Autophagy Function and Dysfunction: Potential Drugs as Anti-Cancer Therapy



Abstract
:1. Introduction
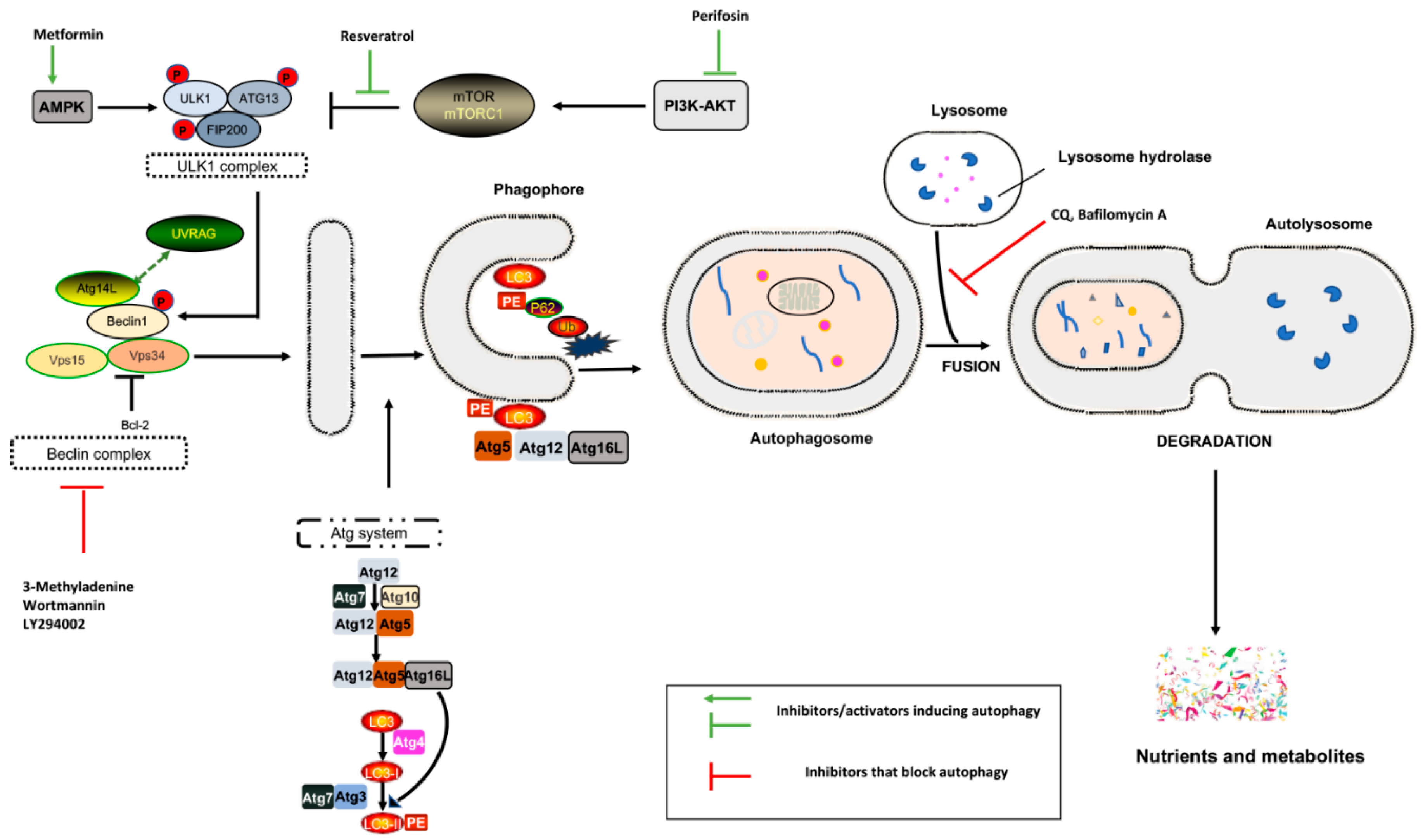
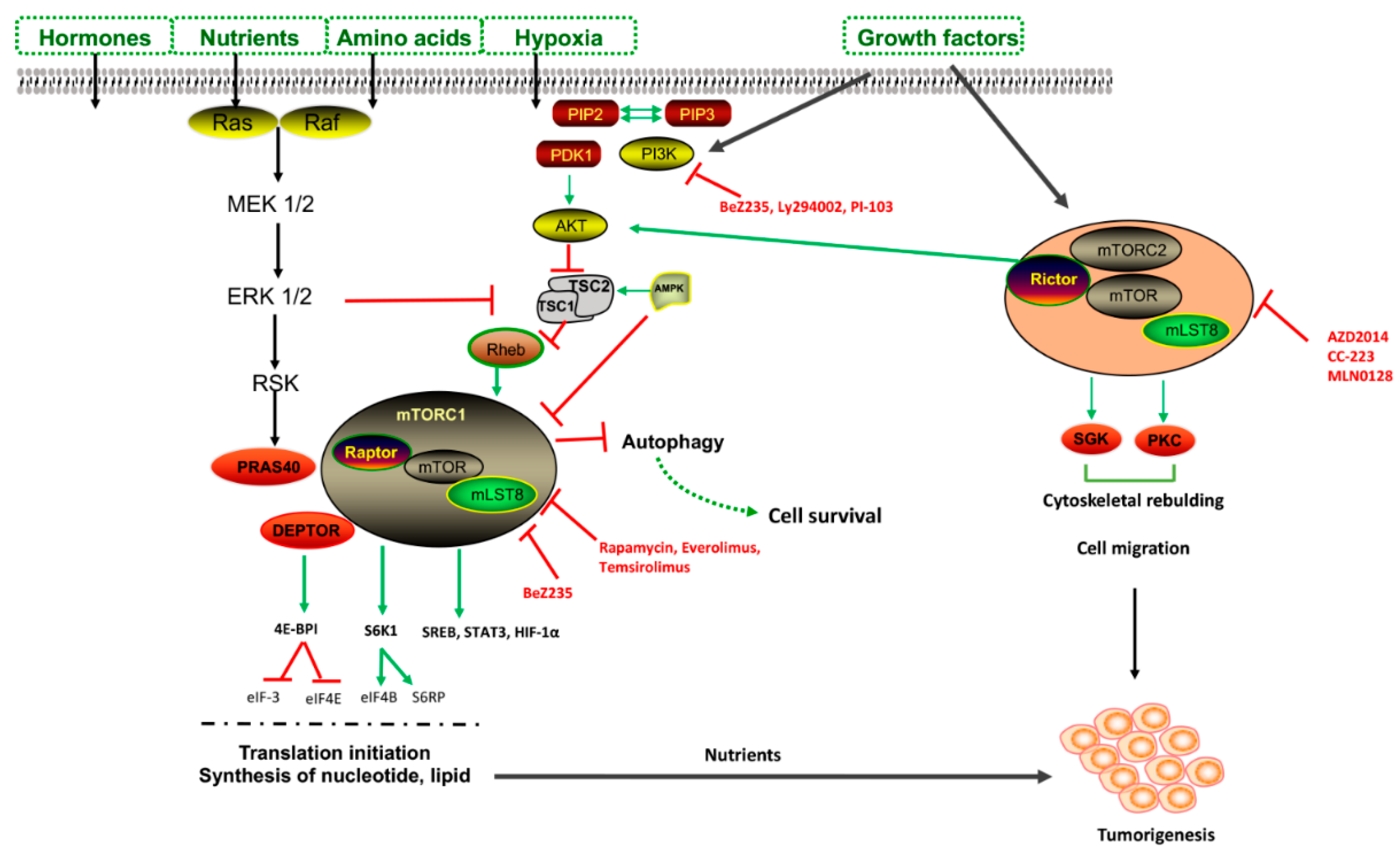
2. Autophagy and Regulatory Machinery
- -
- the formation of a cytoplasmic double-membrane structure called a phagophore
- -
- phagophore elongation, maturation, and closure, followed by the engulfment of long-lived cytosolic proteins and/or damaged organelles (mitochondria, ribosomes, etc.) with the formation of double-membrane vesicles, which are known as autophagosomes
- -
3. Role of Autophagy in Cancer
4. Autophagy and Apoptosis
5. Should We Try to Enhance or Inhibit Autophagy in Cancer?
5.1. Autophagy Inhibitors
5.2. Autophagy Activators
6. Natural Compounds as Inhibitors or Activators of Autophagy
7. mTOR Inhibitors in Clinical Trials
7.1. Breast Cancer
7.2. Ovarian Cancer
7.3. Prostate Cancer
7.4. Thyroid Cancer
7.5. Gastrointestinal Cancer
7.6. Lung Cancer
7.7. Renal Cell Carcinoma
7.8. Leukemia
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S. Autophagy in Cancer Therapy. Front. Oncol. 2017, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Willis, T.L.; Button, R.W.; Strang, C.J.; Fu, Y.; Wen, X.; Grayson, P.R.C.; Evans, T.; Sipthorpe, R.J.; Roberts, S.L.; et al. Cytoplasmic DAXX drives SQSTM1/p62 phase condensation to activate Nrf2-mediated stress response. Nat. Commun. 2019, 10, 3759. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inpanathan, S.; Botelho, R.J. The Lysosome Signaling Platform: Adapting With the Times. Front. Cell Dev. Biol. 2019, 7, 113. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Nnah, I.C.; Wang, B.; Saqcena, C.; Weber, G.F.; Bonder, E.M.; Bagley, D.; De Cegli, R.; Napolitano, G.; Medina, D.L.; Ballabio, A.; et al. TFEB-driven endocytosis coordinates MTORC1 signaling and autophagy. Autophagy 2019, 15, 151–164. [Google Scholar] [CrossRef]
- Yao, C.A.; Ortiz-Vega, S.; Sun, Y.Y.; Chien, C.T.; Chuang, J.H.; Lin, Y. Association of mSin1 with mTORC2 Ras and Akt reveals a crucial domain on mSin1 involved in Akt phosphorylation. Oncotarget 2017, 8, 63392–63404. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.T.; Liu, H.; Mao, M.J.; Tan, Y.; Mo, X.Q.; Meng, X.J.; Cao, M.T.; Zhong, C.Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef]
- Marchand, B.; Arsenault, D.; Raymond-Fleury, A.; Boisvert, F.M.; Boucher, M.J. Glycogen synthase kinase-3 (GSK3) inhibition induces prosurvival autophagic signals in human pancreatic cancer cells. J. Biol. Chem. 2015, 290, 5592–5605. [Google Scholar] [CrossRef]
- Kauffman, E.C.; Ricketts, C.J.; Rais-Bahrami, S.; Yang, Y.; Merino, M.J.; Bottaro, D.P.; Srinivasan, R.; Linehan, W.M. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat. Rev. Urol. 2014, 11, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Wang, Y.; Rao, Q.; Jiang, Y.; Zhang, W.; Li, Y. Xp11.2 translocation renal neoplasm with features of TFE3 rearrangement associated renal cell carcinoma and Xp11 translocation renal mesenchymal tumor with melanocytic differentiation harboring NONO-TFE3 fusion gene. Pathol. Res. Pract. 2019, 215, 152521. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.T.; Grzeskowiak, C.L.; Fradette, J.J.; Gibson, L.A.; Rodriguez, L.B.; Creighton, C.J.; Scott, K.L.; Gibbons, D.L. TMEM106B drives lung cancer metastasis by inducing TFEB-dependent lysosome synthesis and secretion of cathepsins. Nat. Commun. 2018, 9, 2731. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Komatsu, M. Activation of p62/SQSTM1-Keap1-Nuclear Factor Erythroid 2-Related Factor 2 Pathway in Cancer. Front. Oncol. 2018, 8, 210. [Google Scholar] [CrossRef]
- Huang, J.; Duran, A.; Reina-Campos, M.; Valencia, T.; Castilla, E.A.; Muller, T.D.; Tschop, M.H.; Moscat, J.; Diaz-Meco, M.T. Adipocyte p62/SQSTM1 Suppresses Tumorigenesis through Opposite Regulations of Metabolism in Adipose Tissue and Tumor. Cancer Cell 2018, 33, 770–784.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Jeong, E.G.; Soung, Y.H.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Decreased expression of tumour suppressor Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric carcinomas. Pathology 2006, 38, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Marino, G.; Salvador-Montoliu, N.; Fueyo, A.; Knecht, E.; Mizushima, N.; Lopez-Otin, C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J. Biol. Chem. 2007, 282, 18573–18583. [Google Scholar] [CrossRef]
- Pal, R.; Xiong, Y.; Sardiello, M. Abnormal glycogen storage in tuberous sclerosis complex caused by impairment of mTORC1-dependent and -independent signaling pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 2977–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temraz, S.; Mukherji, D.; Shamseddine, A. Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers. Int. J. Mol. Sci. 2015, 16, 22976–22988. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Liu, B.; Oltvai, Z.N.; Bayir, H.; Silverman, G.A.; Pak, S.C.; Perlmutter, D.H.; Bahar, I. Quantitative assessment of cell fate decision between autophagy and apoptosis. Sci. Rep. 2017, 7, 17605. [Google Scholar] [CrossRef]
- Tsapras, P.; Nezis, I.P. Caspase involvement in autophagy. Cell Death Differ. 2017, 24, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.H.; Price, M.; Reiners, J.J., Jr. ATG7 deficiency suppresses apoptosis and cell death induced by lysosomal photodamage. Autophagy 2012, 8, 1333–1341. [Google Scholar] [CrossRef]
- Humphreys, L.; Espona-Fiedler, M.; Longley, D.B. FLIP as a therapeutic target in cancer. FEBS J. 2018, 285, 4104–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordi, M.; Nazio, F.; Campello, S. The Close Interconnection between Mitochondrial Dynamics and Mitophagy in Cancer. Front. Oncol. 2017, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.D.; Liu, S.; Buchan, J.R.; Cho, W.C. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18, 367. [Google Scholar] [CrossRef]
- Luna-Vargas, M.P.; Chipuk, J.E. The deadly landscape of pro-apoptotic BCL-2 proteins in the outer mitochondrial membrane. FEBS J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Frohlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Okamoto, K.; Yu, C.; Sinicrope, F.A. p62/sequestosome-1 up-regulation promotes ABT-263-induced caspase-8 aggregation/activation on the autophagosome. J. Biol. Chem. 2013, 288, 33654–33666. [Google Scholar] [CrossRef]
- Allavena, G.; Cuomo, F.; Baumgartner, G.; Bele, T.; Sellgren, A.Y.; Oo, K.S.; Johnson, K.; Gogvadze, V.; Zhivotovsky, B.; Kaminskyy, V.O. Suppressed translation as a mechanism of initiation of CASP8 (caspase 8)-dependent apoptosis in autophagy-deficient NSCLC cells under nutrient limitation. Autophagy 2018, 14, 252–268. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Youle Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef]
- Al-Bari, M.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef]
- Wang, S.Q.; Zhang, L.W.; Wei, P.; Hua, H. Is hydroxychloroquine effective in treating primary Sjogren’s syndrome: A systematic review and meta-analysis. BMC Musculoskelet. Disord. 2017, 18, 186. [Google Scholar] [CrossRef]
- Alunno, A.; Leone, M.C.; Giacomelli, R.; Gerli, R.; Carubbi, F. Lymphoma and Lymphomagenesis in Primary Sjogren’s Syndrome. Front. Med. 2018, 5, 102. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharm. 2019, 119, 109415. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Hurez, V.; Nawrocki, S.T.; Goros, M.; Michalek, J.; Sarantopoulos, J.; Curiel, T.; Mahalingam, D. Vorinostat and hydroxychloroquine improve immunity and inhibit autophagy in metastatic colorectal cancer. Oncotarget 2016, 7, 59087–59097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef]
- Xu, R.; Ji, Z.; Xu, C.; Zhu, J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: A systematic review and meta-analysis. Medicine 2018, 97, e12912. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zaloudek, C.; Mills, G.B.; Gray, J.; Jaffe, R.B. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin. Cancer Res. 2000, 6, 880–886. [Google Scholar]
- Zhao, J.X.; Liu, H.; Lv, J.; Yang, X.J. Wortmannin enhances cisplatin-induced apoptosis in human ovarian cancer cells in vitro. Eur. Rev. Med. Pharm. Sci. 2014, 18, 2428–2434. [Google Scholar]
- Yuan, N.; Song, L.; Zhang, S.; Lin, W.; Cao, Y.; Xu, F.; Fang, Y.; Wang, Z.; Zhang, H.; Li, X.; et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef]
- Saha, A.; Blando, J.; Tremmel, L.; DiGiovanni, J. Effect of Metformin, Rapamycin and Their Combination on Growth and Progression of Prostate Tumors in HiMyc Mice. Cancer Prev. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gerthofer, V.; Kreutz, M.; Renner, K.; Jachnik, B.; Dettmer, K.; Oefner, P.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Combined Modulation of Tumor Metabolism by Metformin and Diclofenac in Glioma. Int. J. Mol. Sci. 2018, 19, 2586. [Google Scholar] [CrossRef] [PubMed]
- Dallaglio, K.; Bruno, A.; Cantelmo, A.R.; Esposito, A.I.; Ruggiero, L.; Orecchioni, S.; Calleri, A.; Bertolini, F.; Pfeffer, U.; Noonan, D.M.; et al. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis 2014, 35, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Eng, C.; Kolesar, J.; Hideshima, T.; Anderson, K.C. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: Mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Wolf, J.; Jakubowiak, A.; Zonder, J.; Lonial, S.; Irwin, D.; Densmore, J.; Krishnan, A.; Raje, N.; Bar, M.; et al. Perifosine plus bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma previously treated with bortezomib: Results of a multicenter phase I/II trial. J. Clin. Oncol. 2011, 29, 4243–4249. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, A.; Choudhury, D.; Datta, S.; Bhattacharya, S.; Chakrabarti, G. Inhibition of autophagy by chloroquine potentiates synergistically anti-cancer property of artemisinin by promoting ROS dependent apoptosis. Biochimie 2014, 107 Pt B, 338–349. [Google Scholar] [CrossRef]
- Hu, W.; Chen, S.S.; Zhang, J.L.; Lou, X.E.; Zhou, H.J. Dihydroartemisinin induces autophagy by suppressing NF-kappaB activation. Cancer Lett. 2014, 343, 239–248. [Google Scholar] [CrossRef]
- Feng, X.; Li, L.; Jiang, H.; Jiang, K.; Jin, Y.; Zheng, J. Dihydroartemisinin potentiates the anticancer effect of cisplatin via mTOR inhibition in cisplatin-resistant ovarian cancer cells: Involvement of apoptosis and autophagy. Biochem. Biophys. Res. Commun. 2014, 444, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.; Kumar, A.P.; Sethi, G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015, 20, 2728–2769. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Sethi, G.; Aggarwal, B.B. Curcumin potentiates the apoptotic effects of chemotherapeutic agents and cytokines through down-regulation of nuclear factor-kappaB and nuclear factor-kappaB-regulated gene products in IFN-alpha-sensitive and IFN-alpha-resistant human bladder cancer cells. Mol. Cancer Ther. 2007, 6, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Kantara, C.; O’Connell, M.; Sarkar, S.; Moya, S.; Ullrich, R.; Singh, P. Curcumin promotes autophagic survival of a subset of colon cancer stem cells, which are ablated by DCLK1-siRNA. Cancer Res. 2014, 74, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Masuelli, L.; Benvenuto, M.; Di Stefano, E.; Mattera, R.; Fantini, M.; De Feudis, G.; De Smaele, E.; Tresoldi, I.; Giganti, M.G.; Modesti, A.; et al. Curcumin blocks autophagy and activates apoptosis of malignant mesothelioma cell lines and increases the survival of mice intraperitoneally transplanted with a malignant mesothelioma cell line. Oncotarget 2017, 8, 34405–34422. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.C.; Mock, C.D.; Thilagavathi, R.; Selvam, C. Molecular mechanisms of curcumin and its semisynthetic analogues in prostate cancer prevention and treatment. Life Sci. 2016, 152, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, J.; Xu, J.; Lu, Y.; Jiang, J.; Wang, L.; Shen, H.M.; Xia, D. Curcumin targets the TFEB-lysosome pathway for induction of autophagy. Oncotarget 2016, 7, 75659–75671. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.H.; Sethi, G.; Um, J.Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, H.; Peng, X.; Bai, Q.; Yi, L.; Zhou, Y.; Zhu, J.; Mi, M. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/beta-catenin signaling pathway. PLoS ONE 2014, 9, e102535. [Google Scholar]
- Li, J.; Qin, Z.; Liang, Z. The prosurvival role of autophagy in Resveratrol-induced cytotoxicity in human U251 glioma cells. BMC Cancer 2009, 9, 215. [Google Scholar] [CrossRef]
- Scarlatti, F.; Maffei, R.; Beau, I.; Codogno, P.; Ghidoni, R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008, 15, 1318–1329. [Google Scholar] [CrossRef]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.; McGarrigle, S.; Pidgeon, G.P. The next generation of PI3K-Akt-mTOR pathway inhibitors in breast cancer cohorts. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Andre, F.; Jiang, Z.; Shao, Z.; Mano, M.S.; Neciosup, S.P.; Tseng, L.M.; Zhang, Q.; Shen, K.; Liu, D.; et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): A phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015, 16, 816–829. [Google Scholar] [CrossRef]
- Piccart, M.; Hortobagyi, G.N.; Campone, M.; Pritchard, K.I.; Lebrun, F.; Ito, Y.; Noguchi, S.; Perez, A.; Rugo, H.S.; Deleu, I.; et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Overall survival results from BOLERO-2dagger. Ann. Oncol. 2014, 25, 2357–2362. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Thaddeus Beck, J.; Royce, M. Everolimus-based combination therapies for HR+, HER2- metastatic breast cancer. Cancer Treat. Rev. 2018, 69, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Curwen, J.; Bihani, T.; D’Cruz, C.M.; Yates, J.W.; Grondine, M.; Howard, Z.; Davies, B.R.; Bigley, G.; Klinowska, T.; et al. AZD2014, an Inhibitor of mTORC1 and mTORC2, Is Highly Effective in ER+ Breast Cancer When Administered Using Intermittent or Continuous Schedules. Mol. Cancer Ther. 2015, 14, 2508–2518. [Google Scholar] [CrossRef]
- Petrossian, K.; Nguyen, D.; Lo, C.; Kanaya, N.; Somlo, G.; Cui, Y.X.; Huang, C.S.; Chen, S. Use of dual mTOR inhibitor MLN0128 against everolimus-resistant breast cancer. Breast Cancer Res. Treat. 2018, 170, 499–506. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000, 407, 538–541. [Google Scholar] [CrossRef]
- Colon-Otero, G.; Weroha, S.J.; Foster, N.R.; Haluska, P.; Hou, X.; Wahner-Hendrickson, A.E.; Jatoi, A.; Block, M.S.; Dinh, T.A.; Robertson, M.W.; et al. Phase 2 trial of everolimus and letrozole in relapsed estrogen receptor-positive high-grade ovarian cancers. Gynecol. Oncol. 2017, 146, 64–68. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef]
- Gao, N.; Flynn, D.C.; Zhang, Z.; Zhong, X.S.; Walker, V.; Liu, K.J.; Shi, X.; Jiang, B.H. G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am. J. Physiol. Cell Physiol. 2004, 287, C281–C291. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.; Ramirez, J.L.; Pedroza-Torres, A.; Herrera, L.A.; Jimenez-Rios, M.A. The Secret Life of Translation Initiation in Prostate Cancer. Front. Genet. 2019, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Graham, L.; Banda, K.; Torres, A.; Carver, B.S.; Chen, Y.; Pisano, K.; Shelkey, G.; Curley, T.; Scher, H.I.; Lotan, T.L.; et al. A phase II study of the dual mTOR inhibitor MLN0128 in patients with metastatic castration resistant prostate cancer. Investig. New Drugs 2018, 36, 458–467. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Cabanillas, M.E. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- Vaisman, F.; Shaha, A.; Fish, S.; Michael Tuttle, R. Initial therapy with either thyroid lobectomy or total thyroidectomy without radioactive iodine remnant ablation is associated with very low rates of structural disease recurrence in properly selected patients with differentiated thyroid cancer. Clin. Endocrinol. 2011, 75, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Lorch, J.H. A phase II study of everolimus in patients with aggressive RAI refractory (RAIR) thyroid cancer (TC). J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Lim, S.M.; Chang, H.; Yoon, M.J.; Hong, Y.K.; Kim, H.; Chung, W.Y.; Park, C.S.; Nam, K.H.; Kang, S.W.; Kim, M.K.; et al. A multicenter, phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann. Oncol. 2013, 24, 3089–3094. [Google Scholar] [CrossRef] [PubMed]
- Hartgrink, H.H.; Jansen, E.P.; van Grieken, N.C.; van de Velde, C.J. Gastric cancer. Lancet 2009, 374, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Fuereder, T.; Jaeger-Lansky, A.; Hoeflmayer, D.; Preusser, M.; Strommer, S.; Cejka, D.; Koehrer, S.; Crevenna, R.; Wacheck, V. mTOR inhibition by everolimus counteracts VEGF induction by sunitinib and improves anti-tumor activity against gastric cancer in vivo. Cancer Lett. 2010, 296, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, A.; Ajani, J.A.; Bai, Y.X.; Bang, Y.J.; Chung, H.C.; Pan, H.M.; Sahmoud, T.; Shen, L.; Yeh, K.H.; Chin, K.; et al. Everolimus for previously treated advanced gastric cancer: Results of the randomized, double-blind, phase III GRANITE-1 study. J. Clin. Oncol. 2013, 31, 3935–3943. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef]
- Umemura, S.; Mimaki, S.; Makinoshima, H.; Tada, S.; Ishii, G.; Ohmatsu, H.; Niho, S.; Yoh, K.; Matsumoto, S.; Takahashi, A.; et al. Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. J. Thorac. Oncol. 2014, 9, 1324–1331. [Google Scholar] [CrossRef]
- Reungwetwattana, T.; Molina, J.R.; Mandrekar, S.J.; Allen-Ziegler, K.; Rowland, K.M.; Reuter, N.F.; Luyun, R.F.; Dy, G.K.; Marks, R.S.; Schild, S.E.; et al. Brief report: A phase II “window-of-opportunity” frontline study of the MTOR inhibitor, temsirolimus given as a single agent in patients with advanced NSCLC, an NCCTG study. J. Thorac. Oncol. 2012, 7, 919–922. [Google Scholar] [CrossRef]
- Besse, B.; Leighl, N.; Bennouna, J.; Papadimitrakopoulou, V.A.; Blais, N.; Traynor, A.M.; Soria, J.C.; Gogov, S.; Miller, N.; Jehl, V.; et al. Phase II study of everolimus-erlotinib in previously treated patients with advanced non-small-cell lung cancer. Ann. Oncol. 2014, 25, 409–415. [Google Scholar] [CrossRef]
- Mellema, W.W.; Dingemans, A.M.; Thunnissen, E.; Snijders, P.J.; Derks, J.; Heideman, D.A.; Van Suylen, R.; Smit, E.F. KRAS mutations in advanced nonsquamous non-small-cell lung cancer patients treated with first-line platinum-based chemotherapy have no predictive value. J. Thorac. Oncol. 2013, 8, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Shepherd, F.A.; Douillard, J.Y.; Wolf, J.; Giaccone, G.; Crino, L.; Cappuzzo, F.; Sharma, S.; Gross, S.H.; Dimitrijevic, S.; et al. Efficacy of everolimus (RAD001) in patients with advanced NSCLC previously treated with chemotherapy alone or with chemotherapy and EGFR inhibitors. Ann. Oncol. 2009, 20, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Price, K.A.; Azzoli, C.G.; Krug, L.M.; Pietanza, M.C.; Rizvi, N.A.; Pao, W.; Kris, M.G.; Riely, G.J.; Heelan, R.T.; Arcila, M.E.; et al. Phase II trial of gefitinib and everolimus in advanced non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 1623–1629. [Google Scholar] [CrossRef]
- Deutsch, E.; Le Pechoux, C.; Faivre, L.; Rivera, S.; Tao, Y.; Pignon, J.P.; Angokai, M.; Bahleda, R.; Deandreis, D.; Angevin, E.; et al. Phase I trial of everolimus in combination with thoracic radiotherapy in non-small-cell lung cancer. Ann. Oncol. 2015, 26, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.J.; Jac, J.; Giessinger, S.; Saxena, S.; Willis, J.P. A phase 2 study with a daily regimen of the oral mTOR inhibitor RAD001 (everolimus) in patients with metastatic clear cell renal cell cancer. Cancer 2009, 115, 2438–2446. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.J.; Choueiri, T.K.; Fay, A.P.; Rini, B.I.; Thorner, A.R.; de Velasco, G.; Tyburczy, M.E.; Hamieh, L.; Albiges, L.; Agarwal, N.; et al. Mutations in TSC1, TSC2, and MTOR Are Associated with Response to Rapalogs in Patients with Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2016, 22, 2445–2452. [Google Scholar] [CrossRef]
- Gonzalez-Larriba, J.L.; Maroto, P.; Duran, I.; Lambea, J.; Flores, L.; Castellano, D.; The Changing, G. The role of mTOR inhibition as second-line therapy in metastatic renal carcinoma: Clinical evidence and current challenges. Expert Rev. Anticancer Ther. 2017, 17, 217–226. [Google Scholar] [CrossRef]
- Mendes, R.D.; Cante-Barrett, K.; Pieters, R.; Meijerink, J.P. The relevance of PTEN-AKT in relation to NOTCH1-directed treatment strategies in T-cell acute lymphoblastic leukemia. Haematologica 2016, 101, 1010–1017. [Google Scholar] [CrossRef] [Green Version]
- Simioni, C.; Martelli, A.M.; Zauli, G.; Melloni, E.; Neri, L.M. Targeting mTOR in Acute Lymphoblastic Leukemia. Cells 2019, 8, 190. [Google Scholar] [CrossRef]
- Simioni, C.; Martelli, A.M.; Zauli, G.; Vitale, M.; McCubrey, J.A.; Capitani, S.; Neri, L.M. Targeting the phosphatidylinositol 3-kinase/Akt/mechanistic target of rapamycin signaling pathway in B-lineage acute lymphoblastic leukemia: An update. J. Cell Physiol. 2018, 233, 6440–6454. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Chiarini, F.; McCubrey, J.A.; Martelli, A.M. Therapeutic Targeting of mTOR in T-Cell Acute Lymphoblastic Leukemia: An Update. Int. J. Mol. Sci. 2018, 19, 1878. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Alexe, G.; Roti, G.; Conway, A.S.; Furman, A.; Lee, E.S.; Place, A.E.; Kim, S.; Saran, C.; Modiste, R.; et al. Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hua, C.; Cheng, H.; Wang, W.; Hao, S.; Xu, J.; Wang, X.; Gao, Y.; Zhu, X.; Cheng, T.; et al. Distinct sensitivity of CD8+ CD4− and CD8+ CD4+ leukemic cell subpopulations to cyclophosphamide and rapamycin in Notch1-induced T-ALL mouse model. Leuk. Res. 2013, 37, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Man, L.M.; Morris, A.L.; Keng, M. New Therapeutic Strategies in Acute Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2017, 12, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Gazi, M.; Moharram, S.A.; Marhall, A.; Kazi, J.U. The dual specificity PI3K/mTOR inhibitor PKI-587 displays efficacy against T-cell acute lymphoblastic leukemia (T-ALL). Cancer Lett. 2017, 392, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.P.; Reynolds, C.P.; Kang, M.H. Modulation of Glucocorticoid Resistance in Pediatric T-cell Acute Lymphoblastic Leukemia by Increasing BIM Expression with the PI3K/mTOR Inhibitor BEZ235. Clin. Cancer Res. 2016, 22, 621–632. [Google Scholar] [CrossRef]
- Daver, N.; Boumber, Y.; Kantarjian, H.; Ravandi, F.; Cortes, J.; Rytting, M.E.; Kawedia, J.D.; Basnett, J.; Culotta, K.S.; Zeng, Z.; et al. A Phase I/II Study of the mTOR Inhibitor Everolimus in Combination with HyperCVAD Chemotherapy in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2015, 21, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Rheingold, S.R.; Tasian, S.K.; Whitlock, J.A.; Teachey, D.T.; Borowitz, M.J.; Liu, X.; Minard, C.G.; Fox, E.; Weigel, B.J.; Blaney, S.M. A phase 1 trial of temsirolimus and intensive re-induction chemotherapy for 2nd or greater relapse of acute lymphoblastic leukaemia: A Children’s Oncology Group study (ADVL1114). Br. J. Haematol. 2017, 177, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Wang, B.R.; Wang, Y.G. Role of autophagy in tumorigenesis, metastasis, targeted therapy and drug resistance of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 4643–4651. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name | Mode of Action | Structure |
|---|---|---|
| Choloroquine | Endosomal acidification inhibitor |  |
| 3-Methyladenine | PI3K inhibitor |  |
| Bafilomycin A1 | Endosomal acidification inhibitor |  |
| LY294002 | PI3K inhibitor |  |
| SB202190 | MAPK inhibitor |  |
| SB203580 | MAPK inhibitor |  |
| Disease | Trial | Autophagy Inhibitor | No. Patients | Adverse Effects | Dose | Ref. |
|---|---|---|---|---|---|---|
| Colorectal cancer | Phase I, vorinostat + HCQ | HCQ | 19 | Fatigue and gastrointestinal disturbances | VOR 600 mg/daily, HCQ 400 mg/daily | [56] |
| Melanoma | Phase I, HCQ + TMZ | HCQ | 40 | Fatigue, anorexia, nausea, constipation, and diarrhea | HCQ 200-1200 mg/ daily + TMZ 150 mg/m2 | [57] |
| Refractory multiple myeloma | Phase I, HCQ + BOR | HCQ | 25 | None | HCQ 600 mg/daily + standard dose of BOR | [58] |
| Glioblastoma | Phase III, CQ + TMZ + radiation | CQ | 30 | None | 150 mg/daily | [59] |
| Non-small cell lung cancer | Phase II, CQ + whole-brain radiation | CQ | 73 | None | 150 mg/daily | [59] |
| Glioma | Randomized, double-blind phase II, carmustine, radiation, and chloroquine | HCQ | 30 | None | HCQ 150 mg/daily | [59] |
| Breast Cancer | Phase II, everolimus + HCQ | HCQ | 60 | None | HCQ 150 mg/daily | [59] |
| Name | Mode of Action | Structure |
|---|---|---|
| Rapamycin | mTOR inhibition–TLR signaling |  |
| Everolimus | mTOR inhibition |  |
| Metformin | AMPK activation |  |
| Perifosine | AKT inhibition |  |
| Resveratrol | NF-kB inhibition |  |
| Disease | Autophagy Effect | Drug(s) | No. Patients | Refs |
|---|---|---|---|---|
| Breast cancer (HR+) | mTORC1 inhibition mTORC1 and mTORC2 inhibition | Everolimus + aromatase inhibitor exemestane. MLN0128 AZD2014 CC-223 | 724 Experiments in cell lines and xenograft models | [94] [96,97] |
| Ovarian cancer | mTORC1 inhibition PI3K/mTOR inhibition | Everolimus + aromatase inhibitor letrozole BEZ235 + cisplatin | 20 Ovarian cancer stem cells | [100] [101] |
| Prostate cancer | mTORC1 inhibition | MLN0128 BEZ235 | 9 Prostate cancer cells | [106] [107] |
| Thyroid cancer Anaplastic thyroid cancer | mTORC1 inhibition | Everolimus Everolimus | 40 2 | [112] [113] |
| Gastrointestinal cancers | mTORC1 inhibition | Everolimus | 656 | [116] |
| Lung cancer (NSCLC) | mTORC1 inhibition | Everolimus Everolimus Chemoterapy + EGFr inhibition + everolimus Temsirolimus | 92 26 85 63 | [120] [120] [122] [57] |
| Renal cell carcinoma | mTORC1 inhibition | Everolimus (RAD001) Rapamycin + doxorubicin | 41 | [127] |
| Leukemia T-ALL | mTORC1 inhibition | Rapamycin + cyclophosphamide Rapamycin + methotrexate | 7 7 | [134] [135] |
| T-ALL/B-ALL | mTORC1 inhibition | RAD001 (everolimus) + LEE-01 + glucocorticoids | 15 | [134] |
| T-ALL | PI3K/mTOR inhibition | PKI-587 (Gedatolisib) BEZ235 + cytarabine (AraC) or doxorubicin or dexamethasone | [137] [138] | |
| B-ALL/T-ALL | mTORC1 inhibition | Everolimus (RAD001) + vincristine + doxorubicin + cyclophosphamide + dexamethasone | 22 | [139] |
| ALL + Philadelphia chromosome-positive ALL | mTORC1 inhibition | Rapamycin + chemotherapy +/− stem cell transplant in patients | 97 | [140] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuomo, F.; Altucci, L.; Cobellis, G. Autophagy Function and Dysfunction: Potential Drugs as Anti-Cancer Therapy. Cancers 2019, 11, 1465. https://doi.org/10.3390/cancers11101465
Cuomo F, Altucci L, Cobellis G. Autophagy Function and Dysfunction: Potential Drugs as Anti-Cancer Therapy. Cancers. 2019; 11(10):1465. https://doi.org/10.3390/cancers11101465
Chicago/Turabian StyleCuomo, Francesca, Lucia Altucci, and Gilda Cobellis. 2019. "Autophagy Function and Dysfunction: Potential Drugs as Anti-Cancer Therapy" Cancers 11, no. 10: 1465. https://doi.org/10.3390/cancers11101465
APA StyleCuomo, F., Altucci, L., & Cobellis, G. (2019). Autophagy Function and Dysfunction: Potential Drugs as Anti-Cancer Therapy. Cancers, 11(10), 1465. https://doi.org/10.3390/cancers11101465