1. Introduction
Cell migration plays a crucial role in physiological processes such as embryogenesis, immune responses and tissue repair. Moreover, cell migration dysregulation is implicated in various pathologies, like vascular disease, chronic inflammation and cancer [
1]. Indeed, cancer cells subvert the machinery that regulates cell migration and invasion to escape the primary tumor site and form local and distant metastases, which are the leading cause of cancer–related deaths [
2]. Therefore, targeting regulators of cell migration and invasion and understanding the mechanisms by which they control these processes may provide new strategies to delay cancer progression and impair metastasis.
Cell migration is a multistep process that requires spatiotemporal coordination of actin cytoskeleton remodeling and cell adhesion [
1,
3]. Moreover, cell migration relies on the driving force generated by actin polymerization, which allows the extension of protrusions such as lamellipodia and filopodia at the cell’s leading edge. These protrusions are then stabilized by the adhesion to the extracellular matrix (ECM) mediated by focal adhesions (FAs), which link the actin cytoskeleton to the ECM via integrins [
1,
3]. Finally, actomyosin-generated forces allow the forward movement of the cell, while FAs at the cell’s trailing edge are disassembled [
1,
3]. The spatial organization and turnover of FAs is controlled by integrin-mediated signaling and trafficking [
3,
4]. Moreover, increasing evidence indicates that membrane traffic is frequently subverted by cancer cells to enhance tumorigenesis. In fact, many cancers exhibit changes in the expression of membrane traffic regulators [
5,
6]. This raises the need to understand how the membrane traffic machinery is subverted by cancer cells in order to design new therapeutic approaches to impair cancer progression.
Members of the ADP-ribosylation factor (Arf) family of guanosine-5′-triphosphate (GTP)-binding proteins regulate membrane traffic and actin cytoskeleton remodeling, being essential for several cellular functions, including cell spreading and migration [
6,
7]. This family is composed by 6 Arf proteins, 21 Arf-like (Arl) proteins, 2 Secretion-associated and Ras-related (Sar) proteins and Tripartite motif (Trim)23 [
8]. Interestingly, Arfs and Arf GTPase-activating proteins (GAPs) have been shown to regulate cell adhesion by modulating the localization of FA components, the trafficking of integrins and actin cytoskeleton remodeling [
6,
9]. Furthermore, upregulation of Arf1, Arf4 and Arf6, as well as the Arf GAPs AGAP2, ASAP1, ASAP3 and GIT1 correlates with enhanced migration, invasion and metastasis of breast cancer cells [
6,
10].
Arl13b was originally identified as a regulator of ciliogenesis and Sonic hedgehog (Shh) signaling [
11,
12]. Moreover, recent studies showed that Arl13b promotes gastric carcinogenesis, cell migration and invasion [
13], as well as medulloblastoma formation [
14]. However, the molecular mechanism by which Arl13b regulates cancer cell migration and invasion is not understood. We have established a previously unidentified role for Arl13b in the regulation of endocytic recycling traffic [
15] and cell migration in non-pathological conditions [
16]. Moreover, we reported that Arl13b regulates cell migration through actin cytoskeleton remodeling via the interaction with the non-muscle Myosin IIA (NMIIA) [
16]. Therefore, we hypothesized that Arl13b regulates migration and invasion of cancer cells through actin cytoskeleton-related mechanisms.
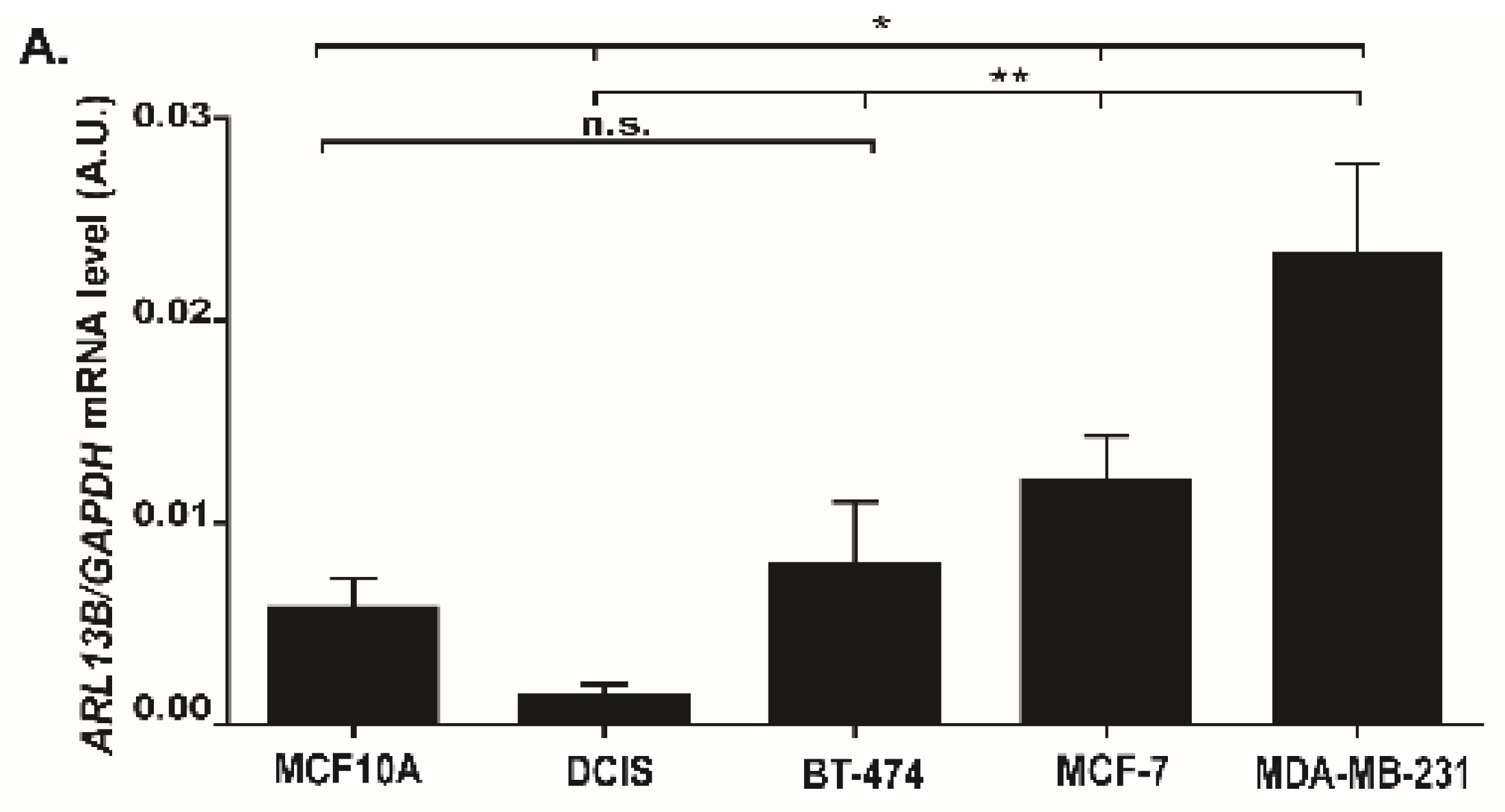
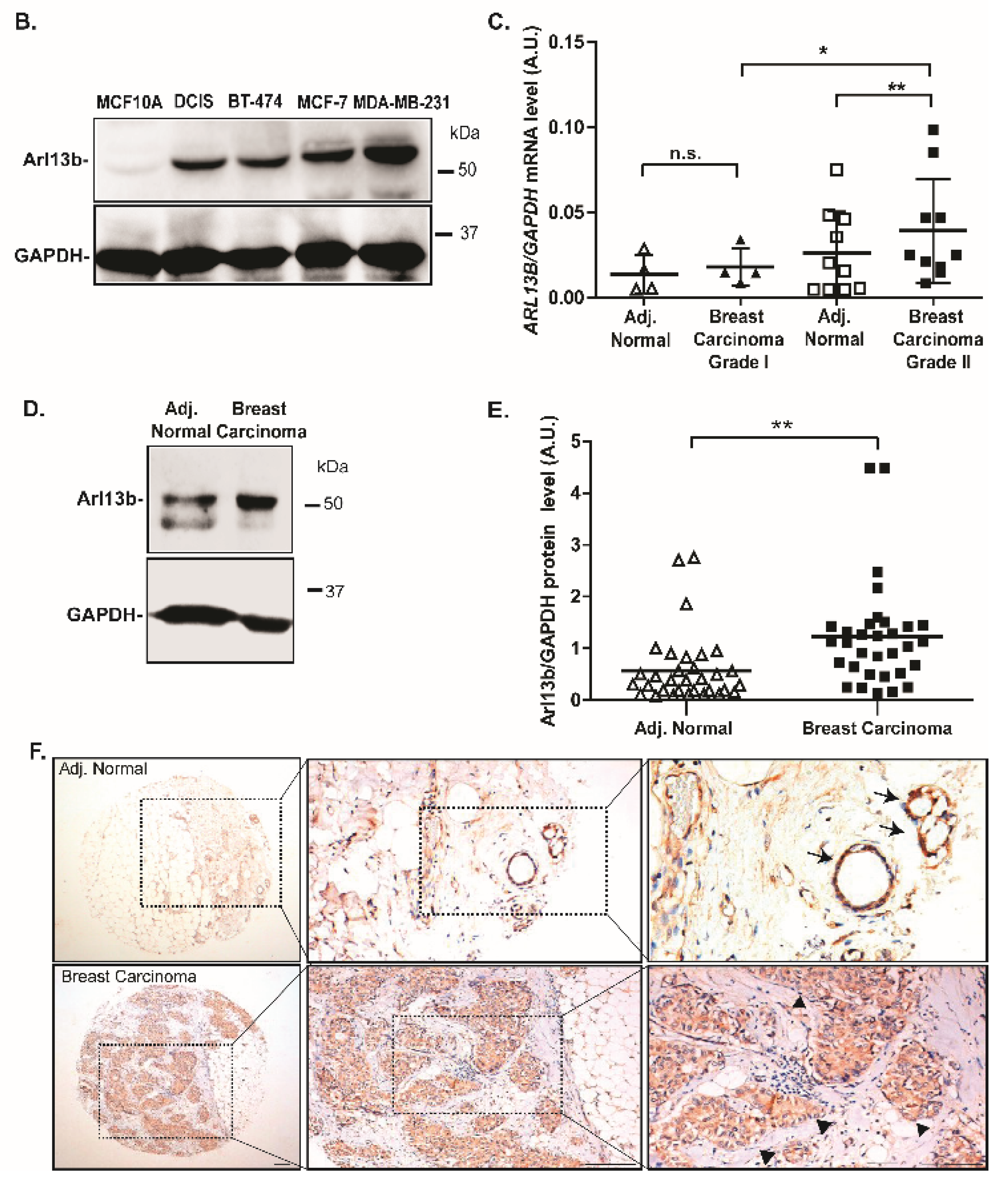
We decided to study this question in breast cancer, which is the most frequent type of cancer in women, with 2.09 million cases estimated worldwide in 2018 and the prime cause of cancer-related deaths among women [
17]. Here, we provide evidence that Arl13b positively regulates breast cancer progression. We observed that Arl13b silencing impairs breast cancer cell migration and invasion in vitro and tumor growth and metastasis in vivo. Moreover, we found that Arl13b interacts with β3-integrin, localizes to FAs and regulates stress fiber (SF) formation and FA size. Furthermore, we observed that Arl13b positively regulates the integrin-mediated signaling involved in the control of FA dynamics. Thus, our observations suggest that Arl13b is involved in cell-ECM adhesion by controlling integrin-mediated signaling. Furthermore, we show that Arl13b is upregulated in breast cancer cell lines and tissues. Thus, our study reinforces previous evidence for the involvement of Arl13b in cancer and suggests that Arl13b is subverted by breast cancer cells to become more motile and invasive, with potential to be targeted for therapeutic purposes.
3. Discussion
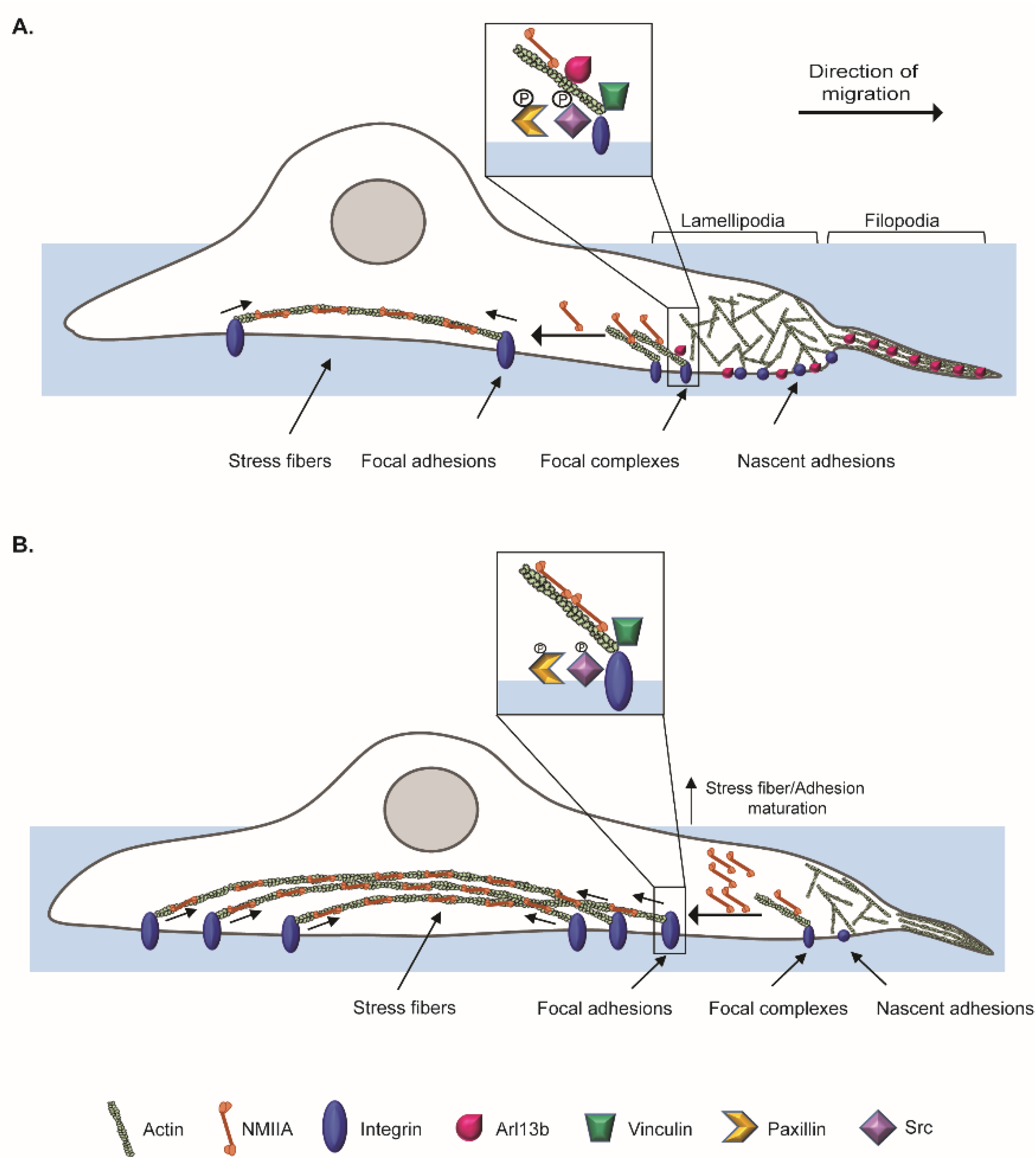
Breast cancer is the most frequent type of cancer in women worldwide and the related deaths result largely from metastatic spread of the primary tumor. In this study, we describe a new role for
ARL13B as an oncogene in breast cancer progression and provide a possible mechanism by which this GTP-binding protein regulates cancer cell migration and invasion. We demonstrate that Arl13b is involved in actin SF formation and the control of FA disassembly through integrin signaling, resulting in a negative regulation of FA size and stability, as well as β3-integrin cell surface levels. This strongly suggests a role for Arl13b in cell-ECM adhesion (
Figure 6). Strikingly, by analyzing breast cancer tissue samples, we found that Arl13b is upregulated in breast carcinomas when compared with the expression in adjacent normal breast tissues. This extends recent reports demonstrating a role for Arl13b in gastric tumorigenesis and medulloblastoma formation [
13,
14]. Thus,
ARL13B can be considered an oncogene with potential to be targeted in anticancer therapies.
Cell migration relies on spatiotemporal coordination of actin cytoskeleton remodeling and cell-ECM adhesion. Arl13b could link these two processes since it binds to actin via NMIIA and plays a role in actin cytoskeleton remodeling [
16]. Indeed, we observed colocalization of endogenous Arl13b with actin in structures such as lamellipodia, filopodia, SFs and CDRs of breast cancer cells. Besides actin, NMIIA is a major component of SFs, which we previously identified as an Arl13b effector [
16]. Interestingly, we observed that Arl13b interacts with NMIIA in a GTP-dependent manner in breast cancer cells. Moreover, we found an inverse correlation between Arl13b and NMIIA expression. As to whether this phenotype results from an Arl13b-dependent regulation of NMIIA expression levels, protein degradation or another mechanism, needs to be further explored. Furthermore, the increased expression of NMIIA may lead to the enhanced formation of SFs. Although the function of SFs in cell migration is not fully established, our results reinforce studies showing that SFs are more prominent in non-migrating cells, while highly motile cells show fewer, thinner and more dynamic actin bundles [
25,
26].
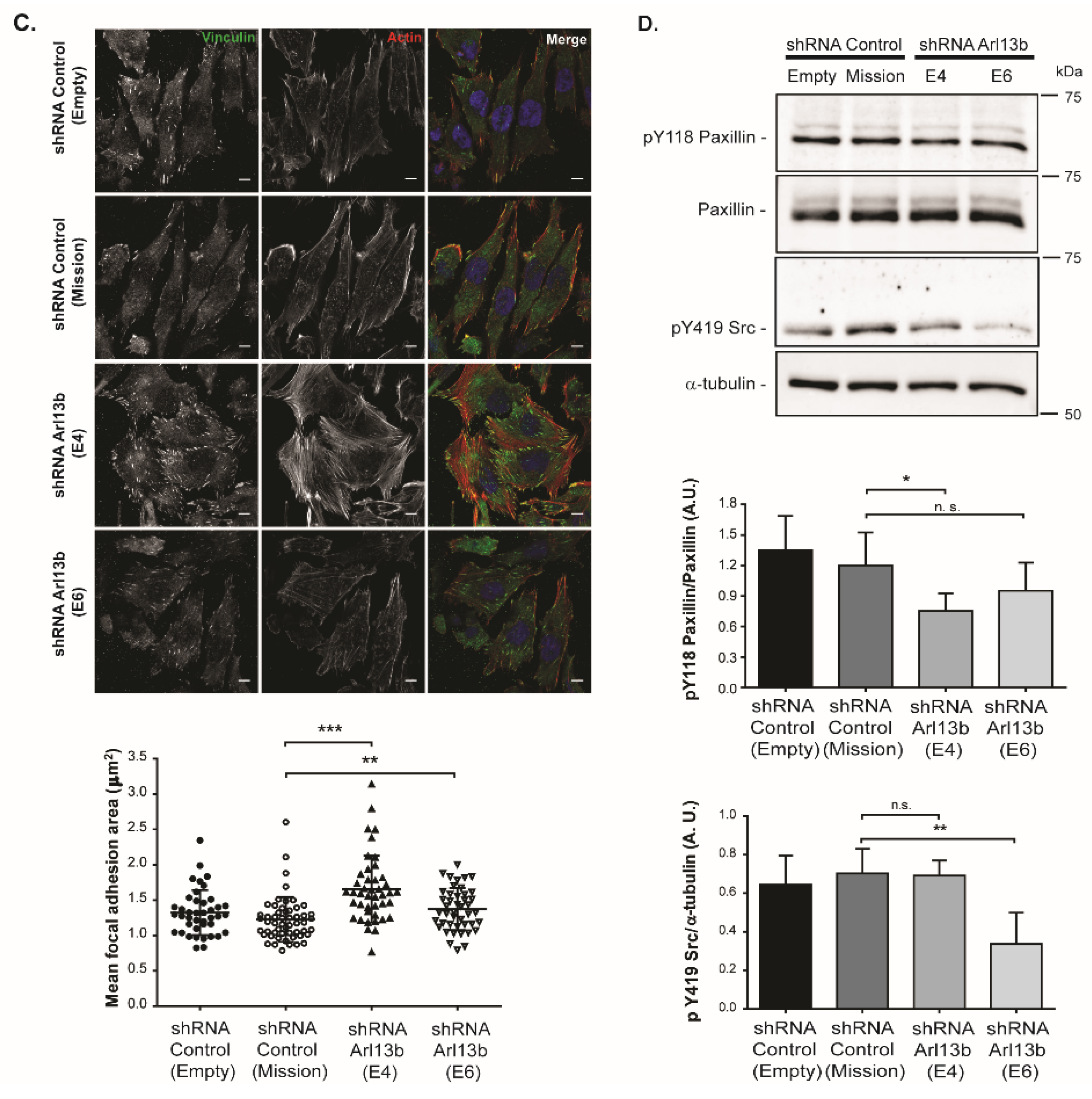
SFs can regulate cell-ECM adhesion by controlling FA size. Supporting the observation of an altered pattern of SFs in Arl13b-silenced cells, we observed that Arl13b depletion leads to larger FAs. Upon the engagement of integrins with the ECM, integrin signaling at the FA complex is activated. The activation of the FAK/Src complex and phosphorylation of Paxillin have an established role in FA turnover [
20,
21]. When Arl13b is silenced, activation of Src is attenuated and a decrease in Paxillin phosphorylation is observed. This suggests that the increase in FA size observed in Arl13b-silenced cells may result from defects in the signaling involved in FA disassembly. Consistent with a role for Arl13b in FA dynamics, we also observed that Arl13b localizes to these structures in fixed and live cells. Since cell migration requires efficient assembly and disassembly of FAs [
27], we propose that a decrease in the disassembly rate of FAs in Arl13b-silenced cells contributes to an abnormal cell-ECM adhesion and decreased cell migration.
FA maturation and cell-ECM adhesion are not only controlled by actomyosin dynamics but also by integrin trafficking [
4]. Indeed, in growth factor-stimulated cell migration, β3-integrin can be endocytosed through CDR-mediated macropinocytosis and then trafficked through the endocytic recycling compartment to newly-established FAs on the ventral surface, at the leading edge of migrating cells [
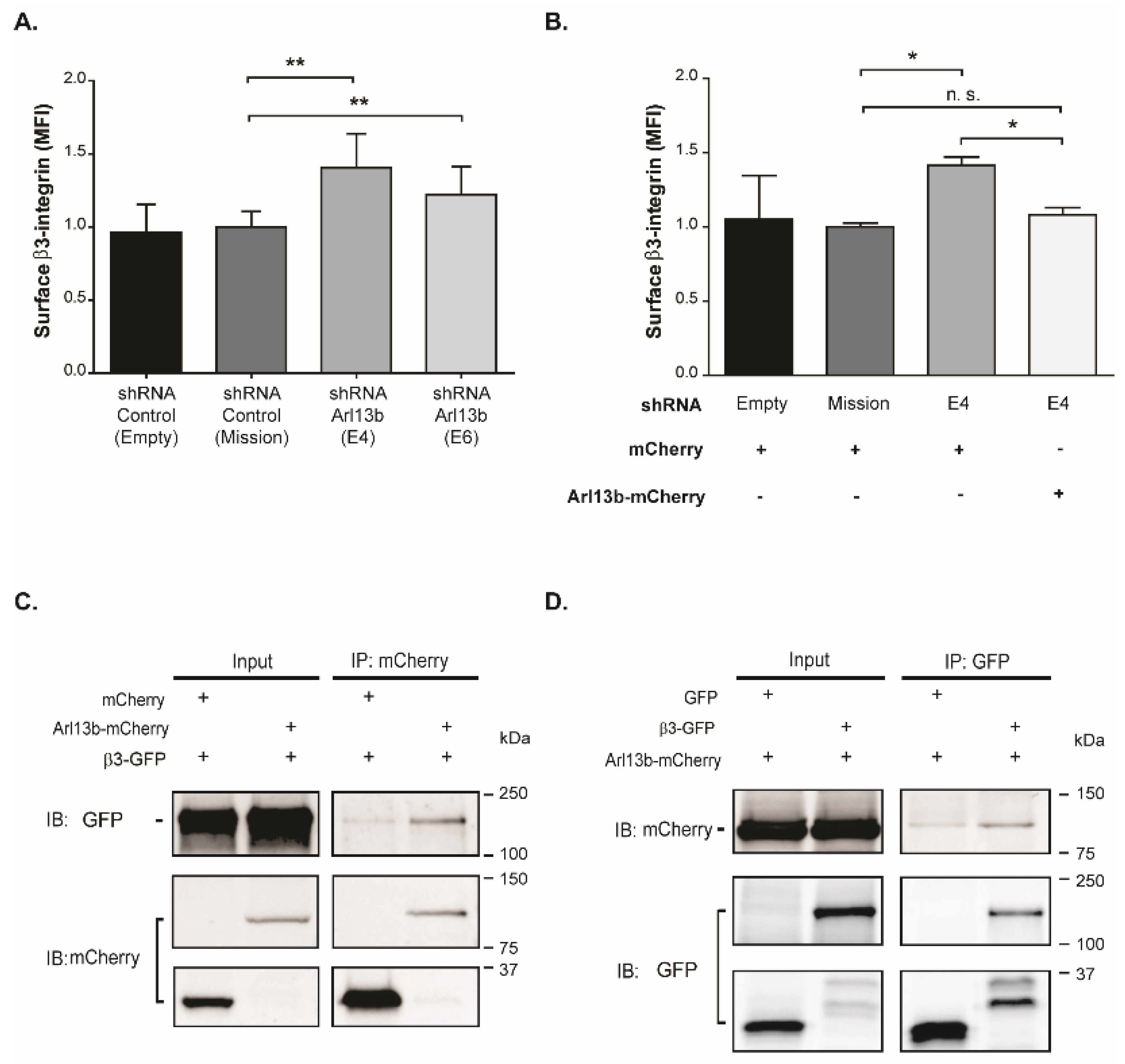
28]. We observed that Arl13b interacts with β3-integrin and downregulates its cell surface levels. Our group has also shown that Arl13b is required for CDR formation [
16]. Therefore, it is tempting to speculate that Arl13b controls β3-integrin surface levels by regulating the internalization and/or recycling of integrins through CDRs.
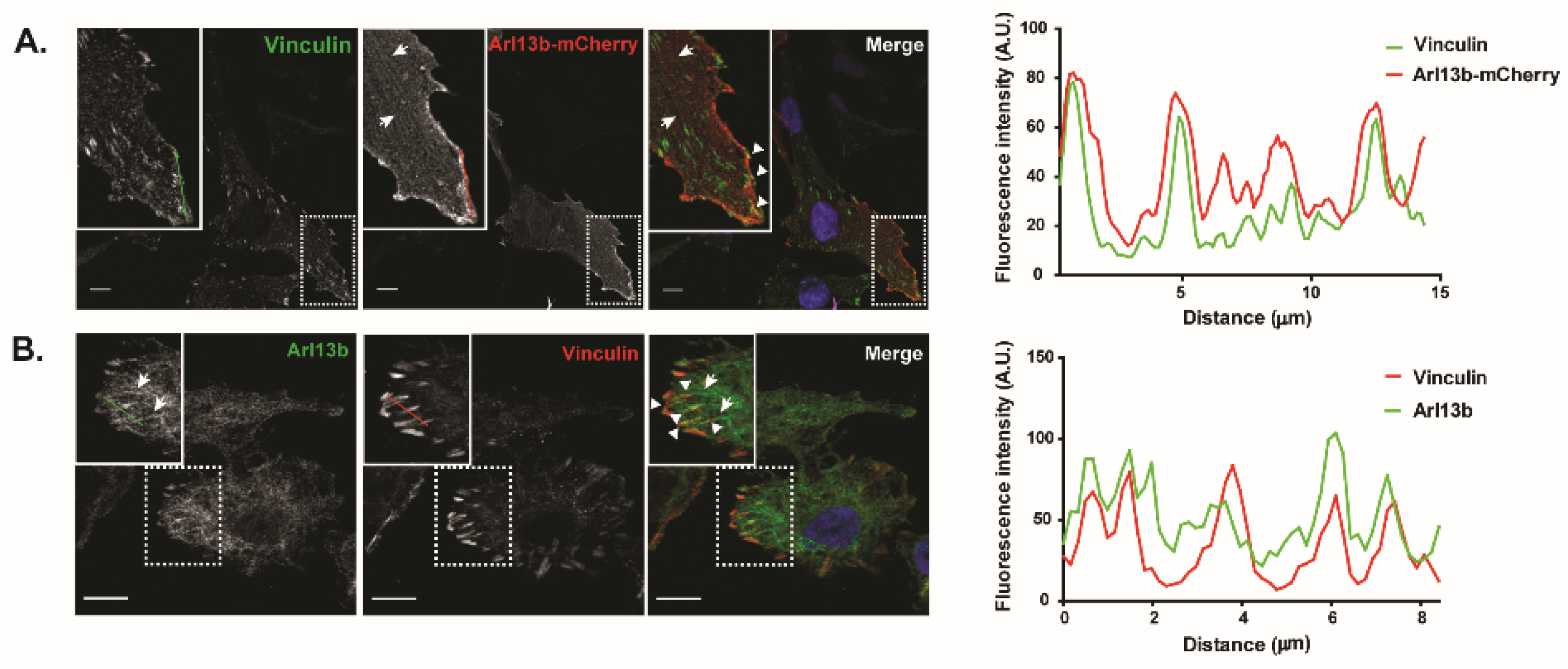
Arl13b has a well-established role in the trafficking of ciliary proteins and ciliogenesis [
11,
12]. Notably, we reported its interaction with actin through NMIIA, its localization to actin-rich structures [
16] and now the interaction of Arl13b with β3-integrin and co-localization with FA components, namely Vinculin and Paxillin. Interestingly, it is now appreciated that many ciliary proteins have other localizations besides primary cilia and that they can play a role in the formation of polarized cellular structures [
29]. Furthermore, it was described that FA proteins associate with basal bodies of ciliated cells and form ciliary adhesion complexes that interact with the actin cytoskeleton [
30]. Therefore, Arl13b functions in primary cilia and FA dynamics further suggest that primary cilia and FAs might share protein components that are recruited to the appropriate location depending on the cellular context or cell transformation status.
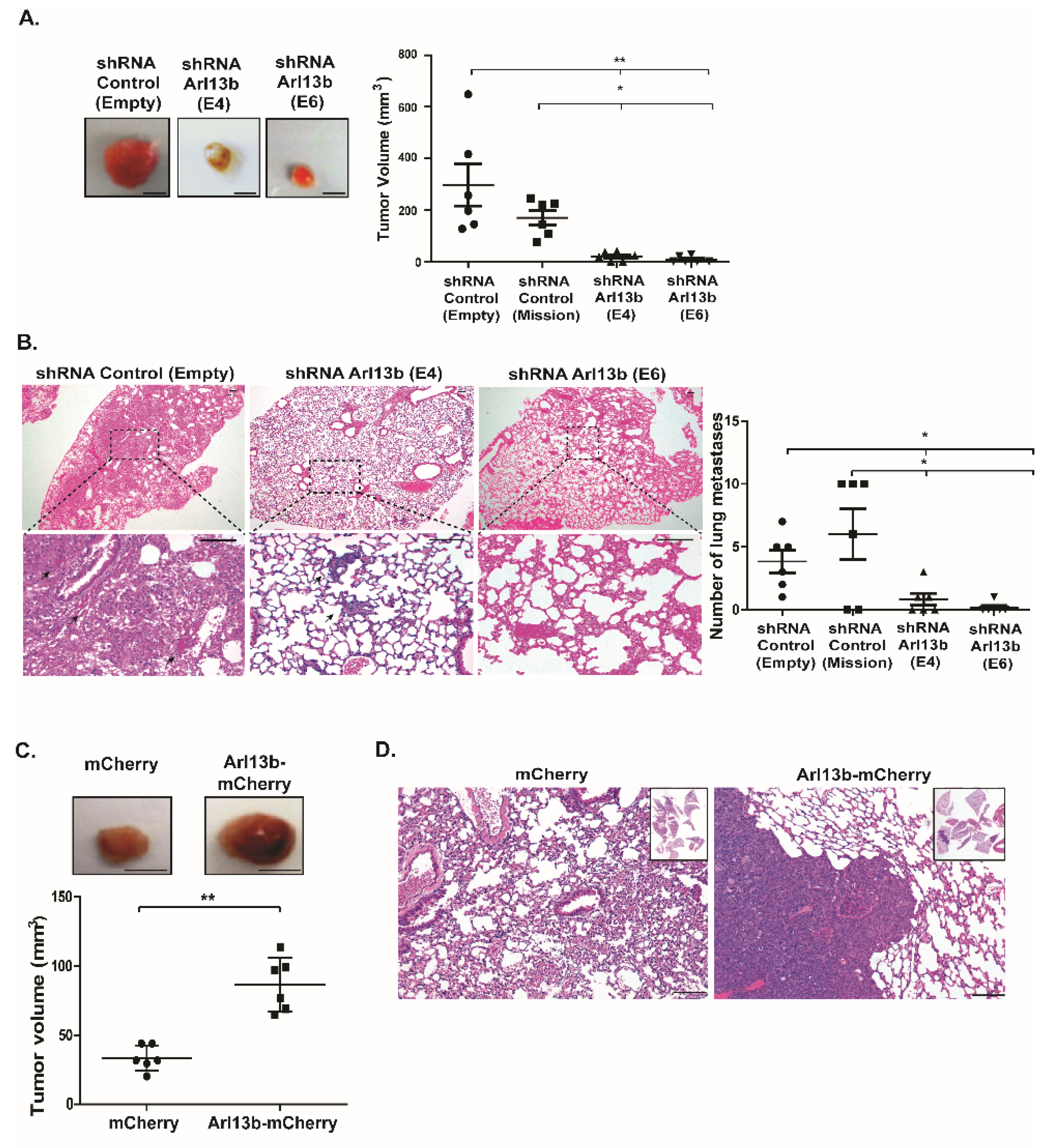
Our results also confirmed the role of Arl13b in cell growth suggested by other studies [
13,
14,
18]. Indeed, mice injected with Arl13b-depleted breast cancer cells hardly develop primary tumors and when they do, these are very small. In contrast, mice injected with Arl13b-overexpressing breast cancer cells develop large tumors. Although the impairment in the formation of cancer metastases in mice injected with Arl13b-depleted breast cancer cells is consistent with in vitro studies pointing to Arl13b being essential for cancer cell migration and invasion, further studies should be performed where primary tumors are allowed to develop and only then Arl13b is depleted, by using an inducible system. This would enable the uncoupling of the effects of Arl13b depletion in cell proliferation and migration/invasion.
Importantly, breast cancer is the third type of cancer whose progression or formation has been shown to be promoted by Arl13b and we also have evidence that the same occurs in colon cancer (our unpublished results). Indeed, recently published studies on gastric cancer and medulloblastoma proposed a Shh signaling-dependent mechanism [
13,
14]. However, breast cancer is a type of cancer where tumor cells show a decrease or loss of primary cilia [
31,
32,
33], contrary to medulloblastoma and gastric cancer cells, where cilia are present [
34,
35] and cilia-dependent Shh signaling can play an important role. Therefore, the mechanisms governing Arl13b-induced tumorigenesis and cancer progression might differ between different cancer types. Nevertheless, we cannot exclude the influence of non-canonical Shh signaling, which is cilia-independent, in breast cancer formation and progression. Thus, future studies should investigate the contribution of Arl13b-regulated actin remodeling and FA dynamics, as well as canonical and non-canonical Shh signaling for tumorigenesis and cancer progression.
4. Materials and Methods
4.1. Cell Culture
Cell culture media, supplements and antibiotics were purchased from GIBCO (Waltham, MA, USA). MDA-MB-231 and MCF-7 human breast adenocarcinoma cell lines (obtained from ATCC, Manassas, VA, USA) were maintained at 37 °C and 5% CO2, in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 µg/mL streptomycin, 2 mM L-glutamine and 15 mM HEPES (DMEM complete medium) for up to 1 month. BT-474 human breast adenocarcinoma cell lines (obtained from ATCC) were maintained at 37 °C and 5% CO2, in RPMI supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 µg/mL streptomycin, 2 mM L-glutamine and 15 mM HEPES. MCF10A-ER-Src cell line (MCF10A), kindly provided by F. Janody (i3S, Universidade do Porto, Porto, Portugal) [
36], was grown in DMEM/F12, supplemented with 5% horse serum, 20 ng/mL epidermal growth factor (EGF), 10 μg/mL insulin, 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin, 100 U/mL penicillin and 100 µg/mL streptomycin. MCF10DCIS.com cell line (DCIS) was purchased from the Animal Model & Therapeutic Evaluation Core (AMTEC), Barbara Ann Karmanos Cancer Institute, Wayne State University (Detroit, MI, USA). DCIS cells were grown in the same media as MCF10A cells.
4.2. Cell Transduction and Lentivirus Production
MDA-MB-231 or MCF-7 were plated at 1-2 × 10
5 cells/well on 6-well plates and transduced on the next day with lentiviral particles in the presence of 6–8 µg/mL polybrene (hexadimethrine bromide, Sigma-Aldrich, St. Louis, MO, USA). After 24 hours, 1.5 µg/mL puromycin, 10 µg/mL blasticidin (MDA-MB-231) or 8 µg/mL blasticidin (MCF-7) were added to select transduced cells. Cells were selected for at least 5 days before assayed. In the rescue experiment, 1 µg/mL of puromycin and 10 µg/mL of blasticidin were used. The lentiviral vectors pLKO.1 and pLenti6/V5-DEST Gateway were used to achieve stable shRNA-mediated gene silencing or overexpression, respectively. For lentivirus production, HEK-293T grown in DMEM supplemented with 10% FBS, 2 mM GlutaMAX and 15 mM HEPES were co-transfected using FuGENE (Promega, Madison, WI, USA) with 1 µg of lentiviral vector, 900 ng packaging plasmid (pCMV-dR8.91 or pxPAX2) and 100 ng envelope plasmid (pCMV-VSV-G). Media containing lentiviral particles were harvested 40- or 64-hours post-transfection and stored in aliquots at 80 °C until use. The target sequences for Arl13b hairpin E4 and the negative control Mission are described elsewhere [
15]. Target sequence for hairpin E6 is: 5′-CCTATATTGGTGTTGGCAAAT-3′. shRNAs were obtained from the Broad Institute RNAi Consortium (Cambridge, MA, USA).
4.3. RNA Extraction, cDNA Synthesis and Real-Time Quantitative PCR
Total RNA was isolated from 14 samples of freshly-collected invasive ductal breast carcinoma tissues and their adjacent parenchyma tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. The extracted RNA was pretreated with DNAse I (ThermoFisher, Waltham, MA, USA) before cDNA synthesis. In the case of cell lines, total RNA was isolated using the RNeasy Mini Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. RNA was used as template for cDNA synthesis using SuperScript II Reverse Transcriptase (Invitrogen), according to the manufacturer’s instructions. Real-time quantitative PCR (RT-qPCR) was performed using the FastStart Essential DNA Green Master kit (Roche, Basel, Switzerland) and a LightCycler 96 System (Roche), according to the manufacturer’s instructions. All
ARL13B expression levels were normalized to the level of
GAPDH expression, which was used as a housekeeping gene. The primer sequences used for the human genes are listed in
Table S1.
4.4. Transwell Migration/Invasion and Scratch Assays
Scratch assays were performed as described elsewhere [
16]. For scratch assays, confluent cell monolayers grown on plates coated with 10 µg/mL fibronectin in PBS were scratched with a pipette tip and induced to migrate by adding DMEM with 10% FBS. Images were taken from each well immediately (time 0) and after 4, 8 and 21 hours. The area of cell migration was measured in 10 randomly-chosen fields using ImageJ software. Percentage of gap closure was determined as follows: [1 − (gap area at
t = 4–18 hours/gap area at
t = 0 hours) × 100]. For transwell migration and invasion assays, 5 × 10
4 cells were seeded into the upper chamber of 8 µm-pore transwells (Corning, Corning, NY, USA) in DMEM with 0.5% bovine serum albumin and allowed to migrate for 6 hours or to invade for 21 hours, towards the lower chamber containing DMEM with 10% FBS. In the case of the invasion assays, cells were seeded into the upper chamber of transwells coated with Matrigel (Corning). In the case of migration assays, cells were seeded into the upper chamber of transwells with the lower side of the membrane coated with 10 µg/mL fibronectin in DMEM with 10% FBS. At the end of the incubation period, cells that migrated/invaded were fixed with 4% paraformaldehyde (PFA) in PBS and stained with 0.1% (w/v) crystal violet in 20% methanol for 15 minutes, whereas the cells remaining in the upper chamber of the transwells were removed with a cotton swab. Images of the cells that migrated/invaded through the membranes were taken using an Axiovert 40 (Zeiss, Oberkochen, Germany) microscope equipped with a digital camera (AxioCam MRc, Zeiss) and ZEN 2 software (blue edition, Zeiss). The number of cells that migrated/invaded in at least 10 randomly-selected fields in duplicate for each condition were counted using ImageJ software. The number of cells that migrated/invaded through the transwell membrane were represented as the percentage of migration/invasion relative to control cells, which was considered 100%.
4.5. Immunofluorescence
Cells were grown for 24–48 hours on glass coverslips, coated with 10 µg/mL fibronectin in DMEM with 10% FBS when indicated, washed with PBS and fixed in 4% PFA in PBS for 15–20 minutes at room temperature and immunostained for Vinculin and F-actin as described [
16], except that in the case of MDA-MB-231 cells transduced with mCherry or Arl13b-mCherry, permeabilization was performed with PBS with 0.1–0.2% Triton X-100 for 30 minutes and blocking with 0.2% BSA in PBS with 100 mM glycine for 1 hour. Antibodies and concentrations used are listed in
Table S2. Intensity plot profiles of Vinculin, Arl13b-mCherry and endogenous Arl13b were obtained using the plot profile function of ImageJ software. In the case of cells transduced with Arl13b-mCherry, a moving average method was applied.
4.6. Quantification of Focal Adhesions
For FA quantification, Vinculin-positive FAs located at the ventral side of well spread and isolated cells were quantified using ImageJ software, essentially as described by Nardone et al. [
37] with the exception that images were not subjected to automatic brightness/contrast and were binarized using MaxEntropy threshold command. Particles were analyzed (size = 0.30–15; circularity = 0.00–0.99) and the size of detected particles was used to calculate FA mean size. Images were acquired using an LSM 710 (Zeiss) confocal microscope equipped with a Plan-Apochromat 63/1.40 Oil Ph3 lens and Zen Blue 2010b SP1software.
4.7. Immunoprecipitation and Immunoblotting
Immunoblotting was essentially performed as described previously [
16], except that cells were lysed in cold lysis buffer containing 50 mM Tris-HCl, pH = 7.5; 1% IGEPAL; 150 mM NaCl; 1 mM EDTA; 1mM EGTA; 2 mM MgCl
2; 1 mM DTT and 0.1% SDS. Antibodies and concentrations used are listed in
Table S2. Uncropped immunoblots are shown in
Figures S6 and S7.
Cells transduced with lentiviruses encoding mCherry or Arl13b-mCherry were plated in tissue culture dishes coated with 10 µg/mL fibronectin in DMEM with 10% FBS and transfected with integrin β3–EGFP, a kind gift of B. Wehrle-Haller (University of Geneva, Geneva, Switzerland) and Z. Gu (Brigham & Women’s Hospital, Harvard Medical School, Boston, MA, USA) [
28,
38] and pEGFP-C3 plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were lysed in cold lysis buffer containing 0.1% Triton X-100, in the presence of protease and phosphatase inhibitors for 30 minutes on ice, followed by centrifugation at 21,100×
g, for 30 minutes at 4 °C. Immunoprecipitation was essentially performed as described previously [
16]. Antibodies and concentrations used are listed in
Table S2. Uncropped immunoblots are shown in
Figures S6 and S7.
4.8. Flow Cytometry
Cells grown on wells coated with 10 µg/mL fibronectin in DMEM with 10% FBS were resuspended in flow cytometry buffer (PBS with 1% FBS and 2 mM EDTA), incubated with anti-β3-integrin antibody for 1 hour, washed twice with flow cytometry buffer and incubated with secondary Alexa Fluor 488-conjugated anti-mouse antibody for 30 minutes, at 4 °C. Antibodies and concentrations used are listed in
Table S2. To exclude dead cells, samples were incubated with 0.5 µg/mL of propidium iodide (PI, Sigma-Aldrich) just before acquisition. Cells were analyzed using a FACS Canto II (Becton Dickinson, Franklin Lakes, NJ, USA) flow cytometer analyzer. In the case of MDA-MB-231 cells transduced with pLenti6-Arl13b-mCherry or mCherry, the analysis was performed in a FACS Aria III High Speed Cell Sorter (Becton-Dickinson) in order to gate and analyze only mCherry-positive cells. Experiments were analyzed using FlowJo software (version 10.1r7).
4.9. Human Tissue Samples
Freshly-collected ductal breast carcinoma samples and paired adjacent parenchyma tissues in TRIzol were obtained during surgery by pathologists of Hospital Beatriz Ângelo (HBA, Loures, Portugal). The study was approved by the ethics committee of HBA (Ref. #0174). The clinicopathological characteristics of breast carcinomas used in the study are described in
Table S3. Frozen breast tumor samples and normal adjacent tissues were obtained from the tumor bank of the Pathology Department of Instituto Português de Oncologia de Lisboa Francisco Gentil (IPOLFG). The study was approved by the ethics committee of the Institute (Ref. #UIC/1050). The clinicopathological characteristics of breast carcinomas used in the study are described in
Table S4. Frozen tissues were reduced to powder with a mortar and pestle in liquid nitrogen and total protein extracts obtained by solubilization in extraction buffer (50 mM Tris-HCl, pH = 7.9; 150 mM NaCl; 1 mM EDTA; 2 mM MgCl
2; 0.1% SDS; 0.5% sodium deoxycholate; 1% IGEPAL) in the presence of protease and phosphatase inhibitors, for 1 hour on ice, followed by centrifugation at 21,100×
g, for 20 minutes at 4 °C. Total protein amounts in the supernatants were quantified and analyzed by immunoblotting as described [
16].
4.10. Murine Breast Cancer Orthotopic Model
Mouse procedures were approved by the national regulatory authority, DGAV (Ref. # 0421/000/000/2016) and the institutional ethics committee (Ref. #36/2016/CEFCM), under the rules of Federation for Animal Science Associations (FELASA), accomplishing the 3Rs through evidence-based guidelines. Six mice were used in each condition, since the strain is syngeneic. In order to establish breast orthotopic tumors, MDA-MB-231 cells (2 × 106), stably transduced with lentiviruses encoding shRNAs targeting Arl13b (E4 and E6), Arl13b-mCherry or the corresponding controls (Empty, Mission or mCherry, respectively) were washed once with PBS and resuspended in 100 µl PBS for subcutaneous injection into the mammary fat pads of 4–6 weeks old BALB/c-SCID female mice. After 6–8 weeks, primary tumors were surgically excised from anesthetized mice and relapse was allowed for more 4–6 weeks. Primary and secondary tumors were collected from euthanized mice, as well as lungs to score metastases. All tissues were formalin-fixed and paraffin-embedded, cut in 3 µm sections and stained with hematoxylin and eosin. Tumor volume was determined by the following formula: v=(ab2)/2 (a = long diameter of the tumor; b = short diameter of the tumor; and v = volume).
4.11. Histology and Immunohistochemistry
Normal and breast tumor formalin-fixed (10%) and paraffin-embedded samples were selected for tissue microarray construction. Briefly, tissue cylinders with 1.4 mm were punched from tumor areas of each donor tissue block and placed in a recipient paraffin block (tissue microarray), using a manual Tissue Puncher/Arrayer (Beecher Instruments, Sun Prairie, WI, USA). Endogenous peroxidase on de-paraffinized sections was blocked with EnVision TM Flex Peroxidase-Blocking reagent (Dako, Glostrup, Denmark). Antigen retrieval was performed by microwaving in citrate buffer (pH = 6.0) for 20 minutes. Slides were incubated with protein block serum-free (Dako) for 30 minutes and then with anti-Arl13b antibody (Sigma-Aldrich) in EnVisionTM Flex Antibody Diluent (Dako) for 60 minutes at room temperature. The slides were washed and incubated with EnVision/HRP rabbit (ChemMate Envision Kit, Dako) and developed with 3, 3´diaminobenzidine-hydrochloride (DAB, Dako) and counterstained with Harris hematoxylin. Slides were mounted with Entellan and analyzed by light microscopy.
4.12. Statistical Analysis
Numerical data are presented as mean ± standard deviation (SD). Statistical tests used for comparing different data sets are indicated. GraphPad Prism software (version 5.00, San Diego, CA, USA) was used to perform the statistical tests.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







