Exploiting ING2 Epigenetic Modulation as a Therapeutic Opportunity for Non-Small Cell Lung Cancer


Abstract
:1. Introduction
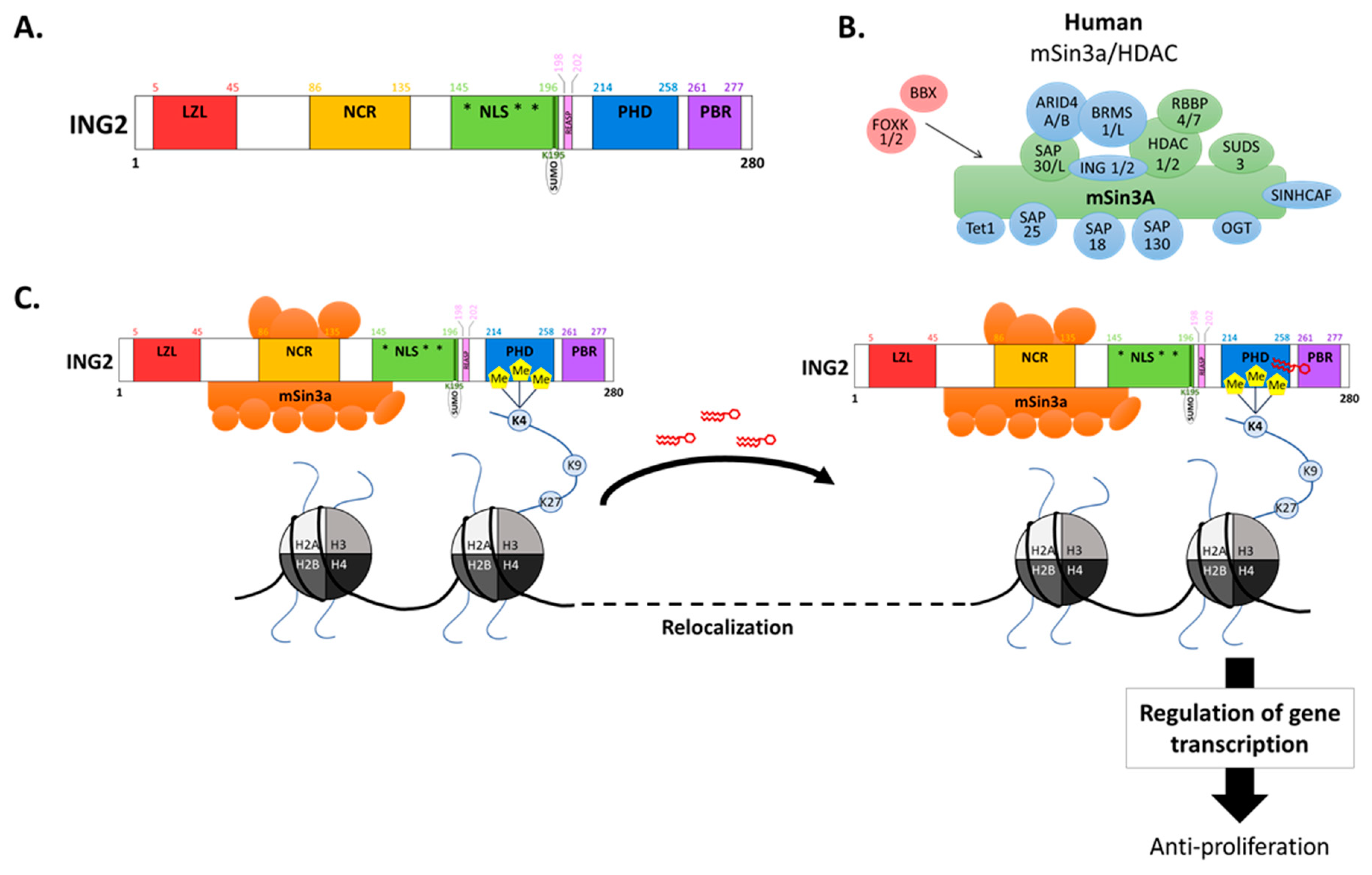
2. ING2 Modulates Transcriptional Activity Through Chromatin Remodeling
3. ING2 Status in Human Tumors
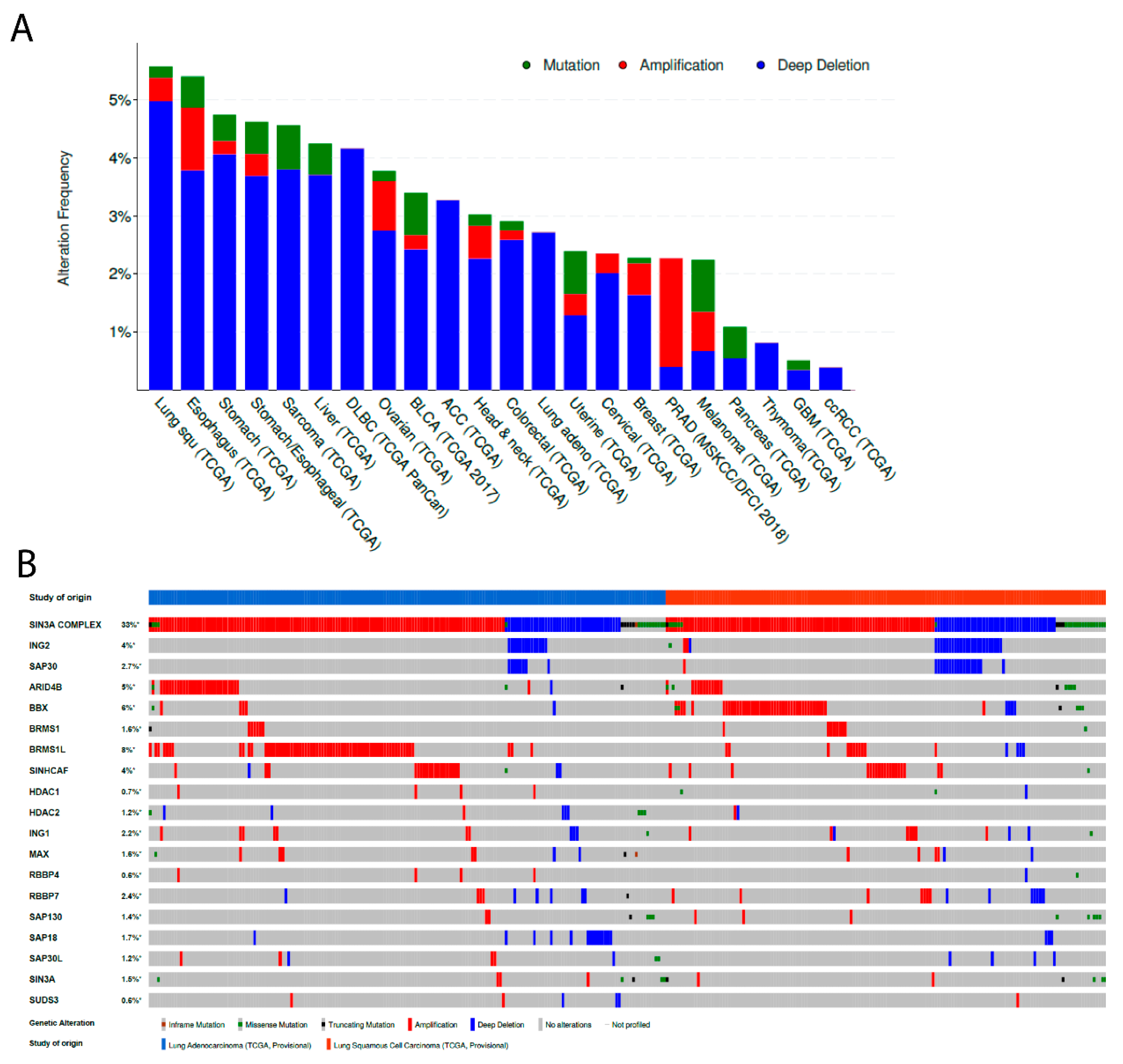
3.1. ING2 Alterations in Human Tumors
3.2. ING2 Alterations in Non-Small Cell Lung Cancer
4. Potential Role of the ING2 Epigenetic Modulation in Lung Cancer Treatment
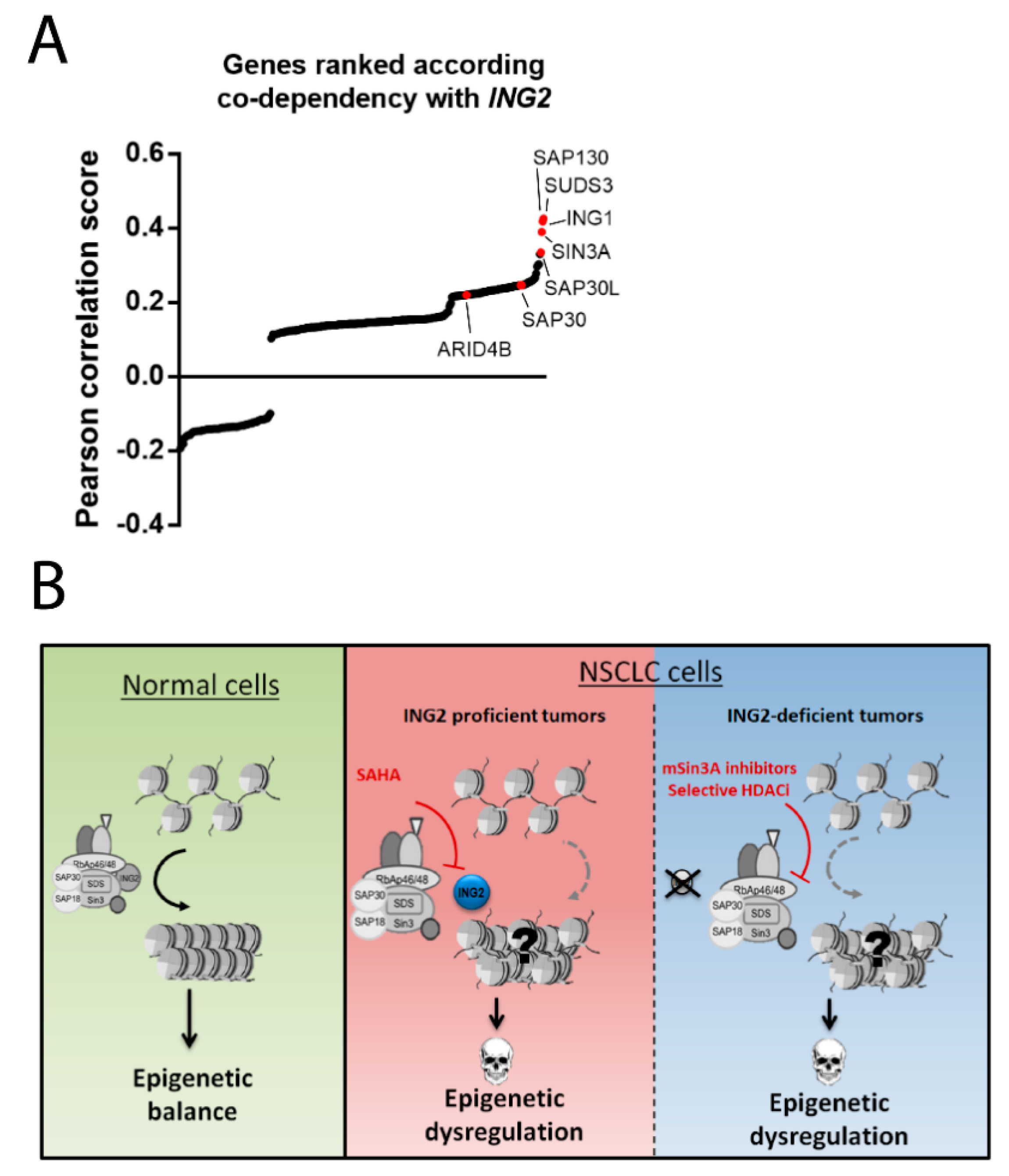
4.1. Exploring ING2 Dependencies in Cancer Cells
4.2. Targeting SIN3A-Mediated ING2 Functions in Cancer Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Peña, P.; Lan, F.; Kaadige, M.R.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Peña, P.V.; Davrazou, F.; Shi, X.; Walter, K.L.; Verkhusha, V.V.; Gozani, O.; Zhao, R.; Kutateladze, T.G. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 2006, 442, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Cayrou, C.; Ullah, M.; Landry, A.-J.; Côté, V.; Selleck, W.; Lane, W.S.; Tan, S.; Yang, X.-J.; Côté, J. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell 2006, 21, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Pile, L.A.; Spellman, P.T.; Katzenberger, R.J.; Wassarman, D.A. The SIN3 deacetylase complex represses genes encoding mitochondrial proteins: Implications for the regulation of energy metabolism. J. Biol. Chem. 2003, 278, 37840–37848. [Google Scholar] [CrossRef]
- Larrieu, D.; Ythier, D.; Binet, R.; Brambilla, C.; Brambilla, E.; Sengupta, S.; Pedeux, R. ING2 controls the progression of DNA replication forks to maintain genome stability. EMBO Rep. 2009, 10, 1168–1174. [Google Scholar] [CrossRef] [Green Version]
- Pedeux, R.; Sengupta, S.; Shen, J.C.; Demidov, O.N.; Saito, S.; Onogi, H.; Kumamoto, K.; Wincovitch, S.; Garfield, S.H.; McMenamin, M.; et al. ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation. Mol. Cell. Biol. 2005, 25, 6639–6648. [Google Scholar] [CrossRef]
- Wang, J.; Chin, M.Y.; Li, G. The novel tumor suppressor p33ING2 enhances nucleotide excision repair via inducement of histone H4 acetylation and chromatin relaxation. Cancer Res. 2006, 66, 1906–1911. [Google Scholar] [CrossRef]
- Saito, M.; Kumamoto, K.; Robles, A.I.; Horikawa, I.; Furusato, B.; Okamura, S.; Goto, A.; Yamashita, T.; Nagashima, M.; Lee, T.-L.; et al. Targeted disruption of Ing2 results in defective spermatogenesis and development of soft-tissue sarcomas. PLoS ONE 2010, 5, e15541. [Google Scholar] [CrossRef]
- Garkavtsev, I.; Kazarov, A.; Gudkov, A.; Riabowol, K. Suppression of the novel growth inhibitor p33ING1 promotes neoplastic transformation. Nat. Genet. 1996, 14, 415–420. [Google Scholar] [CrossRef]
- Shimada, Y.; Saito, A.; Suzuki, M.; Takahashi, E.; Horie, M. Cloning of a novel gene (ING1L) homologous to ING1, a candidate tumor suppressor. Cytogenet. Cell Genet. 1998, 83, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, M.; Shiseki, M.; Miura, K.; Hagiwara, K.; Linke, S.P.; Pedeux, R.; Wang, X.W.; Yokota, J.; Riabowol, K.; Harris, C.C. DNA damage-inducible gene p33ING2 negatively regulates cell proliferation through acetylation of p53. Proc. Natl. Acad. Sci. USA 2001, 98, 9671–9676. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, M.; Shiseki, M.; Pedeux, R.M.; Okamura, S.; Kitahama-Shiseki, M.; Miura, K.; Yokota, J.; Harris, C.C. A novel PHD-finger motif protein, p47ING3, modulates p53-mediated transcription, cell cycle control, and apoptosis. Oncogene 2003, 22, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiseki, M.; Nagashima, M.; Pedeux, R.M.; Kitahama-Shiseki, M.; Miura, K.; Okamura, S.; Onogi, H.; Higashimoto, Y.; Appella, E.; Yokota, J.; et al. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003, 63, 2373–2378. [Google Scholar]
- Unoki, M.; Kumamoto, K.; Robles, A.I.; Shen, J.C.; Zheng, Z.-M.; Harris, C.C. A novel ING2 isoform, ING2b, synergizes with ING2a to prevent cell cycle arrest and apoptosis. FEBS Lett. 2008, 582, 3868–3874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Murray, K. The occurrence of epsilon-n-methyl lysine in histones. Biochemistry 1964, 3, 10–15. [Google Scholar] [CrossRef]
- Fischle, W.; Franz, H.; Jacobs, S.A.; Allis, C.D.; Khorasanizadeh, S. Specificity of the chromodomain Y chromosome family of chromodomains for lysine-methylated ARK(S/T) motifs. J. Biol. Chem. 2008, 283, 19626–19635. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.K.; Kulbokas, E.J.; Gingeras, T.R.; et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 2005, 120, 169–181. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.T.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Gozani, O.; Karuman, P.; Jones, D.R.; Ivanov, D.; Cha, J.; Lugovskoy, A.A.; Baird, C.L.; Zhu, H.; Field, S.J.; Lessnick, S.L.; et al. The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell 2003, 114, 99–111. [Google Scholar] [CrossRef]
- Kaadige, M.R.; Ayer, D.E. The polybasic region that follows the plant homeodomain zinc finger 1 of Pf1 is necessary and sufficient for specific phosphoinositide binding. J. Biol. Chem. 2006, 281, 28831–28836. [Google Scholar] [CrossRef] [PubMed]
- Bua, D.J.; Martin, G.M.; Binda, O.; Gozani, O. Nuclear phosphatidylinositol-5-phosphate regulates ING2 stability at discrete chromatin targets in response to DNA damage. Sci. Rep. 2013, 3, 2137. [Google Scholar] [CrossRef] [PubMed]
- Ohkouchi, C.; Kumamoto, K.; Saito, M.; Ishigame, T.; Suzuki, S.-I.; Takenoshita, S.; Harris, C.C. ING2, a tumor associated gene, enhances PAI-1 and HSPA1A expression with HDAC1 and mSin3A through the PHD domain and C-terminal. Mol. Med. Rep. 2017, 16, 7367–7374. [Google Scholar] [CrossRef] [Green Version]
- Ythier, D.; Larrieu, D.; Binet, R.; Binda, O.; Brambilla, C.; Gazzeri, S.; Pedeux, R. Sumoylation of ING2 regulates the transcription mediated by Sin3A. Oncogene 2010, 29, 5946–5956. [Google Scholar] [CrossRef]
- Zhang, Y.; Iratni, R.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell 1997, 89, 357–364. [Google Scholar] [CrossRef]
- Parthun, M.R.; Widom, J.; Gottschling, D.E. The major cytoplasmic histone acetyltransferase in yeast: Links to chromatin replication and histone metabolism. Cell 1996, 87, 85–94. [Google Scholar] [CrossRef]
- Laherty, C.D.; Billin, A.N.; Lavinsky, R.M.; Yochum, G.S.; Bush, A.C.; Sun, J.M.; Mullen, T.M.; Davie, J.R.; Rose, D.W.; Glass, C.K.; et al. SAP30, a component of the mSin3 corepressor complex involved in N-CoR-mediated repression by specific transcription factors. Mol. Cell 1998, 2, 33–42. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.W.; Iratni, R.; Erdjument-Bromage, H.; Tempst, P.; Hampsey, M.; Reinberg, D. SAP30, a novel protein conserved between human and yeast, is a component of a histone deacetylase complex. Mol. Cell 1998, 1, 1021–1031. [Google Scholar] [CrossRef]
- Alland, L.; David, G.; Shen-Li, H.; Potes, J.; Muhle, R.; Lee, H.-C.; Hou, H.; Chen, K.; DePinho, R.A. Identification of mammalian Sds3 as an integral component of the Sin3/histone deacetylase corepressor complex. Mol. Cell. Biol. 2002, 22, 2743–2750. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, F.; Kudlow, J.E. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: Coupling protein O-GlcNAcylation to transcriptional repression. Cell 2002, 110, 69–80. [Google Scholar] [CrossRef]
- Muñoz, I.M.; MacArtney, T.; Sanchez-Pulido, L.; Ponting, C.P.; Rocha, S.; Rouse, J. Family with sequence similarity 60A (FAM60A) protein is a cell cycle-fluctuating regulator of the SIN3-HDAC1 histone deacetylase complex. J. Biol. Chem. 2012, 287, 32346–32353. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, T.C.; Yun, U.J.; Ayer, D.E. Identification and characterization of three new components of the mSin3A corepressor complex. Mol. Cell. Biol. 2003, 23, 3456–3467. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, Q.; Wang, W.; Wade, P.; Wong, J. Specific targeting and constitutive association of histone deacetylase complexes during transcriptional repression. Genes Dev. 2002, 16, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Kumamoto, K.; Fujita, K.; Kurotani, R.; Saito, M.; Unoki, M.; Hagiwara, N.; Shiga, H.; Bowman, E.D.; Yanaihara, N.; Okamura, S.; et al. ING2 is upregulated in colon cancer and increases invasion by enhanced MMP13 expression. Int. J. Cancer 2009, 125, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Sardiu, M.E.; Smith, K.T.; Groppe, B.D.; Gilmore, J.M.; Saraf, A.; Egidy, R.; Peak, A.; Seidel, C.W.; Florens, L.; Workman, J.L.; et al. Suberoylanilide hydroxamic acid (SAHA)-induced dynamics of a human histone deacetylase protein interaction network. Mol. Cell Proteomics 2014, 13, 3114–3125. [Google Scholar] [CrossRef]
- Kumamoto, K.; Spillare, E.A.; Fujita, K.; Horikawa, I.; Yamashita, T.; Appella, E.; Nagashima, M.; Takenoshita, S.; Yokota, J.; Harris, C.C. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res. 2008, 68, 3193–3203. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Lotem, J.; Sachs, L. Hematopoietic cells from mice deficient in wild-type p53 are more resistant to induction of apoptosis by some agents. Blood 1993, 82, 1092–1096. [Google Scholar] [CrossRef] [Green Version]
- Yonish-Rouach, E.; Resnitzky, D.; Lotem, J.; Sachs, L.; Kimchi, A.; Oren, M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991, 352, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Di Leonardo, A.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994, 8, 2540–2551. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Dai, D.L.; Martinka, M.; Ho, V.; Li, G. Nuclear ING2 expression is reduced in human cutaneous melanomas. Br. J. Cancer 2006, 95, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pan, K.; Wang, H.; Weng, D.; Song, H.; Zhou, J.; Huang, W.; Li, J.; Chen, M.; Xia, J. Decreased expression of ING2 gene and its clinicopathological significance in hepatocellular carcinoma. Cancer Lett. 2008, 261, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Yang, X.-F.; Gou, W.-F.; Lu, H.; Li, H.; Zhu, Z.-T.; Sun, H.-Z.; Zheng, H.-C. Expression profiles of inhibitor of growth protein 2 in normal and cancer tissues: An immunohistochemical screening analysis. Mol. Med. Rep. 2016, 13, 1881–1887. [Google Scholar] [CrossRef]
- Han, X.-R.; Bai, X.-Z.; Sun, Y.; Yang, Y. Nuclear ING2 expression is reduced in osteosarcoma. Oncol. Rep. 2014, 32, 1967–1972. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Liu, Y.; Zhao, Y.; Tian, F.; Wang, G. miR-153-3p Suppresses Inhibitor of Growth Protein 2 Expression to Function as Tumor Suppressor in Acute Lymphoblastic Leukemia. Technol. Cancer Res. Treat. 2019, 18, 1533033819852990. [Google Scholar] [CrossRef]
- Temel, M.; Turkmen, A.; Dokuyucu, R.; Cevik, C.; Oztuzcu, S.; Cengiz, B.; Mutaf, M. A novel tumor suppressor gene in basal cell carcinoma: Inhibition of growth factor-2. Tumour Biol. 2015, 36, 4611–4616. [Google Scholar] [CrossRef]
- Okano, T.; Gemma, A.; Hosoya, Y.; Hosomi, Y.; Nara, M.; Kokubo, Y.; Yoshimura, A.; Shibuya, M.; Nagashima, M.; Harris, C.C.; et al. Alterations in novel candidate tumor suppressor genes, ING1 and ING2 in human lung cancer. Oncol. Rep. 2006, 15, 545–549. [Google Scholar] [CrossRef] [Green Version]
- Walzak, A.A.; Veldhoen, N.; Feng, X.; Riabowol, K.; Helbing, C.C. Expression profiles of mRNA transcript variants encoding the human inhibitor of growth tumor suppressor gene family in normal and neoplastic tissues. Exp. Cell Res. 2008, 314, 273–285. [Google Scholar] [CrossRef]
- Gao, Y.; Ma, H.; Gao, C.; Lv, Y.; Chen, X.; Xu, R.; Sun, M.; Liu, X.; Lu, X.; Pei, X.; et al. Tumor-promoting properties of miR-8084 in breast cancer through enhancing proliferation, suppressing apoptosis and inducing epithelial-mesenchymal transition. J. Transl. Med. 2018, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Gournay, M.; Paineau, M.; Archambeau, J.; Pedeux, R. Regulat-INGs in tumors and diseases: Focus on ncRNAs. Cancer Lett. 2019, 447, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Sironi, E.; Cerri, A.; Tomasini, D.; Sirchia, S.M.; Porta, G.; Rossella, F.; Grati, F.R.; Simoni, G. Loss of heterozygosity on chromosome 4q32-35 in sporadic basal cell carcinomas: Evidence for the involvement of p33ING2/ING1L and SAP30 genes. J. Cutan. Pathol. 2004, 31, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Borkosky, S.S.; Gunduz, M.; Nagatsuka, H.; Beder, L.B.; Gunduz, E.; Ali, M.A.L.S.; Rodriguez, A.P.; Cilek, M.Z.; Tominaga, S.; Yamanaka, N.; et al. Frequent deletion of ING2 locus at 4q35.1 associates with advanced tumor stage in head and neck squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2009, 135, 703–713. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, H.; Wang, Q.; Chen, M.; Weng, D.; Wang, H.; Zhou, J.; Li, Y.; Sun, J.; Chen, Y.; et al. Analysis of loss of heterozygosity on chromosome 4q in hepatocellular carcinoma using high-throughput SNP array. Oncol. Rep. 2010, 23, 445–455. [Google Scholar] [PubMed]
- Borkosky, S.S.; Gunduz, M.; Beder, L.; Tsujigiwa, H.; Tamamura, R.; Gunduz, E.; Katase, N.; Rodriguez, A.P.; Sasaki, A.; Nagai, N.; et al. Allelic loss of the ING gene family loci is a frequent event in ameloblastoma. Oncol. Res. 2010, 18, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Cetin, E.; Cengiz, B.; Gunduz, E.; Gunduz, M.; Nagatsuka, H.; Bekir-Beder, L.; Fukushima, K.; Pehlivan, D.; Nishizaki, K.; Shimizu, K.; et al. Deletion mapping of chromosome 4q22-35 and identification of four frequently deleted regions in head and neck cancers. Neoplasma 2008, 55, 299–304. [Google Scholar]
- Mittal, K.R.; Chen, F.; Wei, J.J.; Rijhvani, K.; Kurvathi, R.; Streck, D.; Dermody, J.; Toruner, G.A. Molecular and immunohistochemical evidence for the origin of uterine leiomyosarcomas from associated leiomyoma and symplastic leiomyoma-like areas. Mod. Pathol. 2009, 22, 1303–1311. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-Q.; Yang, C.-Y.; Wang, S.-Y.; Wang, T.; Han, J.-L.; Wei, K.; Liu, F.-C.; Xu, J.; Peng, X.-Z.; Wang, J.-M. Cell-free plasma hypermethylated CASZ1, CDH13 and ING2 are promising biomarkers of esophageal cancer. J. Biomed. Res. 2018, 32, 424–433. [Google Scholar]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Ythier, D.; Brambilla, E.; Binet, R.; Nissou, D.; Vesin, A.; de Fraipont, F.; Moro-Sibilot, D.; Lantuejoul, S.; Brambilla, C.; Gazzeri, S.; et al. Expression of candidate tumor suppressor gene ING2 is lost in non-small cell lung carcinoma. Lung Cancer 2010, 69, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.Q.; Zhang, X.; Xu, D.P.; Bao, W.G.; Lin, A.F.; Xu, H.H.; Yan, W.H. Decreased expression of ING2 gene and its clinicopathological significance in Chinese NSCLC patients. Neoplasma 2014, 61, 468–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Zeyadi, M.; Dimova, I.; Ranchich, V.; Rukova, B.; Nesheva, D.; Hamude, Z.; Georgiev, S.; Petrov, D.; Toncheva, D. Whole genome microarray analysis in non-small cell lung cancer. Biotechnol. Biotechnol. Equip. 2015, 29, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Liu, L.; Wu, M.; Xing, G.; He, S.; Yin, Y.; Tian, C.; He, F.; Zhang, L. HECT ubiquitin ligase Smurf1 targets the tumor suppressor ING2 for ubiquitination and degradation. FEBS Lett. 2010, 584, 3005–3012. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xiao, N.; Wang, Y.; Wang, R.; Chen, Y.; Pan, W.; Liu, D.; Li, S.; Sun, J.; Zhang, K.; et al. Smurf1 regulates lung cancer cell growth and migration through interaction with and ubiquitination of PIPKIγ. Oncogene 2017, 36, 5668–5680. [Google Scholar] [CrossRef]
- Tan, W.-L.; Jain, A.; Takano, A.; Newell, E.W.; Iyer, N.G.; Lim, W.-T.; Tan, E.-H.; Zhai, W.; Hillmer, A.M.; Tam, W.-L.; et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016, 17, e347–e362. [Google Scholar] [CrossRef]
- Smolle, E.; Fink-Neuboeck, N.; Lindenmann, J.; Smolle-Juettner, F.; Pichler, M. The Biological and Clinical Relevance of Inhibitor of Growth (ING) Genes in Non-Small Cell Lung Cancer. Cancers 2019, 11, 1118. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef]
- Laherty, C.D.; Yang, W.M.; Sun, J.M.; Davie, J.R.; Seto, E.; Eisenman, R.N. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 1997, 89, 349–356. [Google Scholar] [CrossRef]
- Cowley, S.M.; Iritani, B.M.; Mendrysa, S.M.; Xu, T.; Cheng, P.F.; Yada, J.; Liggitt, H.D.; Eisenman, R.N. The mSin3A chromatin-modifying complex is essential for embryogenesis and T-cell development. Mol. Cell. Biol. 2005, 25, 6990–7004. [Google Scholar] [CrossRef]
- Dannenberg, J.-H.; David, G.; Zhong, S.; van der Torre, J.; Wong, W.H.; Depinho, R.A. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005, 19, 1581–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; Carraro, G.; Konda, B.; Guan, X.; Mizuno, T.; Chiba, N.; Kostelny, M.; Kurkciyan, A.; David, G.; McQualter, J.L.; et al. Sin3a regulates epithelial progenitor cell fate during lung development. Development 2017, 144, 2618–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, N.; David, G.; Farias, E.; Waxman, S. Emerging Roles of Epigenetic Regulator Sin3 in Cancer. Adv. Cancer Res. 2016, 130, 113–135. [Google Scholar] [PubMed]
- Kwon, Y.-J.; Petrie, K.; Leibovitch, B.A.; Zeng, L.; Mezei, M.; Howell, L.; Gil, V.; Christova, R.; Bansal, N.; Yang, S.; et al. Selective Inhibition of SIN3 Corepressor with Avermectins as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2015, 14, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- McDonel, P.; Demmers, J.; Tan, D.W.M.; Watt, F.; Hendrich, B.D. Sin3a is essential for the genome integrity and viability of pluripotent cells. Dev. Biol. 2012, 363, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.T.; Martin-Brown, S.A.; Florens, L.; Washburn, M.P.; Workman, J.L. Deacetylase inhibitors dissociate the histone-targeting ING2 subunit from the Sin3 complex. Chem. Biol. 2010, 17, 65–74. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef]
- Larrieu, D.; Ythier, D.; Brambilla, C.; Pedeux, R. ING2 controls the G1 to S-phase transition by regulating p21 expression. Cell Cycle 2010, 9, 3984–3990. [Google Scholar] [CrossRef]
- Jones, P.A.; Issa, J.-P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef]
- Loewith, R.; Meijer, M.; Lees-Miller, S.P.; Riabowol, K.; Young, D. Three yeast proteins related to the human candidate tumor suppressor p33(ING1) are associated with histone acetyltransferase activities. Mol. Cell. Biol. 2000, 20, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Kichina, J.V.; Zeremski, M.; Aris, L.; Gurova, K.V.; Walker, E.; Franks, R.; Nikitin, A.Y.; Kiyokawa, H.; Gudkov, A.V. Targeted disruption of the mouse ing1 locus results in reduced body size, hypersensitivity to radiation and elevated incidence of lymphomas. Oncogene 2006, 25, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Roca, M.S.; Di Gennaro, E.; Budillon, A. Implication for Cancer Stem Cells in Solid Cancer Chemo-Resistance: Promising Therapeutic Strategies Based on the Use of HDAC Inhibitors. J. Clin. Med. 2019, 8, 912. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.; Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Mishra, S.R.; Behera, B.P.; Jena, M.; Bhutia, S.K. Dysregulation of histone deacetylases in carcinogenesis and tumor progression: A possible link to apoptosis and autophagy. Cell. Mol. Life Sci. 2019, 76, 3263–3282. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tissue Type | Origin | Mutation Type/Expression Change | Methods | Position | Coding | Frequency | Ref. |
|---|---|---|---|---|---|---|---|
| Lung cancer | Cell lines | Downregulation | RT-QPCR | 7/8 | [49] | ||
| Patient | Substitution | PCR-SSCP, Sequencing | LZL (13) | Ala -> Ala | 6/31 | ||
| Patient | Substitution | PCR-SSCP, Sequencing | 6bp downstream exon 1 | 6/31 | |||
| Lung cancer | Cell lines | Downregulation | Q-PCR | 2/2 | [50] | ||
| Lung cancer | Patient | Downregulation | IHC | 70/120 | [62] | ||
| Patient | No LOH | MM | 0/12 | ||||
| Patient | Substitution | Sequencing | 39 | Ala -> Ala | 21/22 | ||
| Patient | Downregulation | Q-PCR | 15/22 | ||||
| No change | Q-PCR | 6/22 | |||||
| Upregulation | Q-PCR | 1/22 | |||||
| NSCLC | Patient | Downregulation, aberrantly localization | IHC, RT-PCR, WB | 21/64 (32.8%) | [63] | ||
| Adenocarcinoma | Patient | Downregulation, aberrantly localization | IHC, RT-PCR, WB | 11/24 (45.8%) | |||
| Squamous cell carcinoma | Patient | Downregulation, aberrantly localization | IHC, RT-PCR, WB | 10/38 (26.3%) | |||
| NSCLC | Patient | Chromosomal deletion | cDNA Microarray | 4q34.2–q35.1 | 2/10 (20%) | [64] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blondel, A.; Benberghout, A.; Pedeux, R.; Ricordel, C. Exploiting ING2 Epigenetic Modulation as a Therapeutic Opportunity for Non-Small Cell Lung Cancer. Cancers 2019, 11, 1601. https://doi.org/10.3390/cancers11101601
Blondel A, Benberghout A, Pedeux R, Ricordel C. Exploiting ING2 Epigenetic Modulation as a Therapeutic Opportunity for Non-Small Cell Lung Cancer. Cancers. 2019; 11(10):1601. https://doi.org/10.3390/cancers11101601
Chicago/Turabian StyleBlondel, Alice, Amine Benberghout, Rémy Pedeux, and Charles Ricordel. 2019. "Exploiting ING2 Epigenetic Modulation as a Therapeutic Opportunity for Non-Small Cell Lung Cancer" Cancers 11, no. 10: 1601. https://doi.org/10.3390/cancers11101601
APA StyleBlondel, A., Benberghout, A., Pedeux, R., & Ricordel, C. (2019). Exploiting ING2 Epigenetic Modulation as a Therapeutic Opportunity for Non-Small Cell Lung Cancer. Cancers, 11(10), 1601. https://doi.org/10.3390/cancers11101601