FADD in Cancer: Mechanisms of Altered Expression and Function, and Clinical Implications

 , and
, and
Abstract
:1. Regulation of FADD Gene Expression
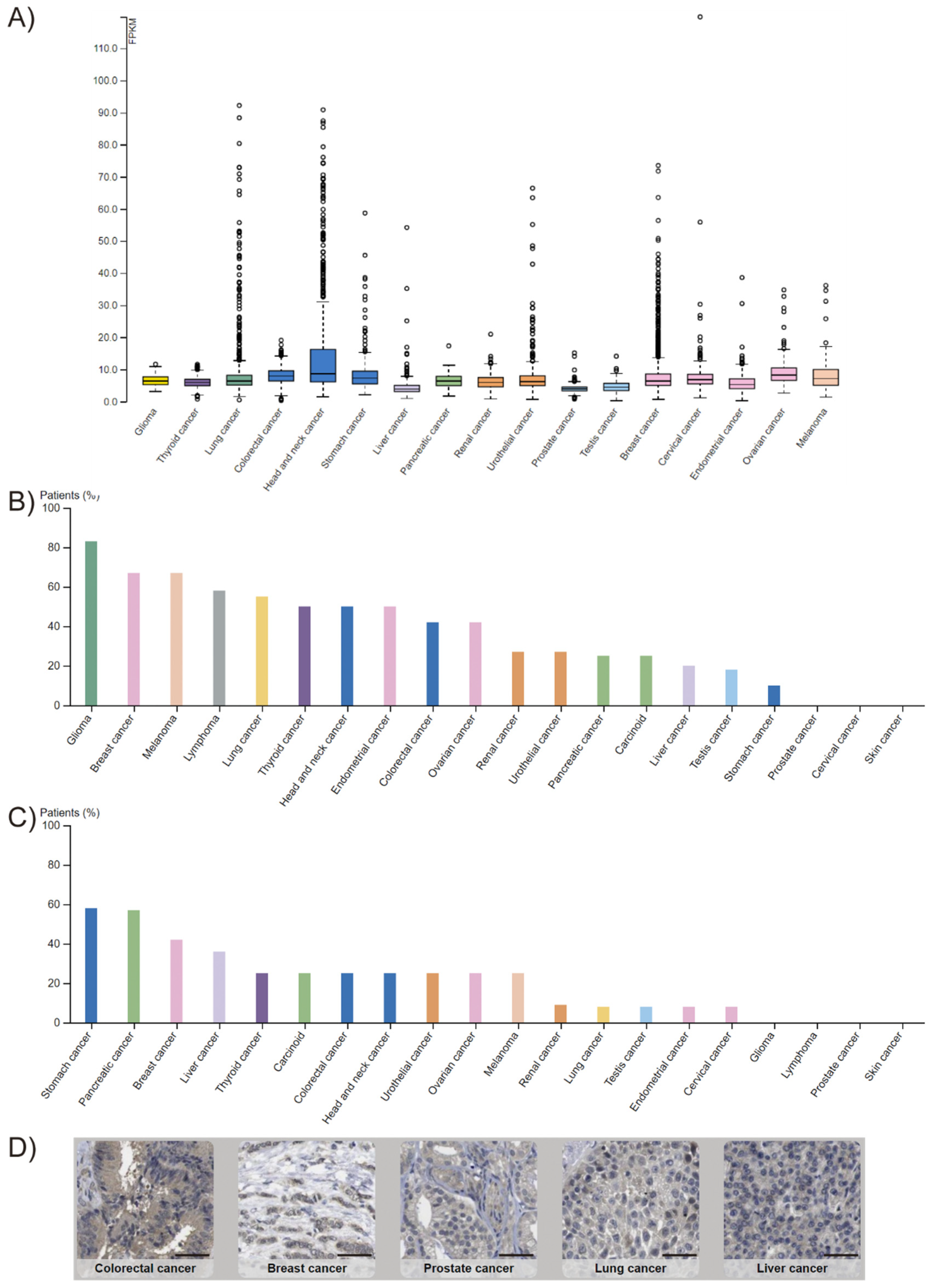
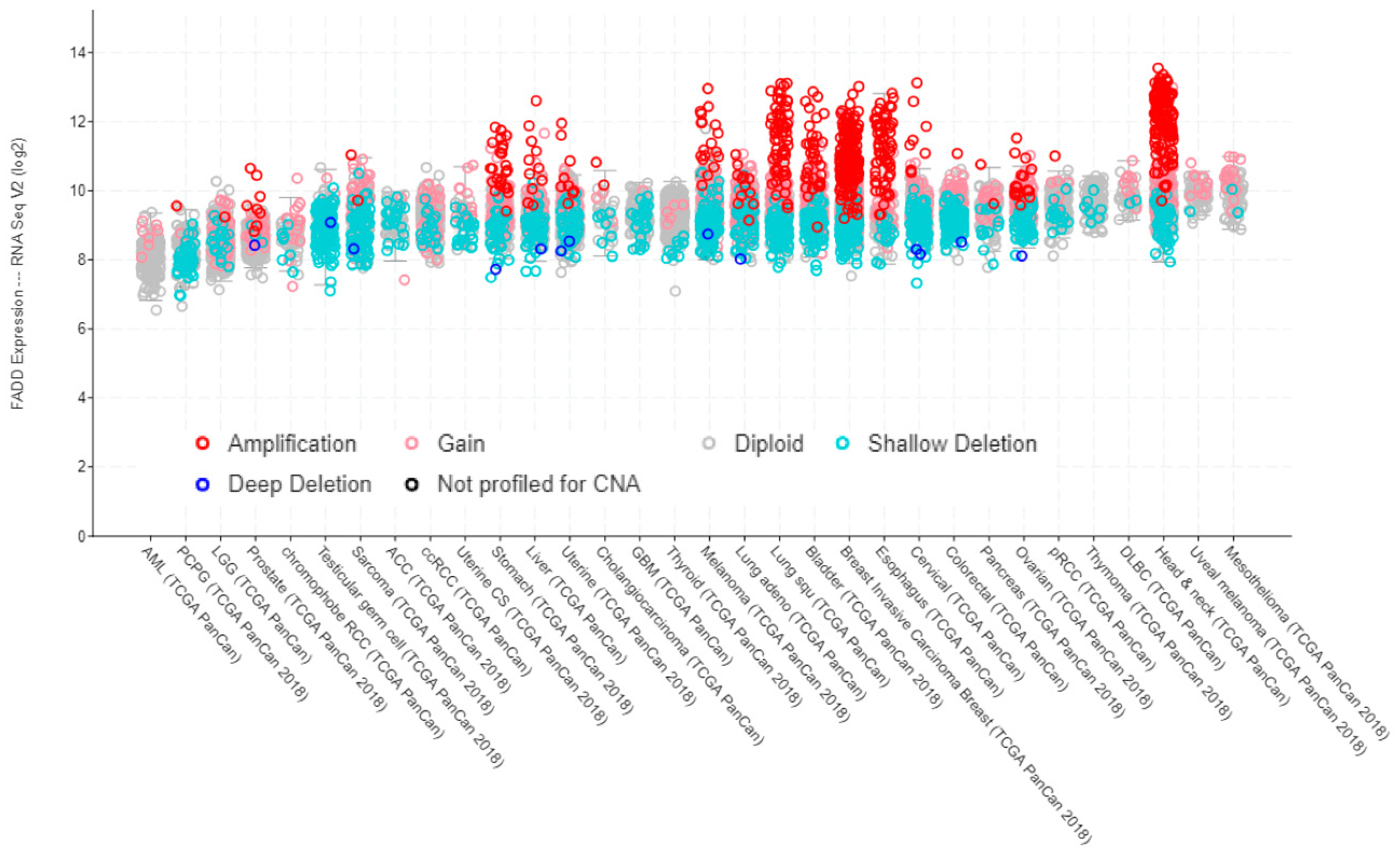
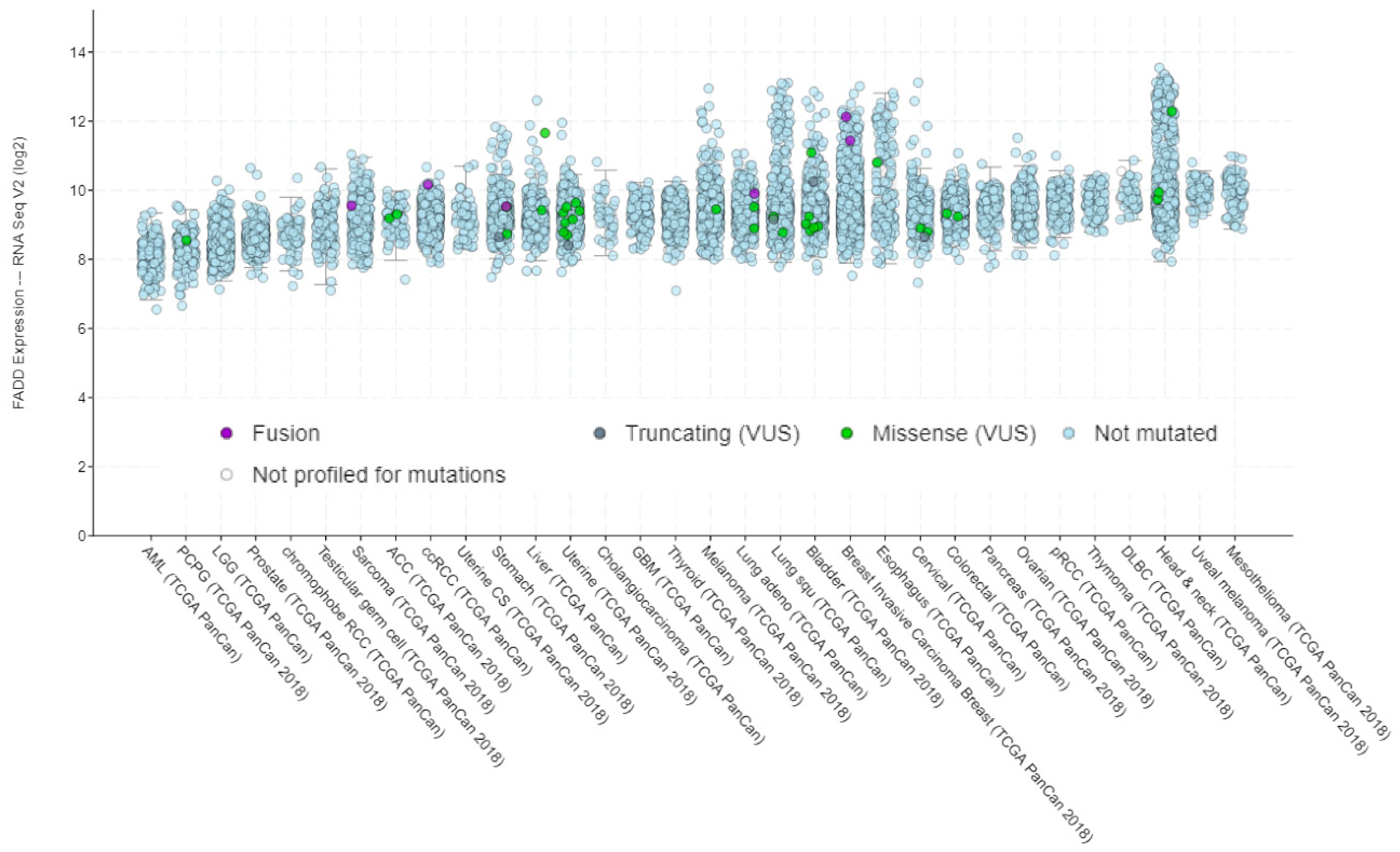
1.1. Genetic and Chromosomal Alterations Affecting FADD
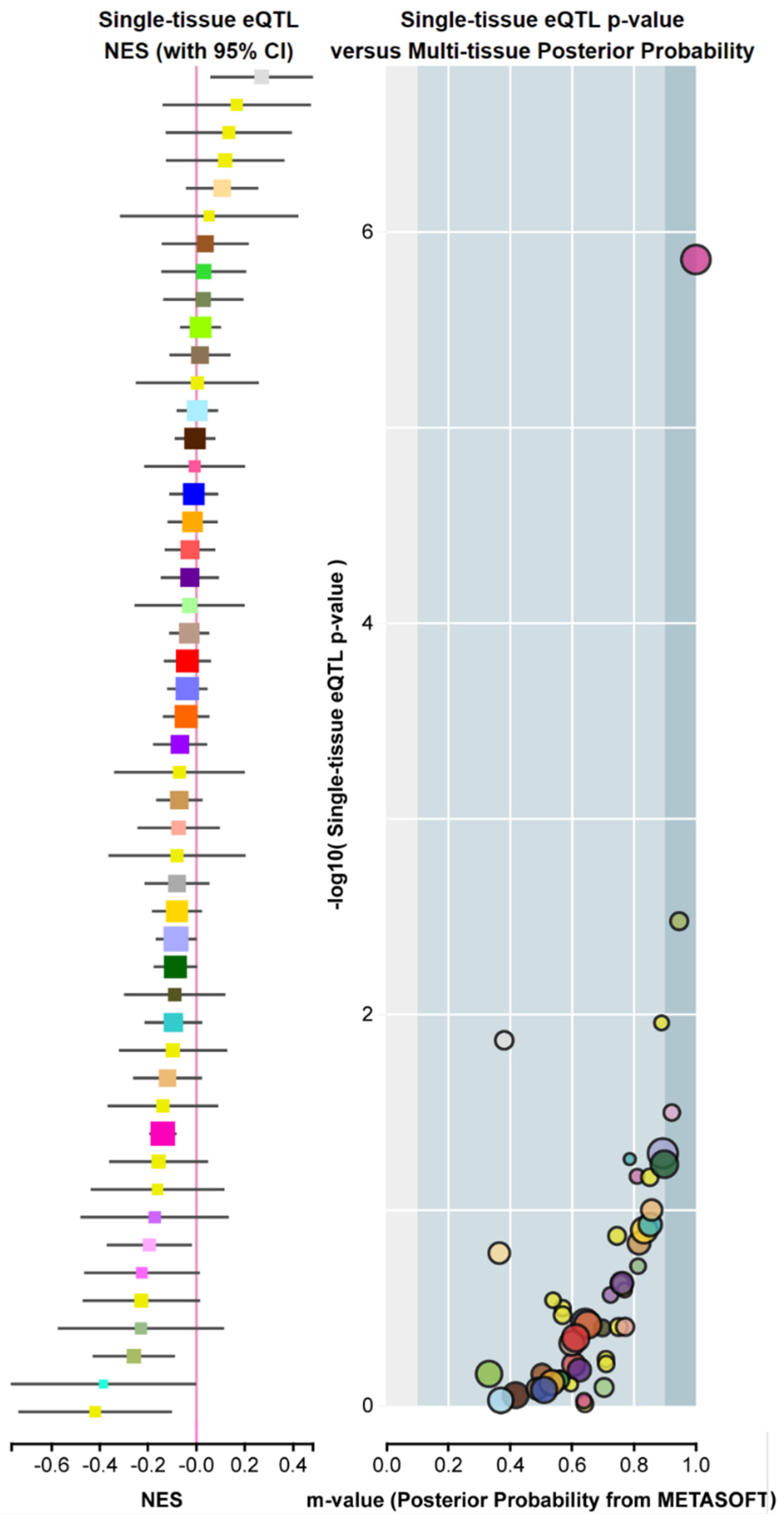
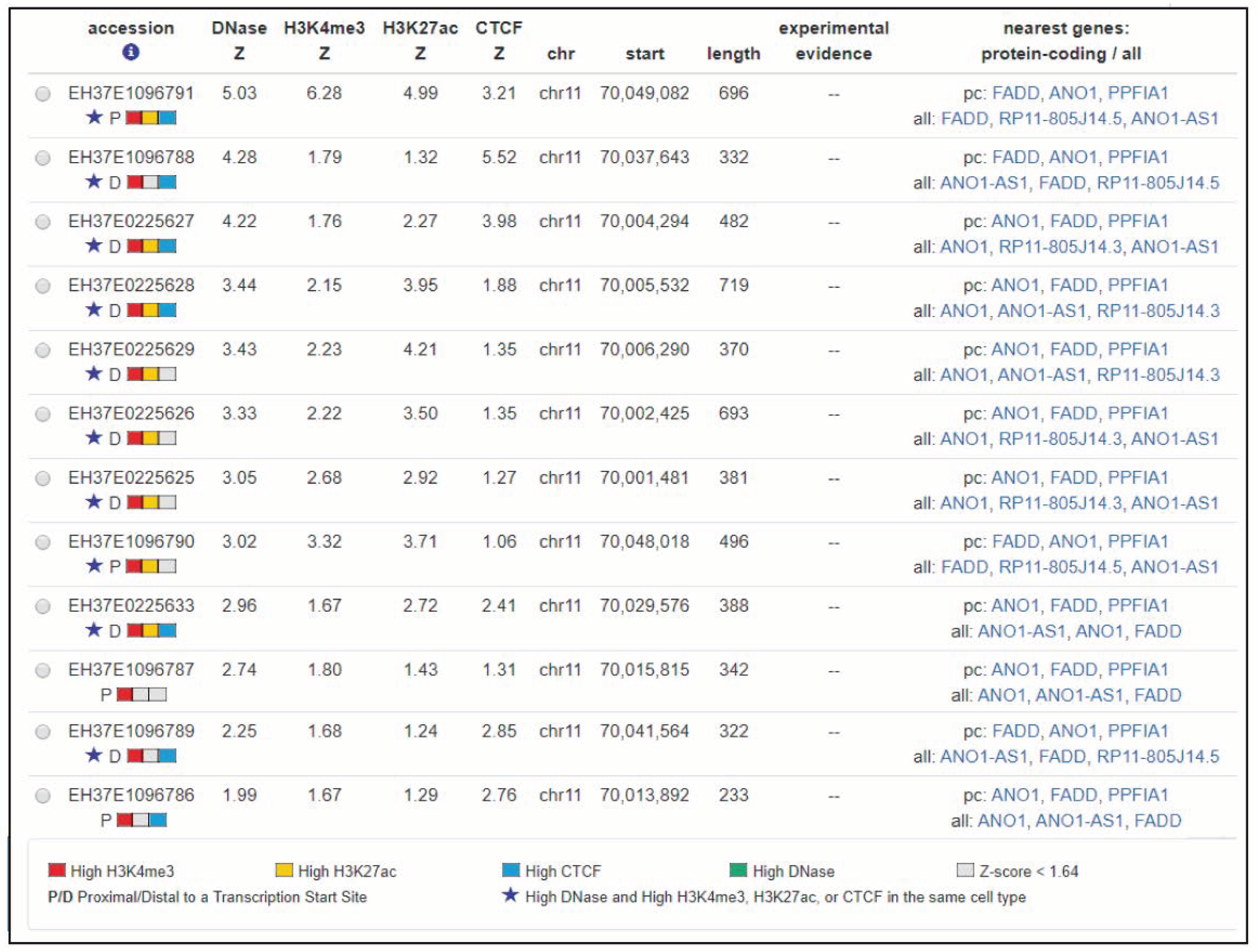
1.2. Transcription Factors Affecting FADD
1.3. Epigenetic Regulation of FADD Expression
1.3.1. Methylation of FADD Promoter Region
1.3.2. Histone Modification of FADD
1.3.3. microRNAs Controlling FADD
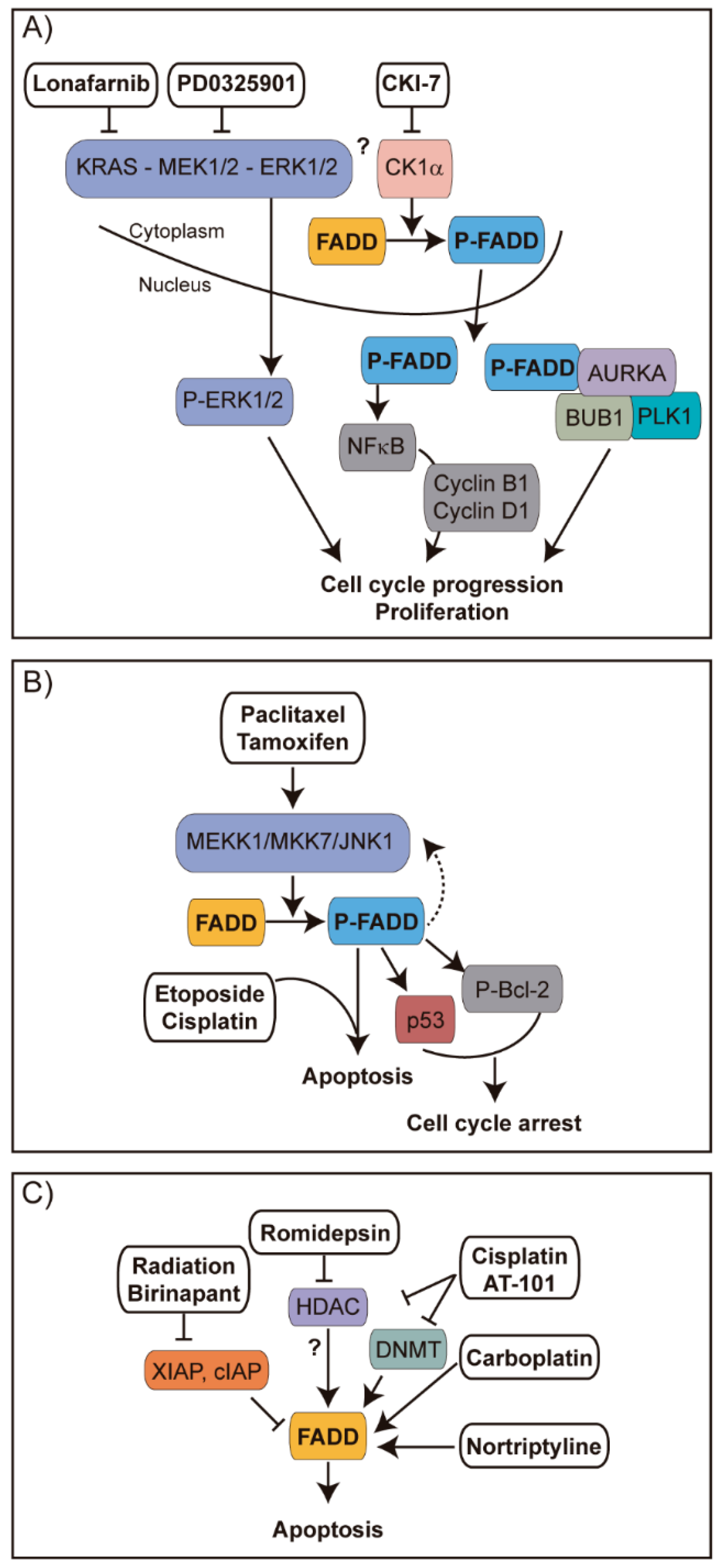
2. Posttranslational Modifications of FADD Protein
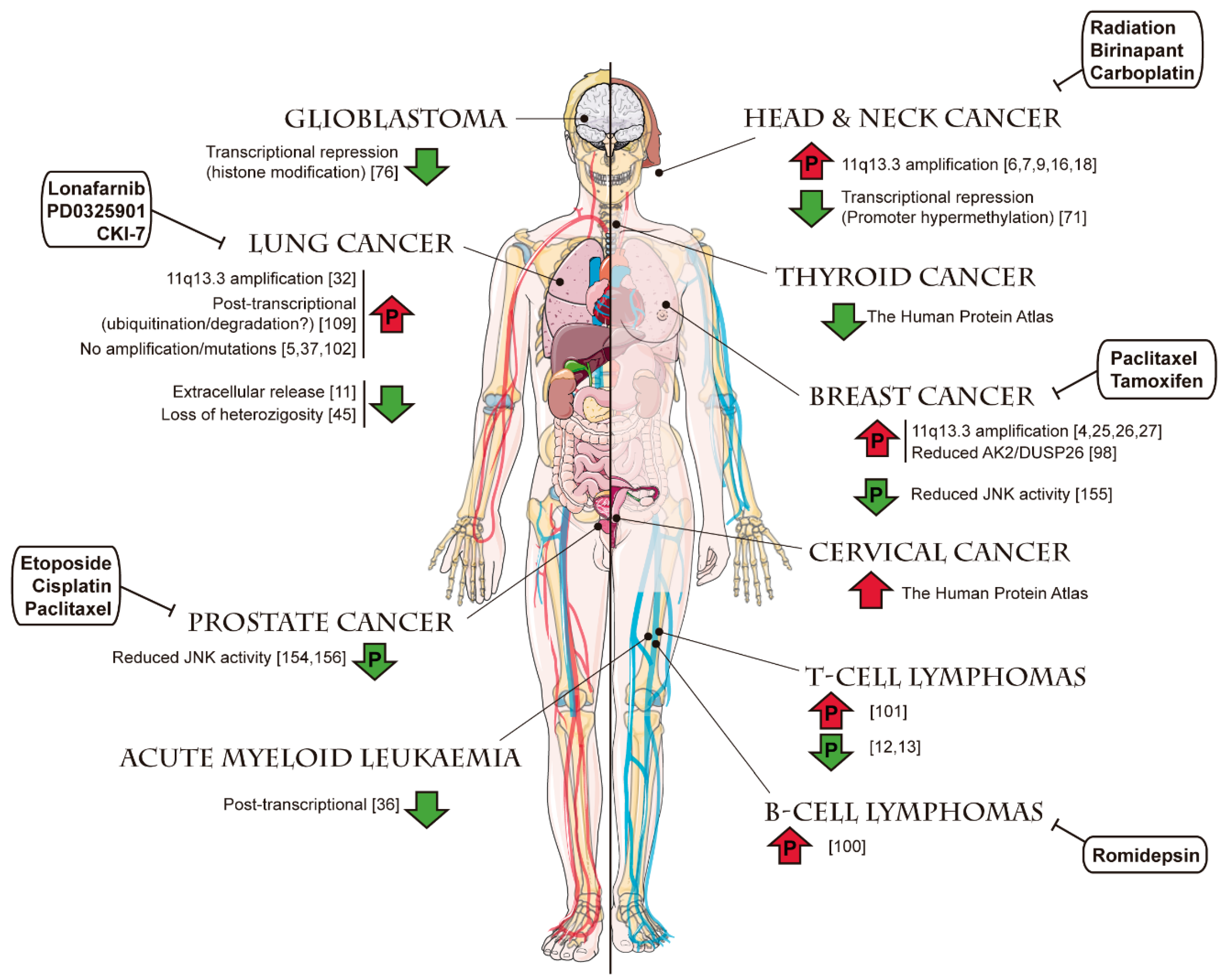
3. The Roles of FADD and Their Participation in Cancer
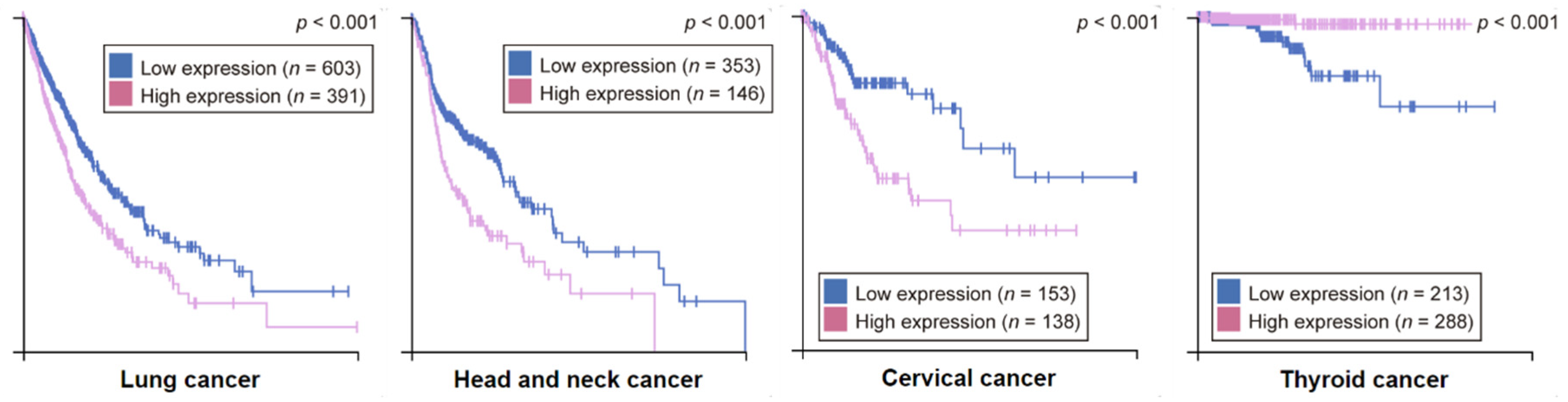
4. Clinical Implications of FADD Alterations in Cancer
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Yeh, W.C.; de la Pompa, J.L.; McCurrach, M.E.; Shu, H.B.; Elia, A.J.; Shahinian, A.; Ng, M.; Wakeham, A.; Khoo, W.; Mitchell, K.; et al. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 1998, 279, 1954–1958. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Callegari, C.C.; Cavalli, I.J.; Lima, R.S.; Jucoski, T.S.; Torresan, C.; Urban, C.A.; Kuroda, F.; Anselmi, K.F.; Cavalli, L.R.; Ribeiro, E.M. Copy number and expression analysis of FOSL1, GSTP1, NTSR1, FADD and CCND1 genes in primary breast tumors with axillary lymph node metastasis. Cancer Genet. 2016, 209, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Bhojani, M.S.; Heaford, A.C.; Chang, D.C.; Laxman, B.; Thomas, D.G.; Griffin, L.B.; Yu, J.; Coppola, J.M.; Giordano, T.J.; et al. Phosphorylated FADD induces NF-kappaB, perturbs cell cycle, and is associated with poor outcome in lung adenocarcinomas. Proc. Natl. Acad. Sci. USA 2005, 102, 12507–12512. [Google Scholar] [CrossRef] [PubMed]
- Chien, H.T.; Cheng, S.D.; Chuang, W.Y.; Liao, C.T.; Wang, H.M.; Huang, S.F. Clinical Implications of FADD Gene Amplification and Protein Overexpression in Taiwanese Oral Cavity Squamous Cell Carcinomas. PLoS ONE 2016, 11, e0164870. [Google Scholar] [CrossRef] [PubMed]
- Gibcus, J.H.; Menkema, L.; Mastik, M.F.; Hermsen, M.A.; de Bock, G.H.; van Velthuysen, M.L.; Takes, R.P.; Kok, K.; Alvarez Marcos, C.A.; van der Laan, B.F.; et al. Amplicon mapping and expression profiling identify the Fas-associated death domain gene as a new driver in the 11q13.3 amplicon in laryngeal/pharyngeal cancer. Clin. Cancer Res. 2007, 13, 6257–6266. [Google Scholar] [CrossRef]
- He, L.; Ren, Y.; Zheng, Q.; Wang, L.; Lai, Y.; Guan, S.; Zhang, X.; Zhang, R.; Wang, J.; Chen, D.; et al. Fas-associated protein with death domain (FADD) regulates autophagy through promoting the expression of Ras homolog enriched in brain (Rheb) in human breast adenocarcinoma cells. Oncotarget 2016, 7, 24572–24584. [Google Scholar] [CrossRef] [Green Version]
- Pattje, W.J.; Melchers, L.J.; Slagter-Menkema, L.; Mastik, M.F.; Schrijvers, M.L.; Gibcus, J.H.; Kluin, P.M.; Hoegen-Chouvalova, O.; van der Laan, B.F.; Roodenburg, J.L.; et al. FADD expression is associated with regional and distant metastasis in squamous cell carcinoma of the head and neck. Histopathology 2013, 63, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Schrijvers, M.L.; Pattje, W.J.; Slagter-Menkema, L.; Mastik, M.F.; Gibcus, J.H.; Langendijk, J.A.; van der Wal, J.E.; van der Laan, B.F.; Schuuring, E. FADD expression as a prognosticator in early-stage glottic squamous cell carcinoma of the larynx treated primarily with radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1220–1226. [Google Scholar] [CrossRef]
- Cimino, Y.; Costes, A.; Damotte, D.; Validire, P.; Mistou, S.; Cagnard, N.; Alifano, M.; Regnard, J.F.; Chiocchia, G.; Sautes-Fridman, C.; et al. FADD protein release mirrors the development and aggressiveness of human non-small cell lung cancer. Br. J. Cancer 2012, 106, 1989–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Rubio, J.L.; de Arriba, M.C.; Cobos-Fernandez, M.A.; Gonzalez-Sanchez, L.; Ors, I.; Sastre, I.; Fernandez-Piqueras, J.; Villa-Morales, M. Deregulated FADD expression and phosphorylation in T-cell lymphoblastic lymphoma. Oncotarget 2016. [Google Scholar] [CrossRef]
- Marin-Rubio, J.L.; Perez-Gomez, E.; Fernandez-Piqueras, J.; Villa-Morales, M. S194-P-FADD as a marker of aggressiveness and poor prognosis in human T-cell lymphoblastic lymphoma. Carcinogenesis 2019. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Schuchmann, M.; Galle, P.R. Cell death and hepatocarcinogenesis: Dysregulation of apoptosis signaling pathways. J. Gastroenterol Hepatol. 2011, 26 (Suppl. 1), 213–219. [Google Scholar] [CrossRef]
- Tourneur, L.; Mistou, S.; Michiels, F.M.; Devauchelle, V.; Renia, L.; Feunteun, J.; Chiocchia, G. Loss of FADD protein expression results in a biased Fas-signaling pathway and correlates with the development of tumoral status in thyroid follicular cells. Oncogene 2003, 22, 2795–2804. [Google Scholar] [CrossRef] [Green Version]
- Eytan, D.F.; Snow, G.E.; Carlson, S.; Derakhshan, A.; Saleh, A.; Schiltz, S.; Cheng, H.; Mohan, S.; Cornelius, S.; Coupar, J.; et al. SMAC Mimetic Birinapant plus Radiation Eradicates Human Head and Neck Cancers with Genomic Amplifications of Cell Death Genes FADD and BIRC2. Cancer Res. 2016, 76, 5442–5454. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Yang, X.; Si, H.; Saleh, A.D.; Xiao, W.; Coupar, J.; Gollin, S.M.; Ferris, R.L.; Issaeva, N.; Yarbrough, W.G.; et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Rep. 2018, 25, 1332–1345. [Google Scholar] [CrossRef]
- Reddy, R.B.; Bhat, A.R.; James, B.L.; Govindan, S.V.; Mathew, R.; Ravindra, D.R.; Hedne, N.; Illiayaraja, J.; Kekatpure, V.; Khora, S.S.; et al. Meta-Analyses of Microarray Datasets Identifies ANO1 and FADD as Prognostic Markers of Head and Neck Cancer. PLoS ONE 2016, 11, e0147409. [Google Scholar] [CrossRef]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 6, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Yun, J.A.; Jabeen, S.; Jeon, E.K.; Won, H.S.; Ko, Y.H.; Kim, S.Y. Prognostic significance of TMEM16A, PPFIA1, and FADD expression in invasive ductal carcinoma of the breast. World J. Surg. Oncol. 2014, 12, 137. [Google Scholar] [CrossRef]
- Lundgren, K.; Holm, K.; Nordenskjold, B.; Borg, A.; Landberg, G. Gene products of chromosome 11q and their association with CCND1 gene amplification and tamoxifen resistance in premenopausal breast cancer. Breast Cancer Res. 2008, 10, R81. [Google Scholar] [CrossRef]
- Norton, N.; Advani, P.P.; Serie, D.J.; Geiger, X.J.; Necela, B.M.; Axenfeld, B.C.; Kachergus, J.M.; Feathers, R.W.; Carr, J.M.; Crook, J.E.; et al. Assessment of Tumor Heterogeneity, as Evidenced by Gene Expression Profiles, Pathway Activation, and Gene Copy Number, in Patients with Multifocal Invasive Lobular Breast Tumors. PLoS ONE 2016, 11, e0153411. [Google Scholar] [CrossRef]
- Matsuda, R.; Enokida, H.; Chiyomaru, T.; Kikkawa, N.; Sugimoto, T.; Kawakami, K.; Tatarano, S.; Yoshino, H.; Toki, K.; Uchida, Y.; et al. LY6K is a novel molecular target in bladder cancer on basis of integrate genome-wide profiling. Br. J. Cancer 2011, 104, 376–386. [Google Scholar] [CrossRef]
- Proctor, A.J.; Coombs, L.M.; Cairns, J.P.; Knowles, M.A. Amplification at chromosome 11q13 in transitional cell tumours of the bladder. Oncogene 1991, 6, 789–795. [Google Scholar]
- Qin, S.L.; Chen, X.J.; Xu, X.; Shou, J.Z.; Bi, X.G.; Ji, L.; Han, Y.L.; Cai, Y.; Wei, F.; Ma, J.H.; et al. Detection of chromosomal alterations in bladder transitional cell carcinomas from Northern China by comparative genomic hybridization. Cancer Lett. 2006, 238, 230–239. [Google Scholar] [CrossRef]
- Zaharieva, B.M.; Simon, R.; Diener, P.A.; Ackermann, D.; Maurer, R.; Alund, G.; Knonagel, H.; Rist, M.; Wilber, K.; Hering, F.; et al. High-throughput tissue microarray analysis of 11q13 gene amplification (CCND1, FGF3, FGF4, EMS1) in urinary bladder cancer. J. Pathol. 2003, 201, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.; Aninat-Meyer, M.; Schluns, K.; Gellert, K.; Dietel, M.; Petersen, I. Chromosomal alterations in the clonal evolution to the metastatic stage of squamous cell carcinomas of the lung. Br. J. Cancer 2000, 82, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Irving, J.; Parker, R.; Kim, H.; Press, J.Z.; Longacre, T.A.; Chia, S.; Magliocco, A.; Makretsov, N.; Gilks, B.; et al. Amplification of EMSY, a novel oncogene on 11q13, in high grade ovarian surface epithelial carcinomas. Gynecol. Oncol. 2006, 100, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Kalloger, S.E.; Miller, M.A.; Shih Ie, M.; McKinney, S.E.; Santos, J.L.; Swenerton, K.; Spellman, P.T.; Gray, J.; Gilks, C.B.; et al. Amplification of 11q13 in ovarian carcinoma. Genes Chromosomes Cancer 2008, 47, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Schraml, P.; Schwerdtfeger, G.; Burkhalter, F.; Raggi, A.; Schmidt, D.; Ruffalo, T.; King, W.; Wilber, K.; Mihatsch, M.J.; Moch, H. Combined array comparative genomic hybridization and tissue microarray analysis suggest PAK1 at 11q13.5-q14 as a critical oncogene target in ovarian carcinoma. Am. J. Pathol. 2003, 163, 985–992. [Google Scholar] [CrossRef]
- Tourneur, L.; Delluc, S.; Levy, V.; Valensi, F.; Radford-Weiss, I.; Legrand, O.; Vargaftig, J.; Boix, C.; Macintyre, E.A.; Varet, B.; et al. Absence or low expression of fas-associated protein with death domain in acute myeloid leukemia cells predicts resistance to chemotherapy and poor outcome. Cancer Res. 2004, 64, 8101–8108. [Google Scholar] [CrossRef]
- Bhojani, M.S.; Chen, G.; Ross, B.D.; Beer, D.G.; Rehemtulla, A. Nuclear localized phosphorylated FADD induces cell proliferation and is associated with aggressive lung cancer. Cell Cycle 2005, 4, 1478–1481. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Srivatsan, E.S.; Wood, T.F.; Eubanks, P.J.; Ebrahimi, S.A.; Gatti, R.A.; Passaro, E., Jr.; Sawicki, M.P. Deletion mapping of endocrine tumors localizes a second tumor suppressor gene on chromosome band 11q13. Genes Chromosomes Cancer 1998, 22, 130–137. [Google Scholar] [CrossRef]
- Cheng, Y.; Chakrabarti, R.; Garcia-Barcelo, M.; Ha, T.J.; Srivatsan, E.S.; Stanbridge, E.J.; Lung, M.L. Mapping of nasopharyngeal carcinoma tumor-suppressive activity to a 1.8-megabase region of chromosome band 11q13. Genes Chromosomes Cancer 2002, 34, 97–103. [Google Scholar] [CrossRef]
- Srivatsan, E.S.; Ying, K.L.; Seeger, R.C. Deletion of chromosome 11 and of 14q sequences in neuroblastoma. Genes Chromosomes Cancer 1993, 7, 32–37. [Google Scholar] [CrossRef]
- Venugopalan, M.; Wood, T.F.; Wilczynski, S.P.; Sen, S.; Peters, J.; Ma, G.C.; Evans, G.A.; Srivatsan, E.S. Loss of heterozygosity in squamous cell carcinomas of the head and neck defines a tumor suppressor gene region on 11q13. Cancer Genet. Cytogenet 1998, 104, 124–132. [Google Scholar] [CrossRef]
- Zhuang, Z.; Merino, M.J.; Chuaqui, R.; Liotta, L.A.; Emmert-Buck, M.R. Identical allelic loss on chromosome 11q13 in microdissected in situ and invasive human breast cancer. Cancer Res. 1995, 55, 467–471. [Google Scholar] [PubMed]
- Soares, B.S.; Eguchi, K.; Frohman, L.A. Tumor deletion mapping on chromosome 11q13 in eight families with isolated familial somatotropinoma and in 15 sporadic somatotropinomas. J. Clin. Endocrinol. Metab. 2005, 90, 6580–6587. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, E.S.; Chakrabarti, R.; Zainabadi, K.; Pack, S.D.; Benyamini, P.; Mendonca, M.S.; Yang, P.K.; Kang, K.; Motamedi, D.; Sawicki, M.P.; et al. Localization of deletion to a 300 Kb interval of chromosome 11q13 in cervical cancer. Oncogene 2002, 21, 5631–5642. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.S.; Lam, W.K.; Wong, M.P.; Fu, K.H.; Lee, J.; Yew, W.W.; Chiu, S.W.; Lung, M.L. Chromosomal 11 alterations in non-small-cell lung carcinomas in Hong Kong. Lung Cancer 1996, 15, 51–65. [Google Scholar] [CrossRef]
- Gregory-Evans, C.Y.; Moosajee, M.; Hodges, M.D.; Mackay, D.S.; Game, L.; Vargesson, N.; Bloch-Zupan, A.; Ruschendorf, F.; Santos-Pinto, L.; Wackens, G.; et al. SNP genome scanning localizes oto-dental syndrome to chromosome 11q13 and microdeletions at this locus implicate FGF3 in dental and inner-ear disease and FADD in ocular coloboma. Hum. Mol. Genet. 2007, 16, 2482–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Kim, G.H.; Byeon, J.H.; Eun, S.H.; Eun, B.L. Chromosome 11q13 deletion syndrome. Korean J. Pediatr. 2016, 59, S10–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.S.; Kim, H.S.; Lee, S.H.; Lee, J.W.; Song, Y.H.; Kim, Y.S.; Park, W.S.; Kim, S.Y.; Lee, S.N.; Park, J.Y.; et al. Alterations of Fas-pathway genes associated with nodal metastasis in non-small cell lung cancer. Oncogene 2002, 21, 4129–4136. [Google Scholar] [CrossRef]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Nam, S.W.; Park, W.S.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutation of FADD gene is rare in human colon and stomach cancers. APMIS 2004, 112, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld-Granot, G.; Toren, A.; Amariglio, N.; Brok-Simoni, F.; Rechavi, G. Mutation analysis of the FAS and TNFR apoptotic cascade genes in hematological malignancies. Exp. Hematol. 2001, 29, 228–233. [Google Scholar] [CrossRef]
- Dechant, M.J.; Fellenberg, J.; Scheuerpflug, C.G.; Ewerbeck, V.; Debatin, K.M. Mutation analysis of the apoptotic “death-receptors” and the adaptors TRADD and FADD/MORT-1 in osteosarcoma tumor samples and osteosarcoma cell lines. Int. J. Cancer 2004, 109, 661–667. [Google Scholar] [CrossRef]
- Bolze, A.; Byun, M.; McDonald, D.; Morgan, N.V.; Abhyankar, A.; Premkumar, L.; Puel, A.; Bacon, C.M.; Rieux-Laucat, F.; Pang, K.; et al. Whole-exome-sequencing-based discovery of human FADD deficiency. Am. J. Hum. Genet. 2010, 87, 873–881. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Eun, Y.G.; Chung, D.H.; Kim, S.W.; Lee, Y.C.; Kim, S.K.; Kwon, K.H. A Fas-associated via death domain promoter polymorphism (rs10898853, -16C/T) as a risk factor for papillary thyroid cancer. Eur. Surg. Res. 2014, 52, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Villa-Morales, M.; Gonzalez-Gugel, E.; Shahbazi, M.N.; Santos, J.; Fernandez-Piqueras, J. Modulation of the Fas-apoptosis-signalling pathway by functional polymorphisms at Fas, FasL and Fadd and their implication in T-cell lymphoblastic lymphoma susceptibility. Carcinogenesis 2010, 31, 2165–2171. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Eskin, E. Interpreting meta-analyses of genome-wide association studies. PLoS Genet. 2012, 8, e1002555. [Google Scholar] [CrossRef]
- Kim, P.K.; Dutra, A.S.; Chandrasekharappa, S.C.; Puck, J.M. Genomic structure and mapping of human FADD, an intracellular mediator of lymphocyte apoptosis. J. Immunol. 1996, 157, 5461–5466. [Google Scholar]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.; Whitfield, T.W.; Greven, M.C.; Pierce, B.G.; Dong, X.; Kundaje, A.; Cheng, Y.; et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012, 22, 1798–1812. [Google Scholar] [CrossRef] [Green Version]
- Hindryckx, P.; De Vos, M.; Jacques, P.; Ferdinande, L.; Peeters, H.; Olievier, K.; Bogaert, S.; Brinkman, B.; Vandenabeele, P.; Elewaut, D.; et al. Hydroxylase inhibition abrogates TNF-alpha-induced intestinal epithelial damage by hypoxia-inducible factor-1-dependent repression of FADD. J. Immunol. 2010, 185, 6306–6316. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Nguyen, D.D.; Lee, D.G.; Kim, S.; Kang, K.; Rhee, J.K.; Chang, S. Integrative Bioinformatics and Functional Analyses of GEO, ENCODE, and TCGA Reveal FADD as a Direct Target of the Tumor Suppressor BRCA1. Int. J. Mol. Sci. 2018, 19, 1458. [Google Scholar] [CrossRef] [PubMed]
- Viringipurampeer, I.A.; Ferreira, T.; DeMaria, S.; Yoon, J.J.; Shan, X.; Moosajee, M.; Gregory-Evans, K.; Ngai, J.; Gregory-Evans, C.Y. Pax2 regulates a fadd-dependent molecular switch that drives tissue fusion during eye development. Hum. Mol. Genet. 2012, 21, 2357–2369. [Google Scholar] [CrossRef] [Green Version]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.B.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef] [PubMed]
- Llinas-Arias, P.; Esteller, M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: An update. Open Biol. 2017, 7, 170152. [Google Scholar] [CrossRef]
- Zhao, X.; Yin, H.; Li, N.; Zhu, Y.; Shen, W.; Qian, S.; He, G.; Li, J.; Wang, X. An Integrated Regulatory Network Based on Comprehensive Analysis of mRNA Expression, Gene Methylation and Expression of Long Non-coding RNAs (lncRNAs) in Myelodysplastic Syndromes. Front. Oncol. 2019, 9, 200. [Google Scholar] [CrossRef] [PubMed]
- Wichnieski, C.; Maheshwari, K.; Souza, L.C.; Nieves, F.; Tartari, T.; Garlet, G.P.; Carneiro, E.; Letra, A.; Silva, R.M. DNA methylation profiles of immune response-related genes in apical periodontitis. Int. Endod. J. 2019, 52, 5–12. [Google Scholar] [CrossRef]
- Friedrich, M.G.; Chandrasoma, S.; Siegmund, K.D.; Weisenberger, D.J.; Cheng, J.C.; Toma, M.I.; Huland, H.; Jones, P.A.; Liang, G. Prognostic relevance of methylation markers in patients with non-muscle invasive bladder carcinoma. Eur. J. Cancer. 2005, 41, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Saberi, E.; Kordi-Tamandani, D.M.; Jamali, S.; Rigi-Ladiz, M.A. Analysis of methylation and mRNA expression status of FADD and FAS genes in patients with oral squamous cell carcinoma. Med. Oral Patol. Oral. Cir. Bucal. 2014, 19, e562–e568. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Kim, J.; Yu, J. Influence of oncogenic transcription factors on chromatin conformation and implications in prostate cancer. Appl. Clin. Genet. 2014, 7, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Wu, M.; Liu, Q.; Lv, H.; Jiang, R. DeepHistone: A deep learning approach to predicting histone modifications. BMC Genom. 2019, 20, 193. [Google Scholar] [CrossRef] [PubMed]
- A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [CrossRef]
- An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef] [PubMed]
- Tamannai, M.; Farhangi, S.; Truss, M.; Sinn, B.; Wurm, R.; Bose, P.; Henze, G.; Riabowol, K.; von Deimling, A.; Tallen, G. The inhibitor of growth 1 (ING1) is involved in trichostatin A-induced apoptosis and caspase 3 signaling in p53-deficient glioblastoma cells. Oncol. Res. 2010, 18, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.; Sugita, F.; Nakamura, Y.; Sasaki, K.; Mitani, K. Fadd and Skp2 are possible downstream targets of RUNX1-EVI1. Int. J. Hematol. 2013, 97, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Tay, Y.M.; Tam, W.L.; Ang, Y.S.; Gaughwin, P.M.; Yang, H.; Wang, W.; Liu, R.; George, J.; Ng, H.H.; Perera, R.J.; et al. MicroRNA-134 modulates the differentiation of mouse embryonic stem cells, where it causes post-transcriptional attenuation of Nanog and LRH1. Stem cells 2008, 26, 17–29. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.F.; Yang, L.X.; Guo, R.W.; Liu, H.; Shi, Y.K.; Wang, H.; Ye, J.S.; Yang, Z.H.; Liang, X. miR-155 inhibits oxidized low-density lipoprotein-induced apoptosis of RAW264.7 cells. Mol. Cell Biochem. 2013, 382, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Yu, X.D.; Liu, Z.H.; Cheng, X.; Samartzis, D.; Jia, L.T.; Wu, S.X.; Huang, J.; Chen, J.; Luo, Z.J. Deregulated miR-155 promotes Fas-mediated apoptosis in human intervertebral disc degeneration by targeting FADD and caspase-3. J. Pathol. 2011, 225, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Liu, Z.; Li, B.S.; Tang, B.; Li, W.; Guo, G.; Shi, Y.; Wang, F.; Wu, Y.; Tong, W.D.; et al. Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J. Infect. Dis. 2009, 200, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Noguchi, S.; Kumazaki, M.; Shinohara, H.; Miki, K.; Naoe, T.; Akao, Y. Epigenetic regulation of microRNA-128a expression contributes to the apoptosis-resistance of human T-cell leukaemia jurkat cells by modulating expression of fas-associated protein with death domain (FADD). Biochim. Biophys. Acta 2014, 1843, 590–602. [Google Scholar] [CrossRef]
- Tili, E.; Croce, C.M.; Michaille, J.J. miR-155: On the crosstalk between inflammation and cancer. Int. Rev. Immunol. 2009, 28, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Mouasni, S.; Tourneur, L. FADD at the Crossroads between Cancer and Inflammation. Trends Immunol. 2018, 39, 1036–1053. [Google Scholar] [CrossRef] [PubMed]
- Tourneur, L.; Chiocchia, G. FADD: A regulator of life and death. Trends Immunol. 2010, 31, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, D.; Hua, Z. FADD and its phosphorylation. IUBMB Life 2004, 56, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.S.; Lee, S.J.; Kang, N.S.; Kim, E. Cooperative phosphorylation of FADD by Aur-A and Plk1 in response to taxol triggers both apoptotic and necrotic cell death. Cancer Res. 2011, 71, 7207–7215. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Volkland, J.; Blomberg, I.; Hoffmann, I.; Krammer, P.H.; Peter, M.E. Phosphorylation of FADD/ MORT1 at serine 194 and association with a 70-kDa cell cycle-regulated protein kinase. J. Immunol. 2000, 164, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Vilmont, V.; Filhol, O.; Hesse, A.M.; Coute, Y.; Hue, C.; Remy-Tourneur, L.; Mistou, S.; Cochet, C.; Chiocchia, G. Modulatory role of the anti-apoptotic protein kinase CK2 in the sub-cellular localization of Fas associated death domain protein (FADD). Biochim. Biophys. Acta 2015, 1853, 2885–2896. [Google Scholar] [CrossRef] [Green Version]
- Hua, Z.C.; Sohn, S.J.; Kang, C.; Cado, D.; Winoto, A. A function of Fas-associated death domain protein in cell cycle progression localized to a single amino acid at its C-terminal region. Immunity 2003, 18, 513–521. [Google Scholar] [CrossRef]
- Yao, C.; Zhuang, H.; Cheng, W.; Lin, Y.; Du, P.; Yang, B.; Huang, X.; Chen, S.; Hu, Q.; Hua, Z.C. FADD phosphorylation impaired islet morphology and function. J. Cell Physiol. 2015, 230, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Foger, N.; Bulfone-Paus, S.; Chan, A.C.; Lee, K.H. Subcellular compartmentalization of FADD as a new level of regulation in death receptor signaling. FEBS J. 2009, 276, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.W.; Chandra, J.; Loegering, D.; Van Becelaere, K.; Kottke, T.J.; Gore, S.D.; Karp, J.E.; Sebolt-Leopold, J.; Kaufmann, S.H. Central role of Fas-associated death domain protein in apoptosis induction by the mitogen-activated protein kinase kinase inhibitor CI-1040 (PD184352) in acute lymphocytic leukemia cells in vitro. J. Biol. Chem. 2003, 278, 47326–47339. [Google Scholar] [CrossRef] [PubMed]
- Screaton, R.A.; Kiessling, S.; Sansom, O.J.; Millar, C.B.; Maddison, K.; Bird, A.; Clarke, A.R.; Frisch, S.M. Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: A potential link between genome surveillance and apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 5211–5216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Vallabhaneni, R.; Loughran, P.A.; Shapiro, R.; Yin, X.M.; Yuan, Y.; Billiar, T.R. Changes in FADD levels, distribution, and phosphorylation in TNFalpha-induced apoptosis in hepatocytes is caspase-3, caspase-8 and BID dependent. Apoptosis 2008, 13, 983–992. [Google Scholar] [CrossRef]
- Kim, H.; Lee, H.J.; Oh, Y.; Choi, S.G.; Hong, S.H.; Kim, H.J.; Lee, S.Y.; Choi, J.W.; Su Hwang, D.; Kim, K.S.; et al. The DUSP26 phosphatase activator adenylate kinase 2 regulates FADD phosphorylation and cell growth. Nat. Commun. 2014, 5, 3351. [Google Scholar] [CrossRef]
- Zhang, J.; Winoto, A. A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Mol. Cell Biol. 1996, 16, 2756–2763. [Google Scholar] [CrossRef] [Green Version]
- Drakos, E.; Leventaki, V.; Atsaves, V.; Schlette, E.J.; Lin, P.; Vega, F.; Miranda, R.N.; Claret, F.X.; Medeiros, L.J.; Rassidakis, G.Z. Expression of serine 194-phosphorylated Fas-associated death domain protein correlates with proliferation in B-cell non-Hodgkin lymphomas. Hum. Pathol. 2011, 42, 1117–1124. [Google Scholar] [CrossRef]
- Patel, S.; Murphy, D.; Haralambieva, E.; Abdulla, Z.A.; Wong, K.K.; Chen, H.; Gould, E.; Roncador, G.; Hatton, C.S.; Anderson, A.P.; et al. Increased Expression of Phosphorylated FADD in Anaplastic Large Cell and Other T-Cell Lymphomas. Biomark. insights 2014, 9, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.M.; Sebolt, K.A.; Hoff, B.A.; Boes, J.L.; Daniels, D.L.; Heist, K.A.; Galban, C.J.; Patel, R.M.; Zhang, J.; Beer, D.G.; et al. Phosphorylation of FADD by the kinase CK1alpha promotes KRASG12D-induced lung cancer. Sci. Signal. 2015, 8, ra9. [Google Scholar] [CrossRef] [PubMed]
- Schinske, K.A.; Nyati, S.; Khan, A.P.; Williams, T.M.; Johnson, T.D.; Ross, B.D.; Perez Tomas, R.; Rehemtulla, A. A Novel Kinase Inhibitor of FADD Phosphorylation Chemosensitizes through the Inhibition of NF-κB. Mol. Cancer Ther. 2011, 10, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.W.; Kim, J.H.; Ahn, Y.H.; Seo, J.; Ko, A.; Jeong, M.; Kim, S.J.; Ro, J.Y.; Park, K.M.; Lee, H.W.; et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat. Commun. 2012, 3, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feltham, R.; Silke, J. The small molecule that packs a punch: Ubiquitin-mediated regulation of RIPK1/FADD/caspase-8 complexes. Cell Death Differ. 2017, 24, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Lee, E.W.; Shin, J.; Seong, D.; Nam, Y.W.; Jeong, M.; Lee, S.H.; Lee, C.; Song, J. K6 linked polyubiquitylation of FADD by CHIP prevents death inducing signaling complex formation suppressing cell death. Oncogene 2018, 37, 4994–5006. [Google Scholar] [CrossRef] [PubMed]
- Goto, E.; Tokunaga, F. Decreased linear ubiquitination of NEMO and FADD on apoptosis with caspase-mediated cleavage of HOIP. Biochem. Biophys. Res. Commun. 2017, 485, 152–159. [Google Scholar] [CrossRef]
- Tan, P.; Wang, A.; Chen, H.; Du, Y.; Qian, B.; Shi, H.; Zhang, Y.; Xia, X.; Fu, W. SPOP inhibits mice pancreatic stellate cell activation by promoting FADD degradation in cerulein-induced chronic pancreatitis. Exp. Cell Res. 2019, 111606. [Google Scholar] [CrossRef]
- Luo, J.; Chen, B.; Gao, C.X.; Xie, H.K.; Han, C.N.; Zhou, C.C. SPOP promotes FADD degradation and inhibits NF-kappaB activity in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2018, 504, 289–294. [Google Scholar] [CrossRef]
- Chen, L.; Pei, H.; Lu, S.J.; Liu, Z.J.; Yan, L.; Zhao, X.M.; Hu, B.; Lu, H.G. SPOP suppresses osteosarcoma invasion via PI3K/AKT/NF-kappaB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 609–615. [Google Scholar] [CrossRef]
- Boldin, M.P.; Varfolomeev, E.E.; Pancer, Z.; Mett, I.L.; Camonis, J.H.; Wallach, D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 1995, 270, 7795–7798. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Oberst, A.; Pop, C.; Tremblay, A.G.; Blais, V.; Denault, J.B.; Salvesen, G.S.; Green, D.R. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J. Biol. Chem. 2010, 285, 16632–16642. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.P.; Nair, A.; Zhang, J. A novel function of RIP1 in postnatal development and immune homeostasis by protecting against RIP3-dependent necroptosis and FADD-mediated apoptosis. Front. Cell Dev. Biol. 2015, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Osborn, S.L.; Diehl, G.; Han, S.J.; Xue, L.; Kurd, N.; Hsieh, K.; Cado, D.; Robey, E.A.; Winoto, A. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc. Natl. Acad. Sci. USA 2010, 107, 13034–13039. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J. Immunol. 2015, 194, 1938–1944. [Google Scholar] [CrossRef]
- Peter, M.E.; Hadji, A.; Murmann, A.E.; Brockway, S.; Putzbach, W.; Pattanayak, A.; Ceppi, P. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015, 22, 885–886. [Google Scholar] [CrossRef]
- Alappat, E.C.; Feig, C.; Boyerinas, B.; Volkland, J.; Samuels, M.; Murmann, A.E.; Thorburn, A.; Kidd, V.J.; Slaughter, C.A.; Osborn, S.L.; et al. Phosphorylation of FADD at serine 194 by CKIalpha regulates its nonapoptotic activities. Mol. cell 2005, 19, 321–332. [Google Scholar] [CrossRef]
- Gomez-Angelats, M.; Cidlowski, J.A. Molecular evidence for the nuclear localization of FADD. Cell Death Differ. 2003, 10, 791–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beisner, D.R.; Chu, I.H.; Arechiga, A.F.; Hedrick, S.M.; Walsh, C.M. The requirements for Fas-associated death domain signaling in mature T cell activation and survival. J. Immunol. 2003, 171, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Osborn, S.L.; Sohn, S.J.; Winoto, A. Constitutive phosphorylation mutation in Fas-associated death domain (FADD) results in early cell cycle defects. J. Biol. Chem. 2007, 282, 22786–22792. [Google Scholar] [CrossRef] [PubMed]
- Papoff, G.; Trivieri, N.; Crielesi, R.; Ruberti, F.; Marsilio, S.; Ruberti, G. FADD-calmodulin interaction: A novel player in cell cycle regulation. Biochim. Biophys. Acta 2010, 1803, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Alappat, E.C.; Volkland, J.; Peter, M.E. Cell cycle effects by C-FADD depend on its C-terminal phosphorylation site. J. Biol. Chem. 2003, 278, 41585–41588. [Google Scholar] [CrossRef] [PubMed]
- Kabra, N.H.; Kang, C.; Hsing, L.C.; Zhang, J.; Winoto, A. T cell-specific FADD-deficient mice: FADD is required for early T cell development. Proc. Natl. Acad. Sci. USA 2001, 98, 6307–6312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kabra, N.H.; Cado, D.; Kang, C.; Winoto, A. FADD-deficient T cells exhibit a disaccord in regulation of the cell cycle machinery. J. Biol. Chem. 2001, 276, 29815–29818. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Thomas, E.; Barber, G.N. A FADD-dependent innate immune mechanism in mammalian cells. Nature 2004, 432, 401–405. [Google Scholar] [CrossRef]
- Leulier, F.; Vidal, S.; Saigo, K.; Ueda, R.; Lemaitre, B. Inducible expression of double-stranded RNA reveals a role for dFADD in the regulation of the antibacterial response in Drosophila adults. Curr. Biol. 2002, 12, 996–1000. [Google Scholar] [CrossRef]
- Zhang, X.; Zang, S.; Li, C.; Wei, J.; Qin, Q. Molecular cloning and characterization of FADD from the orange-spotted grouper (Epinephelus coioides). Fish. Shellfish. Immunol. 2018, 74, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.P.; Henry, C.M.; Kearney, C.J.; Logue, S.E.; Feoktistova, M.; Tynan, G.A.; Lavelle, E.C.; Leverkus, M.; Martin, S.J. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol. Cell 2013, 49, 1034–1048. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Kawazu, M.; Ueno, T.; Fukumura, K.; Yamato, A.; Soda, M.; Yamashita, Y.; Choi, Y.L.; Yamasoba, T.; Mano, H. Cancer-associated missense mutations of caspase-8 activate nuclear factor-kappaB signaling. Cancer Sci. 2013, 104, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Shikama, Y.; Yamada, M.; Miyashita, T. Caspase-8 and caspase-10 activate NF-kappaB through RIP, NIK and IKKalpha kinases. Eur. J. Immunol. 2003, 33, 1998–2006. [Google Scholar] [CrossRef]
- Freer-Prokop, M.; O’Flaherty, J.; Ross, J.A.; Weyman, C.M. Non-canonical role for the TRAIL receptor DR5/FADD/caspase pathway in the regulation of MyoD expression and skeletal myoblast differentiation. Differentiation 2009, 78, 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Dong, X.; Wang, H.; Li, J.; Yang, B.; Zhang, J.; Hua, Z.C. FADD regulates thymocyte development at the beta-selection checkpoint by modulating Notch signaling. Cell Death Dis. 2014, 5, e1273. [Google Scholar] [CrossRef]
- Pajerowski, A.G.; Nguyen, C.; Aghajanian, H.; Shapiro, M.J.; Shapiro, V.S. NKAP is a transcriptional repressor of notch signaling and is required for T cell development. Immunity 2009, 30, 696–707. [Google Scholar] [CrossRef]
- Song, T.; Liu, J.Y.; Yang, J.J. NKAP plays an oncogenic function partly through AKT signaling pathway in hepatocellular carcinoma. Neoplasma 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Gao, T.; Zhang, L.; Chen, X.; Pang, Q.; Wang, Y.; Wang, D.; Li, J.; Liu, Q. NKAP alters tumor immune microenvironment and promotes glioma growth via Notch1 signaling. J. Exp. Clin. Cancer Res. 2019, 38, 291. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, H.; Yin, Y.; Li, Q.; Zhang, M. NKAP functions as an oncogene and its expression is induced by CoCl2 treatment in breast cancer via AKT/mTOR signaling pathway. Cancer Manag. Res. 2018, 10, 5091–5100. [Google Scholar] [CrossRef] [PubMed]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Yang, B.Y.; Wang, J.Y.; Mo, X.; Zhang, J.; Hua, Z.C. FADD is essential for glucose uptake and survival of thymocytes. Biochem. Biophys. Res. Commun. 2014, 451, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Gan, Z.; Jiang, W.; Zhang, X.; Hua, Z.C. Comparative proteomics analysis reveals roles for FADD in the regulation of energy metabolism and proteolysis pathway in mouse embryonic fibroblast. Proteomics 2013, 13, 2398–2413. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Gan, Z.; Jiang, W.; Zhang, X.; Hua, Z.C. Functional specific roles of FADD: Comparative proteomic analyses from knockout cell lines. Mol. Biosyst. 2013, 9, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Wang, X.; Zha, D.; Gan, Z.; Cai, F.; Du, P.; Yang, Y.; Yang, B.; Zhang, X.; Yao, C.; et al. FADD is a key regulator of lipid metabolism. EMBO Mol. Med. 2016, 8, 895–918. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Muller, S.; Chen, Z.G.; Pan, L.; Tighiouart, M.; Shin, D.M.; Khuri, F.R.; Sun, S.Y. Prognostic impact of Fas-associated death domain, a key component in death receptor signaling, is dependent on the presence of lymph node metastasis in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2013, 14, 365–369. [Google Scholar] [CrossRef]
- Rasamny, J.J.; Allak, A.; Krook, K.A.; Jo, V.Y.; Policarpio-Nicolas, M.L.; Sumner, H.M.; Moskaluk, C.A.; Frierson, H.F., Jr.; Jameson, M.J. Cyclin D1 and FADD as biomarkers in head and neck squamous cell carcinoma. Otolaryngol. Head Neck Surg. 2012, 146, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Y.; Hammache, K.; He, L.; Zhu, B.; Cheng, W.; Hua, Z.C. The role of FADD in pancreatic cancer cell proliferation and drug resistance. Oncol. Lett. 2017, 13, 1899–1904. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Miguel, A.; Garcia-Sevilla, J.A.; Barr, A.M.; Bayer, T.A.; Falkai, P.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A.; Honer, W.G.; Garcia-Fuster, M.J. Decreased cortical FADD protein is associated with clinical dementia and cognitive decline in an elderly community sample. Mol. Neurodegener. 2017, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Tanaka, N.; Shimada, K.; Matsumura, Y.; Miyake, M.; Anai, S.; Tomioka, A.; Okajima, E.; Hirayama, A.; Fujimoto, K.; et al. Phosphorylation status of Fas-associated death domain protein is associated with biochemical recurrence after radical prostatectomy. Urology 2013, 81, 607–610. [Google Scholar] [CrossRef]
- Matsuyoshi, S.; Shimada, K.; Nakamura, M.; Ishida, E.; Konishi, N. FADD phosphorylation is critical for cell cycle regulation in breast cancer cells. Br. J. Cancer 2006, 94, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Nakamura, M.; Ishida, E.; Konishi, N. Molecular roles of MAP kinases and FADD phosphorylation in prostate cancer. Histol. Histopathol. 2006, 21, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Wachters, J.E.; Schrijvers, M.L.; Slagter-Menkema, L.; Mastik, M.; Langendijk, J.A.; de Bock, G.H.; Roodenburg, J.L.; van der Laan, B.; van der Wal, J.E.; Schuuring, E. Phosphorylated FADD is not prognostic for local control in T1-T2 supraglottic laryngeal carcinoma treated with radiotherapy. Laryngoscope 2017, 127, E301–E307. [Google Scholar] [CrossRef]
- Yin, A.; Jiang, Y.; Zhang, X.; Luo, H. Overexpression of FADD enhances 5-fluorouracil-induced apoptosis in colorectal adenocarcinoma cells. Med. Oncol. 2010, 27, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.A.; Ng, W.H.; Lam, P.Y. FasL and FADD delivery by a glioma-specific and cell cycle-dependent HSV-1 amplicon virus enhanced apoptosis in primary human brain tumors. Mol. Cancer 2010, 9, 270. [Google Scholar] [CrossRef]
- Komata, T.; Koga, S.; Hirohata, S.; Takakura, M.; Germano, I.M.; Inoue, M.; Kyo, S.; Kondo, S.; Kondo, Y. A novel treatment of human malignant gliomas in vitro and in vivo: FADD gene transfer under the control of the human telomerase reverse transcriptase gene promoter. Int. J. Oncol. 2001, 19, 1015–1020. [Google Scholar] [CrossRef] [Green Version]
- Mishima, K.; Nariai, Y.; Yoshimura, Y. Carboplatin induces Fas (APO-1/CD95)-dependent apoptosis of human tongue carcinoma cells: Sensitization for apoptosis by upregulation of FADD expression. Int. J. Cancer 2003, 105, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Y.; Cheng, C.L.; Ho, H.C.; Wang, S.S.; Chiu, K.Y.; Su, C.K.; Ou, Y.C.; Lin, C.C. Nortriptyline induces mitochondria and death receptor-mediated apoptosis in bladder cancer cells and inhibits bladder tumor growth in vivo. Eur. J. Pharmacol. 2015, 761, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Karaca, B.; Atmaca, H.; Bozkurt, E.; Kisim, A.; Uzunoglu, S.; Karabulut, B.; Sezgin, C.; Sanli, U.A.; Uslu, R. Combination of AT-101/cisplatin overcomes chemoresistance by inducing apoptosis and modulating epigenetics in human ovarian cancer cells. Mol. Biol. Rep. 2013, 40, 3925–3933. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Harper, N.; Walewska, R.; Dyer, M.J.; Cohen, G.M. Enhanced Fas-associated death domain recruitment by histone deacetylase inhibitors is critical for the sensitization of chronic lymphocytic leukemia cells to TRAIL-induced apoptosis. Mol. Cancer Ther. 2009, 8, 3088–3097. [Google Scholar] [CrossRef]
- Shimada, K.; Matsuyoshi, S.; Nakamura, M.; Ishida, E.; Kishi, M.; Konishi, N. Phosphorylation of FADD is critical for sensitivity to anticancer drug-induced apoptosis. Carcinogenesis 2004, 25, 1089–1097. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Condition(s) | Clinical Significance (Last Reviewed) | GRCh37 Location | GRCh38 Location | Variation ID | Allele ID(s) |
|---|---|---|---|---|---|---|
| NM_003824.3(FADD): c.31G > A (p.Val11Met) | Infections, recurrent, with encephalopathy, hepatic dysfunction, and cardiovascular malformations | Uncertain significance (26 February 2018) | 11: 70049596 | 11: 70203490 | 579410 | 566162 |
| NM_003824.3(FADD): c.93G > T (p.Val31=) | Benign (30 October 2017) | 11: 70049658 | 11: 70203552 | 471687 | 461658 | |
| NM_003824.3(FADD): c.168G > T (p.Glu56Asp) | Uncertain significance (22 September 2017) | 11: 70049733 | 11: 70203627 | 539062 | 526519 | |
| NM_003824.3(FADD): c.287-8C > G | Likely benign (21 July 2017) | 11: 70052231 | 11: 70206125 | 471685 | 462293 | |
| NM_003824.3(FADD): c.315T > G (p.Cys105Trp) | Pathogenic (10 December 2010) | 11: 70052267 | 11: 70206161 | 30267 | 39223 | |
| NM_003824.3(FADD): c.452C > T (p.Thr151Ile) | Uncertain significance (26 June 2018) | 11: 70052404 | 11: 70206298 | 575179 | 564906 | |
| NM_003824.3(FADD): c.475G > A (p.Ala159Thr) | Uncertain significance (3 July 2017) | 11: 70052427 | 11: 70206321 | 471686 | 461663 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marín-Rubio, J.L.; Vela-Martín, L.; Fernández-Piqueras, J.; Villa-Morales, M. FADD in Cancer: Mechanisms of Altered Expression and Function, and Clinical Implications. Cancers 2019, 11, 1462. https://doi.org/10.3390/cancers11101462
Marín-Rubio JL, Vela-Martín L, Fernández-Piqueras J, Villa-Morales M. FADD in Cancer: Mechanisms of Altered Expression and Function, and Clinical Implications. Cancers. 2019; 11(10):1462. https://doi.org/10.3390/cancers11101462
Chicago/Turabian StyleMarín-Rubio, José L, Laura Vela-Martín, José Fernández-Piqueras, and María Villa-Morales. 2019. "FADD in Cancer: Mechanisms of Altered Expression and Function, and Clinical Implications" Cancers 11, no. 10: 1462. https://doi.org/10.3390/cancers11101462
APA StyleMarín-Rubio, J. L., Vela-Martín, L., Fernández-Piqueras, J., & Villa-Morales, M. (2019). FADD in Cancer: Mechanisms of Altered Expression and Function, and Clinical Implications. Cancers, 11(10), 1462. https://doi.org/10.3390/cancers11101462