Epigenetic Mechanisms of Escape from BRAF Oncogene Dependency


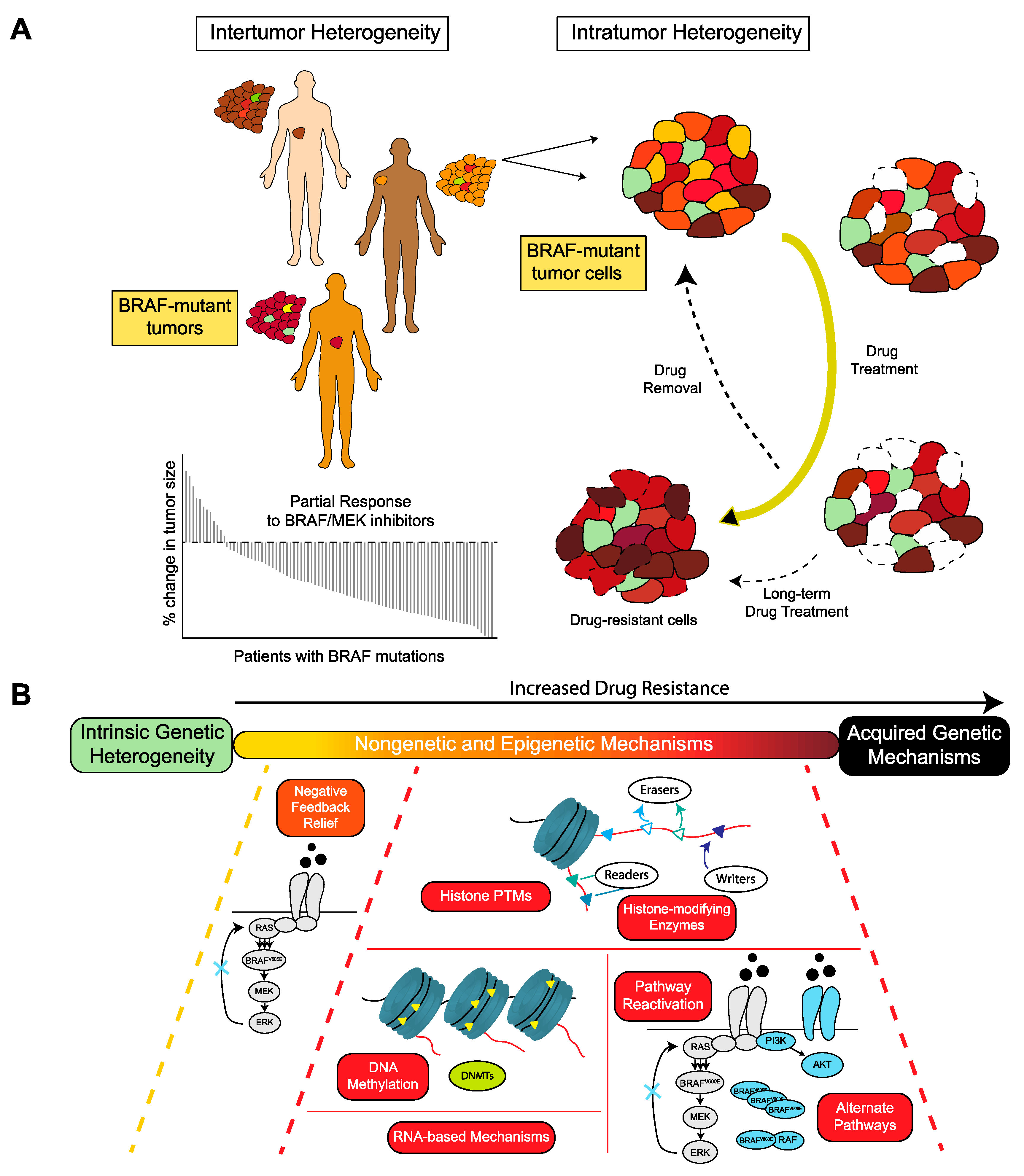{kind=link}
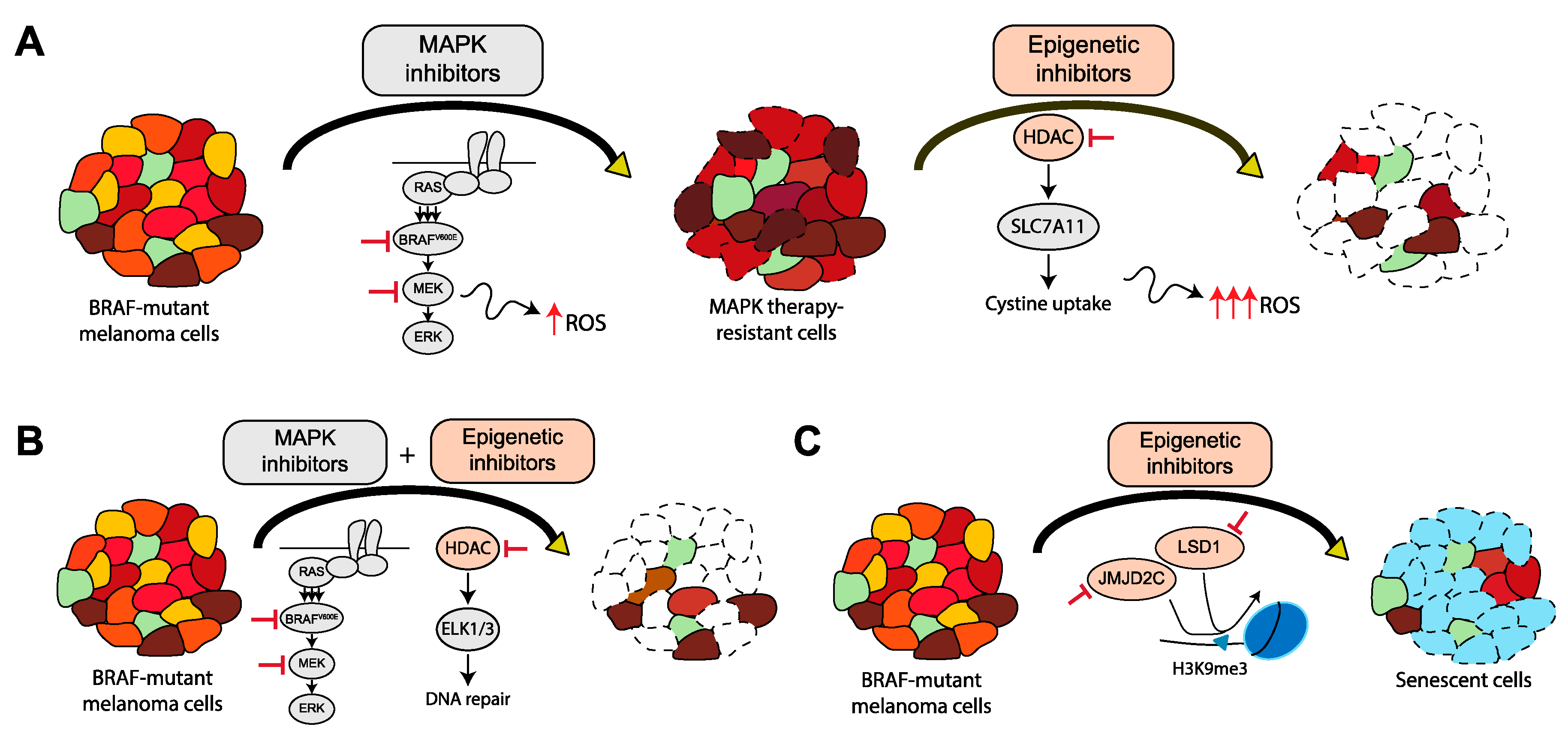{kind=link}
Abstract
:1. Introduction
2. BRAF Oncogene: Role in MAPK Pathway, Dysregulation in Cancer, and Potential as a Therapeutic Target
3. Current Strategies to Target BRAF-Mutated Cancers
4. Limitations of Current Therapies Targeting BRAF-Mutated Cancers
5. Epigenetic Mechanisms of MAPK Dependency in BRAF-Mutated Tumors
6. DNA Methylation in BRAF-Mutated Cancers
7. Histone-Modifying Enzymes and Post-Translational Modifications in BRAF-Mutated Cancers
8. Alternative Combinatorial Treatments to Overcome Therapy-Resistant BRAF-Mutated Cancers
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene Addiction—A Rationale for Molecular Targeting in Cancer Therapy. Nat. Clin. Pract. Oncol 2006, 3, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Doebele, R.C. A Framework for Understanding and Targeting Residual Disease in Oncogene-Driven Solid Cancers. Nat. Med. 2016, 22, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Hazar-Rethinam, M.; Kleyman, M.; Han, G.C.; Liu, D.; Ahronian, L.G.; Shahzade, H.A.; Chen, L.; Parikh, A.R.; Allen, J.N.; Clark, J.W.; et al. Convergent Therapeutic Strategies to Overcome the Heterogeneity of Acquired Resistance in BRAFV600E Colorectal Cancer. Cancer Discov. 2018, 8, 417–427. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [CrossRef]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-Mutated Metastatic Melanoma: A Multicentre, Open-Label, Phase 3 Randomised Controlled Trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP Kinase Pathway Alterations in BRAF-Mutant Melanoma Patients with Acquired Resistance to Combined RAF/MEK Inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas Acquire Resistance to B-RAF(V600E) Inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF Inhibitor Resistance is Mediated by Dimerization of Aberrantly Spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Infante, J.R.; Krepler, C.; Reyes-Uribe, P.; Samanta, M.; Chen, H.-Y.; Li, B.; Swoboda, R.K.; Wilson, M.; Vultur, A.; et al. Concurrent MEK2 Mutation and BRAF Amplification Confer Resistance to BRAF and MEK Inhibitors in Melanoma. Cell Rep. 2013, 4, 1090–1099. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic Melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-Combinatorial Mechanisms of Acquired Resistance Limit the Efficacy of BRAF/MEK Cotargeting But Result in Melanoma Drug Addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK Reactivation in Early Resistance to Dabrafenib/Trametinib Combination Therapy of BRAF-Mutant Metastatic Melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hong, A.; Kong, X.; Koya, R.C.; Song, C.; Moriceau, G.; Hugo, W.; Yu, C.C.; Ng, C.; Chodon, T.; et al. A Novel AKT1 Mutant Amplifies An Adaptive Melanoma Response to BRAF Inhibition. Cancer Discov. 2014, 4, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-Mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Emmons, M.F.; Faião-Flores, F.; Smalley, K.S.M. The Role of Phenotypic Plasticity in the Escape of Cancer Cells from Targeted Therapy. Biochem. Pharmacol. 2016, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [Green Version]
- Perego, M.; Maurer, M.; Wang, J.X.; Shaffer, S.; Müller, A.C.; Parapatics, K.; Li, L.; Hristova, D.; Shin, S.; Keeney, F.; et al. A Slow-Cycling Subpopulation of Melanoma Cells with Highly Invasive Properties. Oncogene 2018, 37, 302–312. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells is Required for Continuous Tumor Growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1B(high) Cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef]
- Lin, L.; Asthana, S.; Chan, E.; Bandyopadhyay, S.; Martins, M.M.; Olivas, V.; Yan, J.J.; Pham, L.; Wang, M.M.; Bollag, G.; et al. Mapping the Molecular Determinants of BRAF Oncogene Dependence in Human Lung Cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E748–E757. [Google Scholar] [CrossRef]
- Ravindran Menon, D.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Bonyadi Rad, E.; et al. A Stress-Induced Early Innate Response Causes Multidrug Tolerance in Melanoma. Oncogene 2015, 34, 4448–4459. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-Diaz, A.; Homet Moreno, B.; Kim, J.; Ng, R.H.; Lee, J.W.; et al. Single-Cell Analysis Resolves the Cell State Transition and Signaling Dynamics Associated with Melanoma Drug-Induced Resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.-R.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive Resistance of Melanoma Cells to RAF Inhibition Via Reversible Induction of a Slowly Dividing De-Differentiated State. Mol. Syst. Biol. 2017, 13, 905. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and Adaptive Resistance to BRAF(V600E) Inhibition in Melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallahi-Sichani, M.; Moerke, N.J.; Niepel, M.; Zhang, T.; Gray, N.S.; Sorger, P.K. Systematic Analysis of BRAF(V600E) Melanomas Reveals a Role for JNK/c-Jun Pathway in Adaptive Resistance to Drug-Induced Apoptosis. Mol. Syst. Biol. 2015, 11, 797. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Fang, B.; Munko, A.C.; Gibney, G.T.; Koomen, J.M.; Smalley, K.S.M. Phosphoproteomic Analysis of Basal and Therapy-Induced Adaptive Signaling Networks in BRAF and NRAS Mutant Melanoma. Proteomics 2015, 15, 327–339. [Google Scholar] [CrossRef]
- Gopal, Y.N.V.; Deng, W.; Woodman, S.E.; Komurov, K.; Ram, P.; Smith, P.D.; Davies, M.A. Basal and Treatment-Induced Activation of AKT Mediates Resistance to Cell Death by AZD6244 (ARRY-142886) in Braf-Mutant Human Cutaneous Melanoma Cells. Cancer Res. 2010, 70, 8736–8747. [Google Scholar] [CrossRef]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-Induced Tumour Secretomes Promote Resistance and Tumour Progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital Imaging Reveals How BRAF Inhibition Generates Drug-Tolerant Microenvironments with High Integrin β1/FAK Signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A Melanocyte Lineage Program Confers Resistance to MAP Kinase Pathway Inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Geukes Foppen, M.H.; et al. Low MITF/AXL Ratio Predicts Early Resistance to Multiple Targeted Drugs In Melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A Melanoma Cell State Distinction Influences Sensitivity to MAPK Pathway Inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare Cell Variability and Drug-Induced Reprogramming As a Mode of Cancer Drug Resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Schleich, K.; Yue, B.; Ji, S.; Lohneis, P.; Kemper, K.; Silvis, M.R.; Qutob, N.; van Rooijen, E.; Werner-Klein, M.; et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell 2018, 33, 322–336.e8. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef]
- Fiziev, P.; Akdemir, K.C.; Miller, J.P.; Keung, E.Z.; Samant, N.S.; Sharma, S.; Natale, C.A.; Terranova, C.J.; Maitituoheti, M.; Amin, S.B.; et al. Systematic Epigenomic Analysis Reveals Chromatin States Associated with Melanoma Progression. Cell Rep. 2017, 19, 875–889. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor Cells can Follow Distinct Evolutionary Paths to Become Resistant to Epidermal Growth Factor Receptor Inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.-H.; Thurley, K.; et al. Diverse Drug-Resistance Mechanisms can Emerge from Drug-Tolerant Cancer Persister Cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP Kinase Signalling Cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP Kinase Signalling Pathways in Cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Weber, C.K.; Slupsky, J.R.; Kalmes, H.A.; Rapp, U.R. Active Ras Induces Heterodimerization of cRaf and BRaf. Cancer Res. 2001, 61, 3595–3598. [Google Scholar]
- Cancer Genome Atlas Network Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic Evidence for Constitutive Activation of the RET/PTC-RAS-BRAF Signaling Pathway in Papillary Thyroid Carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Singer, G.; Oldt, R.; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih, I.-M. Mutations in BRAF and KRAS Characterize the Development of Low-Grade Ovarian Serous Carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [CrossRef]
- Lavoie, H.; Therrien, M. Regulation of RAF Protein Kinases in ERK Signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Cardarella, S.; Ogino, A.; Nishino, M.; Butaney, M.; Shen, J.; Lydon, C.; Yeap, B.Y.; Sholl, L.M.; Johnson, B.E.; Jänne, P.A. Clinical, Pathologic, and Biologic Features Associated with BRAF Mutations in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 4532–4540. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Arcila, M.E.; Fara, M.; Sima, C.S.; Miller, V.A.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Clinical Characteristics of Patients with Lung Adenocarcinomas Harboring BRAF Mutations. J. Clin. Oncol. 2011, 29, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular Testing for BRAF Mutations to Inform Melanoma Treatment Decisions: A Move toward Precision Medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, W.I.; Mahoney, M.R.; Sargent, D.J.; Nelson, G.D.; Alberts, S.R.; Sinicrope, F.A.; Goldberg, R.M.; Limburg, P.J.; Thibodeau, S.N.; Grothey, A.; et al. Patient and Tumor Characteristics and BRAF and KRAS Mutations in Colon Cancer, NCCTG/Alliance N0147. J. Natl. Cancer Inst. 2014, 106, dju106. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Quiros, R.M.; Gattuso, P.; Ain, K.B.; Prinz, R.A. High Prevalence of BRAF Gene Mutation in Papillary Thyroid Carcinomas and Thyroid Tumor Cell Lines. Cancer Res. 2003, 63, 4561–4567. [Google Scholar]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Kaley, T.; Touat, M.; Subbiah, V.; Hollebecque, A.; Rodon, J.; Lockhart, A.C.; Keedy, V.; Bielle, F.; Hofheinz, R.-D.; Joly, F.; et al. BRAF Inhibition in BRAFV600-Mutant Gliomas: Results From the VE-BASKET Study. J. Clin. Oncol. 2018, 36, 3477–3484. [Google Scholar] [CrossRef]
- Röring, M.; Herr, R.; Fiala, G.J.; Heilmann, K.; Braun, S.; Eisenhardt, A.E.; Halbach, S.; Capper, D.; von Deimling, A.; Schamel, W.W.; et al. Distinct Requirement for An Intact Dimer Interface in Wild-Type, V600E and Kinase-Dead B-Raf Signalling. EMBO J. 2012, 31, 2629–2647. [Google Scholar] [CrossRef]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. Effects of Raf Dimerization and Its Inhibition on Normal and Disease-Associated Raf Signaling. Mol. Cell 2013, 49, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with Class 3 BRAF Mutants Are Sensitive to the Inhibition of Activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression Through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Tseng, L.-H.; Chen, G.; Haley, L.; Illei, P.; Gocke, C.D.; Eshleman, J.R.; Lin, M.-T. Clinical Detection and Categorization of Uncommon and Concomitant Mutations Involving BRAF. BMC Cancer 2015, 15, 779. [Google Scholar] [CrossRef] [PubMed]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New Perspectives for Targeting RAF Kinase in Human Cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Nieto, P.; Ambrogio, C.; Esteban-Burgos, L.; Gómez-López, G.; Blasco, M.T.; Yao, Z.; Marais, R.; Rosen, N.; Chiarle, R.; Pisano, D.G.; et al. A Braf Kinase-Inactive Mutant Induces Lung Adenocarcinoma. Nature 2017, 548, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Greulich, H.; Kaplan, B.; Araujo, L.; Amann, J.; Horn, L.; Schiller, J.; Villalona-Calero, M.A.; Meyerson, M.; Carbone, D.P. Oncogenic and Sorafenib-Sensitive ARAF Mutations in Lung Adenocarcinoma. J. Clin. Investig. 2014, 124, 1582–1586. [Google Scholar] [CrossRef]
- Emuss, V.; Garnett, M.; Mason, C.; Marais, R. Mutations of C-RAF are Rare in Human Cancer Because C-RAF Has a Low Basal Kinase Activity Compared with B-RAF. Cancer Res. 2005, 65, 9719–9726. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining Complete Cancer Genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef]
- Stanton, V.P.; Cooper, G.M. Activation of Human Raf Transforming Genes by Deletion of Normal Amino-Terminal Coding Sequences. Mol. Cell. Biol. 1987, 7, 1171–1179. [Google Scholar] [CrossRef]
- Ikawa, S.; Fukui, M.; Ueyama, Y.; Tamaoki, N.; Yamamoto, T.; Toyoshima, K. B-raf, a New Member of the Raf Family, is Activated by DNA Rearrangement. Mol. Cell. Biol. 1988, 8, 2651–2654. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Buchanan, S.; Yadav, V.; Mochalkin, I.; Wong, S.S.; Yue, Y.G.; Huber, L.; et al. Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discov. 2016, 6, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.F.; Wilhelm, S.; Hibner, B.; Bollag, G. Discovery of a Novel Raf Kinase Inhibitor. Endocr. Relat. Cancer 2001, 8, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hamilton, A.; Min, C.; Safarzadeh-Amiri, S.; Goldberg, L.; Yoon, J.; Yee, H.; Buckley, M.; Christos, P.J.; Wright, J.J.; et al. A Phase II Trial of Sorafenib in Metastatic Melanoma with Tissue Correlates. PLoS ONE 2010, 5, e15588. [Google Scholar] [CrossRef]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a Selective Inhibitor of Oncogenic B-Raf Kinase with Potent Antimelanoma Activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical Efficacy of a RAF Inhibitor Needs Broad Target Blockade in BRAF-Mutant Melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients with BRAF-Mutant Melanoma (COLUMBUS): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.M.; Shao, Y.; Mayberry, M.M.; Aplin, A.E. Hyperactivation of MEK-ERK1/2 Signaling and Resistance to Apoptosis Induced by the Oncogenic B-RAF Inhibitor, PLX4720, in Mutant N-RAS Melanoma Cells. Oncogene 2011, 30, 366–371. [Google Scholar] [CrossRef]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS Mutations in Cutaneous Squamous-Cell Carcinomas in Patients Treated with BRAF Inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef]
- Halaban, R.; Zhang, W.; Bacchiocchi, A.; Cheng, E.; Parisi, F.; Ariyan, S.; Krauthammer, M.; McCusker, J.P.; Kluger, Y.; Sznol, M. PLX4032, a Selective BRAF(V600E) Kinase Inhibitor, Activates the ERK Pathway and Enhances Cell Migration and Proliferation of BRAF Melanoma Cells. Pigment Cell Melanoma Res. 2010, 23, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF Inhibitors Prime Wild-Type RAF to Activate the MAPK Pathway and Enhance Growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF Inhibitors Transactivate RAF Dimers and ERK Signalling in Cells With Wild-Type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef]
- Karoulia, Z.; Wu, Y.; Ahmed, T.A.; Xin, Q.; Bollard, J.; Krepler, C.; Wu, X.; Zhang, C.; Bollag, G.; Herlyn, M.; et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 2016, 30, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF Inhibitors that Evade Paradoxical MAPK Pathway Activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef]
- Yao, Z.; Gao, Y.; Su, W.; Yaeger, R.; Tao, J.; Na, N.; Zhang, Y.; Zhang, C.; Rymar, A.; Tao, A.; et al. RAF Inhibitor PLX8394 Selectively Disrupts BRAF Dimers and RAS-Independent BRAF-Mutant-Driven Signaling. Nat. Med. 2019, 25, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Saborowski, A.; Yue, J.; Solomon, M.; Joseph, E.; Gadal, S.; Saborowski, M.; Kastenhuber, E.; Fellmann, C.; Ohara, K.; et al. Disruption of CRAF-Mediated MEK Activation is Required for Effective MEK Inhibition in KRAS Mutant Tumors. Cancer Cell 2014, 25, 697–710. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. GSK1120212 (JTP-74057) is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained in vivo Pathway Inhibition. Clin. Cancer Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition Versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and Trametinib Versus Dabrafenib and Placebo for Val600 BRAF-Mutant Melanoma: A Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Menzies, A.M.; Haydu, L.E.; Carlino, M.S.; Azer, M.W.F.; Carr, P.J.A.; Kefford, R.F.; Long, G.V. Inter- and Intra-Patient Heterogeneity of Response and Progression to Targeted Therapy in Metastatic Melanoma. PLoS ONE 2014, 9, e85004. [Google Scholar] [CrossRef] [PubMed]
- Ahronian, L.G.; Sennott, E.M.; Van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 Mutations Confer Resistance to MEK and B-RAF Inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.R.; Theurillat, J.-P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A Genome-Scale RNA Interference Screen Implicates NF1 Loss in Resistance to RAF Inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN Loss Confers BRAF Inhibitor Resistance to Melanoma Cells through the Suppression of BIM Expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired Resistance to BRAF Inhibitors Mediated by a RAF Kinase Switch in Melanoma can be Overcome by Cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF Receptor or SRC Family Kinase Signaling Overcomes BRAF Inhibitor Resistance in Melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT Drives Resistance to RAF Inhibition Through MAP Kinase Pathway Reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug. Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor Adaptation and Resistance to RAF Inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M.; Cichowski, K.; Elledge, S.J. Tissue-Specificity in Cancer: The Rule, Not the Exception. Science 2019, 363, 1150–1151. [Google Scholar] [CrossRef] [PubMed]
- Sack, L.M.; Davoli, T.; Li, M.Z.; Li, Y.; Xu, Q.; Naxerova, K.; Wooten, E.C.; Bernardi, R.J.; Martin, T.D.; Chen, T.; et al. Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell 2018, 173, 499–514.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Strub, T.; Ghiraldini, F.G.; Carcamo, S.; Li, M.; Wroblewska, A.; Singh, R.; Goldberg, M.S.; Hasson, D.; Wang, Z.; Gallagher, S.J.; et al. SIRT6 Haploinsufficiency Induces BRAFV600E Melanoma Cell Resistance to MAPK Inhibitors Via IGF Signalling. Nat. Commun. 2018, 9, 3440. [Google Scholar] [CrossRef] [PubMed]
- Hinohara, K.; Wu, H.-J.; Vigneau, S.; McDonald, T.O.; Igarashi, K.J.; Yamamoto, K.N.; Madsen, T.; Fassl, A.; Egri, S.B.; Papanastasiou, M.; et al. KDM5 Histone Demethylase Activity Links Cellular Transcriptomic Heterogeneity to Therapeutic Resistance. Cancer Cell 2018, 34, 939–953.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-Tolerant Persister Cancer Cells are Vulnerable to GPX4 Inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Bivona, T.G. Polytherapy and Targeted Cancer Drug Resistance. Trends Cancer 2019, 5, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Phenotype Plasticity As Enabler of Melanoma Progression and Therapy Resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Rowling, E.J.; Miskolczi, Z.; Ferguson, J.; Spoerri, L.; Haass, N.K.; Sloss, O.; McEntegart, S.; Arozarena, I.; von Kriegsheim, A.; et al. Targeting Endothelin Receptor Signalling Overcomes Heterogeneity Driven Therapy Failure. EMBO Mol. Med. 2017, 9, 1011–1029. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Fisher, D.E. The Master Role of Microphthalmia-Associated Transcription Factor in Melanocyte and Melanoma Biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Fisher, D.E. Melanocyte Biology and Skin Pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wölfel, T.; Hölzel, M.; et al. Melanomas Resist T-cell Therapy Through Inflammation-Induced Reversible Dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Mica, Y.; Lee, G.; Chambers, S.M.; Tomishima, M.J.; Studer, L. Modeling Neural Crest Induction, Melanocyte Specification, and Disease-Related Pigmentation Defects in hESCs and Patient-Specific iPSCs. Cell Rep. 2013, 3, 1140–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Fisher, D.E.; Flaherty, K.T. Cell-State Dynamics and Therapeutic Resistance in Melanoma from the Perspective of MITF and IFNγ Pathways. Nat. Rev. Clin. Oncol. 2019, 16, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.-Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425.e14. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, F.; Gundersen, G.; Lopez, R.; Prydz, H. CpG Islands As Gene Markers in the Human Genome. Genomics 1992, 13, 1095–1107. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG Islands and the Regulation of Transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The Human Colon Cancer Methylome Shows Similar Hypo- and Hypermethylation at Conserved Tissue-Specific CpG Island Shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in Regulators of the Epigenome and Their Connections to Global Chromatin Patterns in Cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Jin, S.-G.; Jiang, Y.; Qiu, R.; Rauch, T.A.; Wang, Y.; Schackert, G.; Krex, D.; Lu, Q.; Pfeifer, G.P. 5-Hydroxymethylcytosine is Strongly Depleted in Human Cancers But Its Levels Do Not Correlate with IDH1 Mutations. Cancer Res. 2011, 71, 7360–7365. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A Decade of Exploring the Cancer Epigenome—Biological and Translational Implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5′ CpG Island Methylation is Associated with Transcriptional Silencing of the Tumour Suppressor p16/CDKN2/MTS1 in Human Cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 Promoter Correlates with Lack of Expression of hMLH1 in Sporadic Colon Tumors and Mismatch Repair-Defective Human Tumor Cell Lines. Cancer Res. 1997, 57, 808–811. [Google Scholar]
- Kaneda, A.; Yagi, K. Two Groups of DNA Methylation Markers to Classify Colorectal Cancer into Three Epigenotypes. Cancer Sci. 2011, 102, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K.; Akagi, K.; Hayashi, H.; Nagae, G.; Tsuji, S.; Isagawa, T.; Midorikawa, Y.; Nishimura, Y.; Sakamoto, H.; Seto, Y.; et al. Three DNA Methylation Epigenotypes in Human Colorectal Cancer. Clin. Cancer Res. 2010, 16, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG Island Methylator Phenotype Underlies Sporadic Microsatellite Instability and is Tightly Associated with BRAF Mutation in Colorectal Cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Kambara, T.; Simms, L.A.; Whitehall, V.L.J.; Spring, K.J.; Wynter, C.V.A.; Walsh, M.D.; Barker, M.A.; Arnold, S.; McGivern, A.; Matsubara, N.; et al. BRAF Mutation is Associated with DNA Methylation in Serrated Polyps and Cancers of the Colorectum. Gut 2004, 53, 1137–1144. [Google Scholar] [CrossRef]
- Jin, S.-G.; Xiong, W.; Wu, X.; Yang, L.; Pfeifer, G.P. The DNA Methylation Landscape of Human Melanoma. Genomics 2015, 106, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Hutchinson, L.; Deng, A.; Green, M.R. Common BRAF(V600E)-Directed Pathway Mediates Widespread Epigenetic Silencing in Colorectal Cancer and Melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1250–1255. [Google Scholar] [CrossRef]
- Girault, I.; Tozlu, S.; Lidereau, R.; Bièche, I. Expression Analysis of DNA Methyltransferases 1, 3A, and 3B in Sporadic Breast Carcinomas. Clin. Cancer Res. 2003, 9, 4415–4422. [Google Scholar] [PubMed]
- Saito, Y.; Kanai, Y.; Sakamoto, M.; Saito, H.; Ishii, H.; Hirohashi, S. Expression of mRNA for DNA Methyltransferases and Methyl-CpG-Binding Proteins and DNA Methylation Status on CpG Islands and Pericentromeric Satellite Regions During Human Hepatocarcinogenesis. Hepatology 2001, 33, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Micevic, G.; Muthusamy, V.; Damsky, W.; Theodosakis, N.; Liu, X.; Meeth, K.; Wingrove, E.; Santhanakrishnan, M.; Bosenberg, M. DNMT3b Modulates Melanoma Growth by Controlling Levels of mTORC2 Component RICTOR. Cell Rep. 2016, 14, 2180–2192. [Google Scholar] [CrossRef] [Green Version]
- Al Emran, A.; Marzese, D.M.; Menon, D.R.; Stark, M.S.; Torrano, J.; Hammerlindl, H.; Zhang, G.; Brafford, P.; Salomon, M.P.; Nelson, N.; et al. Distinct Histone Modifications Denote Early Stress-Induced Drug Tolerance in Cancer. Oncotarget 2018, 9, 8206–8222. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, P.; Tee, W.-W.; Reinberg, D. A Double Take on Bivalent Promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global Histone Modification Patterns Predict Risk of Prostate Cancer Recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Laird, P.W. Interplay Between the Cancer Genome and Epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-Genome Sequencing of Liver Cancers Identifies Etiological Influences on Mutation Patterns and Recurrent Mutations in Chromatin Regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent Mutations of Chromatin Remodeling Genes in Transitional Cell Carcinoma of the Bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The Histone Methyltransferase SETDB1 is Recurrently Amplified in Melanoma and Accelerates Its Onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef]
- Wagner, V.; Gil, J. An Epigenetic Switch: From Senescent Melanocytes to Malignant Melanoma (and Back). Cancer Cell 2018, 33, 162–163. [Google Scholar] [CrossRef] [Green Version]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core Epithelial-to-Mesenchymal Transition Interactome Gene-Expression Signature is Associated with Claudin-Low and Metaplastic Breast Cancer Subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef]
- Keren-Paz, A.; Emmanuel, R.; Samuels, Y. YAP and the Drug Resistance Highway. Nat. Genet. 2015, 47, 193–194. [Google Scholar] [CrossRef]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo Effector YAP Promotes Resistance to RAF- and MEK-Targeted Cancer Therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional Addiction in Cancer Cells is Mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Paoluzzi, L.; Hanniford, D.; Sokolova, E.; Osman, I.; Darvishian, F.; Wang, J.; Bradner, J.E.; Hernando, E. BET and BRAF Inhibitors Act Synergistically Against BRAF-Mutant Melanoma. Cancer Med. 2016, 5, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Echevarría-Vargas, I.M.; Reyes-Uribe, P.I.; Guterres, A.N.; Yin, X.; Kossenkov, A.V.; Liu, Q.; Zhang, G.; Krepler, C.; Cheng, C.; Wei, Z.; et al. Co-Targeting BET and MEK As Salvage Therapy for MAPK and Checkpoint Inhibitor-Resistant Melanoma. EMBO Mol. Med. 2018, 10, e8446. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maertens, O.; Kuzmickas, R.; Manchester, H.E.; Emerson, C.E.; Gavin, A.G.; Guild, C.J.; Wong, T.C.; De Raedt, T.; Bowman-Colin, C.; Hatchi, E.; et al. MAPK Pathway Suppression Unmasks Latent DNA Repair Defects and Confers a Chemical Synthetic Vulnerability in BRAF-, NRAS-, and NF1-Mutant Melanomas. Cancer Discov. 2019, 9, 526–545. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 Expression is Associated with High Proliferation Rate and Aggressive Tumor Subgroups in Cutaneous Melanoma and Cancers of the Endometrium, Prostate, and Breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M.; et al. The Epigenetic Modifier EZH2 Controls Melanoma Growth and Metastasis through Silencing of Distinct Tumour Suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef]
- Perotti, V.; Baldassari, P.; Molla, A.; Nicolini, G.; Bersani, I.; Grazia, G.; Benigni, F.; Maurichi, A.; Santinami, M.; Anichini, A.; et al. An Actionable Axis Linking NFATc2 to EZH2 Controls the EMT-Like Program of Melanoma Cells. Oncogene 2019, 38, 4384–4396. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khaliq, M.; Fallahi-Sichani, M. Epigenetic Mechanisms of Escape from BRAF Oncogene Dependency. Cancers 2019, 11, 1480. https://doi.org/10.3390/cancers11101480
Khaliq M, Fallahi-Sichani M. Epigenetic Mechanisms of Escape from BRAF Oncogene Dependency. Cancers. 2019; 11(10):1480. https://doi.org/10.3390/cancers11101480
Chicago/Turabian StyleKhaliq, Mehwish, and Mohammad Fallahi-Sichani. 2019. "Epigenetic Mechanisms of Escape from BRAF Oncogene Dependency" Cancers 11, no. 10: 1480. https://doi.org/10.3390/cancers11101480
APA StyleKhaliq, M., & Fallahi-Sichani, M. (2019). Epigenetic Mechanisms of Escape from BRAF Oncogene Dependency. Cancers, 11(10), 1480. https://doi.org/10.3390/cancers11101480