, 1433–1450. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, V.; Gaboriaud-Kolar, N.; Tallant, C.; Hall, M.-L.; Grigoriou, S.; Brownlee, P.M.; Fedorov, O.; Rogers, C.; Heidenreich, D.; Wanior, M. Discovery and optimization of a selective ligand for the switch/sucrose nonfermenting-related bromodomains of polybromo protein-1 by the use of virtual screening and hydration analysis. J. Med. Chem. 2016, 59, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Leung, K.-H.; Chan, D.S.; Wang, Y.; Ma, D.; Leung, C.H. Identification of a natural product-like STAT3 dimerization inhibitor by structure-based virtual screening. Cell Death Dis. 2014, 5, e1293. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins 1997, 29 (Suppl. 1), 215–220. [Google Scholar] [CrossRef]
- Chan, J.D.; Cupit, P.M.; Gunaratne, G.S.; McCorvy, J.D.; Yang, Y.; Stoltz, K.; Webb, T.R.; Dosa, P.I.; Roth, B.L.; Abagyan, R. The anthelmintic praziquantel is a human serotoninergic G-protein-coupled receptor ligand. Nat. Commun. 2017, 8, 1910. [Google Scholar] [CrossRef]
- Abagyan, R. Computational chemistry in 25 years. J. Comput. Aided Mol. Des. 2012, 26, 9–10. [Google Scholar] [CrossRef]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef]
- Hu, D.; Liu, Y.; Lai, Y.T.; Tong, K.C.; Fung, Y.M.; Lok, C.N.; Che, C.M. Anticancer Gold (III) Porphyrins Target Mitochondrial Chaperone Hsp60. Angew. Chem. Int. Ed. 2016, 55, 1387–1391. [Google Scholar] [CrossRef]
- Al Temimi, A.H.; Belle, R.; Kumar, K.; Poater, J.; Betlem, P.; Pieters, B.J.; Paton, R.S.; Bickelhaupt, F.M.; Mecinović, J. Recognition of shorter and longer trimethyllysine analogues by epigenetic reader proteins. Chem. Commun. 2018, 54, 2409–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, T.; Carceller, E.; Salas, J.; Ortega, A.; Buesa, C. Advances in the development of histone lysine demethylase inhibitors. Curr. Opin. Pharmacol. 2015, 23, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Thinnes, C.C.; England, K.S.; Kawamura, A.; Chowdhury, R.; Schofield, C.J.; Hopkinson, R.J. Targeting histone lysine demethylases—Progress, challenges, and the future. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 1416–1432. [Google Scholar] [CrossRef]
- Korczynska, M.; Le, D.D.; Younger, N.; Gregori-Puigjané, E.; Tumber, A.; Krojer, T.; Velupillai, S.; Gileadi, C.; Nowak, R.P.; Iwasa, E. Docking and linking of fragments to discover jumonji histone demethylase inhibitors. J. Med. Chem. 2015, 59, 1580–1598. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wu, J.; Dombkowski, A.; Zhang, K.; Holowatyj, A.; Boerner, J.L.; Yang, Z.Q. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am. J. Transl. Res. 2012, 4, 247–256. [Google Scholar] [PubMed]
- Liu, L.J.; Lu, L.; Zhong, H.J.; He, B.; Kwong, D.W.; Ma, D.L.; Leung, C.H. An Iridium(III) Complex Inhibits JMJD2 Activities and Acts as a Potential Epigenetic Modulator. J. Med. Chem. 2015, 58, 6697–6703. [Google Scholar] [CrossRef] [PubMed]
- Xhabija, B.; Kidder, B.L. KDM5B is a master regulator of the H3K4-methylome in stem cells, development and cancer. Semin. Cancer Biol. 2018. [Google Scholar] [CrossRef]
- Vallianatos, C.N.; Iwase, S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics 2015, 7, 503–519. [Google Scholar] [CrossRef]
- Paolicchi, E.; Crea, F.; Farrar, W.L.; Green, J.E.; Danesi, R. Histone lysine demethylases in breast cancer. Crit. Rev. Oncol. Hematol. 2013, 86, 97–103. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.; Trojer, P. Targeting histone lysine methylation in cancer. Pharmacol. Ther. 2015, 150, 1–22. [Google Scholar] [CrossRef]
- Wang, Y.; Lee, S.; Ha, Y.; Lam, W.; Chen, S.-R.; Dutschman, G.E.; Gullen, E.A.; Grill, S.P.; Cheng, Y.; Fürstner, A. Tylophorine Analogs Allosterically Regulates Heat Shock Cognate Protein 70 And Inhibits Hepatitis C Virus Replication. Sci. Rep. 2017, 7, 10037. [Google Scholar] [CrossRef]
- Li, G.-B.; Yang, L.-L.; Yuan, Y.; Zou, J.; Cao, Y.; Yang, S.-Y.; Xiang, R.; Xiang, M. Virtual screening in small molecule discovery for epigenetic targets. Methods 2015, 71, 158–166. [Google Scholar] [CrossRef]
- Pandit, B.; Sun, Y.; Chen, P.; Sackett, D.L.; Hu, Z.; Rich, W.; Li, C.; Lewis, A.; Schaefer, K.; Li, P.-K. Structure–activity-relationship studies of conformationally restricted analogs of combretastatin A-4 derived from SU5416. Bioorg. Med. Chem. 2006, 14, 6492–6501. [Google Scholar] [CrossRef]
- Pandit, B.; Hu, Z.; Chettiar, S.N.; Zink, J.; Xiao, Z.; Etter, J.P.; Bhasin, D.; Li, P.-K. Structure–activity relationship studies of thalidomide analogs with a taxol-like mode of action. Bioorg. Med. Chem. Lett. 2013, 23, 6902–6904. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.-I.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhong, H.-J.; Ko, C.-N.; Wong, S.-Y.; Vellaisamy, K.; Ye, M.; Ma, D.-L.; Leung, C.-H. Identification of a Rhodium (III) Complex as a Wee1 Inhibitor Against TP53-mutated Triple-negative Breast Cancer Cells. Chem. Commun. 2018, 54, 2463–2466. [Google Scholar] [CrossRef]
- Zhong, H.-J.; Lu, L.; Leung, K.-H.; Wong, C.C.; Peng, C.; Yan, S.-C.; Ma, D.-L.; Cai, Z.; Wang, H.-M.D.; Leung, C.-H. An iridium (III)-based irreversible protein–protein interaction inhibitor of BRD4 as a potent anticancer agent. Chem. Sci. 2015, 6, 5400–5408. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | ZINC No. | Relative Molecular Weight (Mr) | Scores |
|---|---|---|---|
| 1 | ZINC02140392 | 401.395 | −30.01 |
| 2 | ZINC02155003 | 442.448 | −34.63 |
| 3 | ZINC02113595 | 470.502 | −32.43 |
| 4 | ZINC08791298 | 441.463 | −42.55 |
| 5 | ZINC02091441 | 476.529 | −31.4 |
| 6 | ZINC01062498 | 364.445 | −31.17 |
| 7 | ZINC02113595 | 470.502 | −32.43 |
| 8 | ZINC12874897 | 523.633 | −30.7 |
| 9 | ZINC02128801 | 463.466 | −33.14 |
| 10 | ZINC03999938 | 378.472 | −31.78 |
| 11 | ZINC04012432 | 382.416 | −30.26 |
| 12 | ZINC12889905 | 418.406 | −40.18 |
| 13 | ZINC12893848 | 478.501 | −33.87 |
| 14 | ZINC08877996 | 468.509 | −31.27 |
| 15 | ZINC02334012 | 402.224 | −30.58 |
| 16 | ZINC08764396 | 439.423 | −31.21 |
| 17 | ZINC01475035 | 428.405 | −34.84 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.-J.; Ko, C.-N.; Zhong, H.-J.; Leung, C.-H.; Ma, D.-L. Structure-Based Discovery of a Selective KDM5A Inhibitor that Exhibits Anti-Cancer Activity via Inducing Cell Cycle Arrest and Senescence in Breast Cancer Cell Lines. Cancers 2019, 11, 92. https://doi.org/10.3390/cancers11010092
Yang G-J, Ko C-N, Zhong H-J, Leung C-H, Ma D-L. Structure-Based Discovery of a Selective KDM5A Inhibitor that Exhibits Anti-Cancer Activity via Inducing Cell Cycle Arrest and Senescence in Breast Cancer Cell Lines. Cancers. 2019; 11(1):92. https://doi.org/10.3390/cancers11010092
Chicago/Turabian StyleYang, Guan-Jun, Chung-Nga Ko, Hai-Jing Zhong, Chung-Hang Leung, and Dik-Lung Ma. 2019. "Structure-Based Discovery of a Selective KDM5A Inhibitor that Exhibits Anti-Cancer Activity via Inducing Cell Cycle Arrest and Senescence in Breast Cancer Cell Lines" Cancers 11, no. 1: 92. https://doi.org/10.3390/cancers11010092
APA StyleYang, G. -J., Ko, C. -N., Zhong, H. -J., Leung, C. -H., & Ma, D. -L. (2019). Structure-Based Discovery of a Selective KDM5A Inhibitor that Exhibits Anti-Cancer Activity via Inducing Cell Cycle Arrest and Senescence in Breast Cancer Cell Lines. Cancers, 11(1), 92. https://doi.org/10.3390/cancers11010092