An Integrative Data Mining and Omics-Based Translational Model for the Identification and Validation of Oncogenic Biomarkers of Pancreatic Cancer

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Integrative Screening Approach Suggests 23 Novel Biomarker Candidates for the Diagnosis and Management of Pancreatic Cancer
2.2. Literature- and Experiment-Based Validation of the 23 Selected Candidates
2.3. ADAM9, ANXA2, ITGA2, MET, and LAMC2 Exhibit Substantial Impact on the Survival of PC Patients
2.4. Supervised Machine Learning Classification Demonstrates that ADAM9, ANXA2, LAMC2, and APLP2 Are Important in Differentiating PC Tissue from Normal Tissue
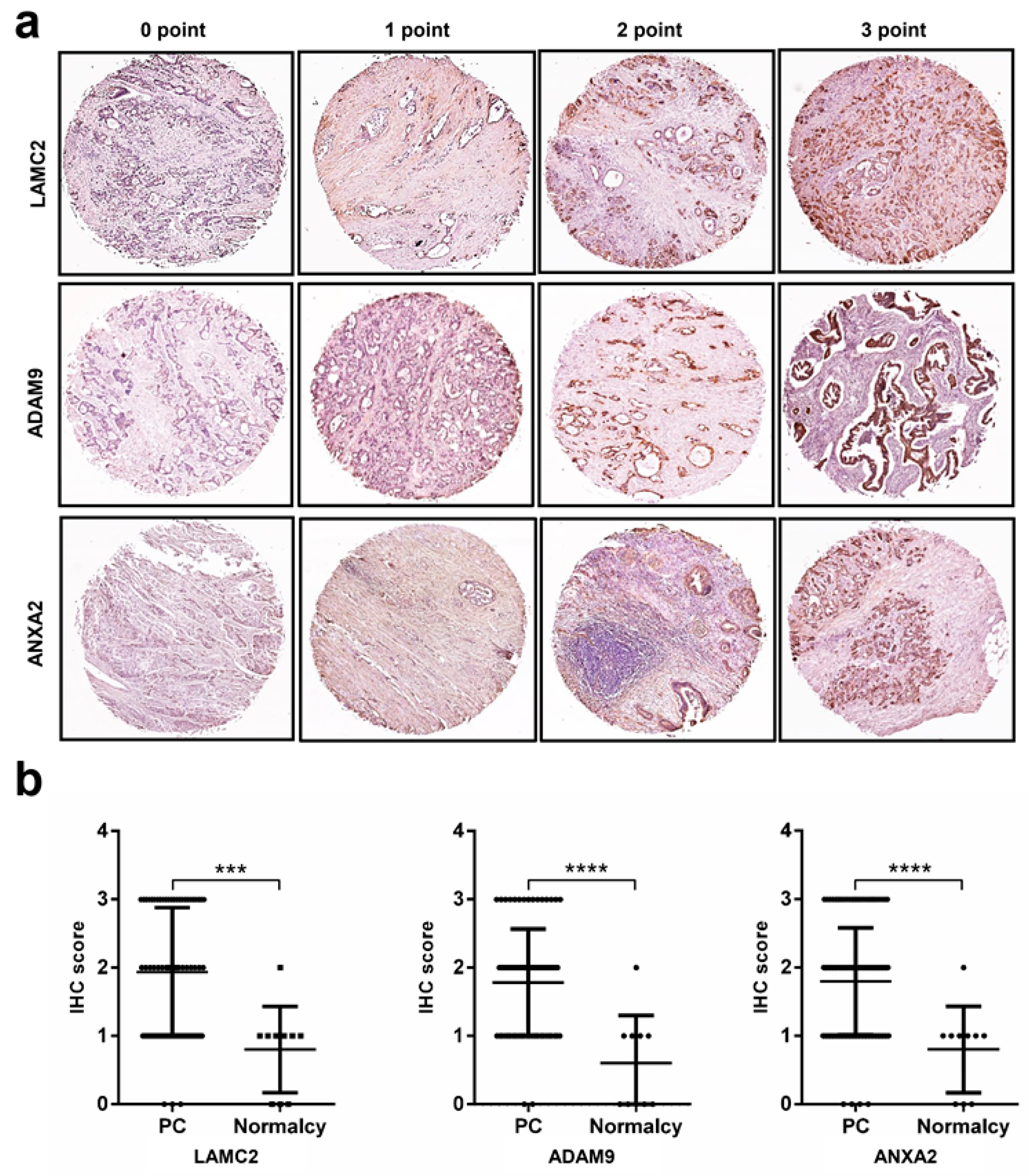
2.5. Immunohistochemistry Analysis Reveals High Protein Expression Levels of LAMC2, ADAM9, ANXA2, and APLP2 in Pancreatic Cancer
2.6. Functional Analysis and Public Data Mining to Create a Comprehensive Picture of the Biological Processes in Pancreatic Cancer
2.7. Drug–Gene Interaction and miRNA–Gene for Further Development of Novel Therapeutics
3. Discussion
4. Materials and Methods
4.1. Ethical Approval and Consent of Patients
4.2. Antibodies
4.3. Pancreatic Cancer and Normal Control Cohorts
4.3.1. NGS Sample Collection
4.3.2. Microarray Sample Collection
4.3.3. Tissue-Array Sample Collection
4.4. RNA Extraction from Formalin-Fixed Paraffin-Embedded Tissue
4.5. RNA Data Processing and Analysis
4.6. Microarray Data Processing and Gene Expression Meta-Analysis
4.7. Immunohistochemistry (IHC) Experiments
4.8. Unsupervised and Supervised Machine Learning Algorithms
4.8.1. Variable Selection
4.8.2. Data Exploration and Visualization
4.8.3. Random Forests Classification Model and Explanation
4.9. Correlation Analysis
4.10. Kaplan-Meier Plots and Cox Regression Analysis
4.11. Database Mining, Pathway Enrichment, STRING, and CHAT Analyses
4.12. Drug-Gene Interaction and miRNA–Gene Interaction for Further Developing Novel Therapeutics
4.13. Statistical Significance Level
4.14. Availability of Data and Materials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Maisonneuve, P.; Lowenfels, A.B. Epidemiology of pancreatic cancer: An overview. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Decker, G.A.; Batheja, M.J.; Collins, J.M.; Silva, A.C.; Mekeel, K.L.; Moss, A.A.; Nguyen, C.C.; Lake, D.F.; Miller, L.J. Risk factors for pancreatic adenocarcinoma and prospects for screening. Gastroenterol. Hepatol. 2010, 6, 246–254. [Google Scholar]
- Semaan, A.; Maitra, A. Pancreatic cancer in 2017: Rebooting pancreatic cancer knowledge and treatment options. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Sant, G.R.; Knopf, K.B.; Albala, D.M. Live-single-cell phenotypic cancer biomarkers-future role in precision oncology? NPJ Precis. Oncol. 2017, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Graham, C.; D’Amato, A.; Gentry-Maharaj, A.; Ryan, A.; Kalsi, J.K.; Ainley, C.; Whetton, A.D.; Menon, U.; Jacobs, I.; et al. A combined biomarker panel shows improved sensitivity for the early detection of ovarian cancer allowing the identification of the most aggressive type II tumours. Br. J. Cancer 2017, 117, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Jang, C.Y.; Kim, S.A.; Park, S.B.; Jung, D.E.; Kim, B.O.; Kim, H.Y.; Chung, M.J.; Park, J.Y.; Bang, S.; et al. Combined use of CEMIP and CA 19-9 enhances diagnostic accuracy for pancreatic cancer. Sci. Rep. 2018, 8, 3383. [Google Scholar] [CrossRef]
- Chan, A.; Prassas, I.; Dimitromanolakis, A.; Brand, R.E.; Serra, S.; Diamandis, E.P.; Blasutig, I.M. Validation of biomarkers that complement CA19.9 in detecting early pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5787–5795. [Google Scholar] [CrossRef]
- Long, N.P.; Yoon, S.J.; Anh, N.H.; Nghi, T.D.; Lim, D.K.; Hong, Y.J.; Hong, S.-S.; Kwon, S.W. A systematic review on metabolomics-based diagnostic biomarker discovery and validation in pancreatic cancer. Metabolomics 2018, 14, 109. [Google Scholar] [CrossRef]
- Zeh, H.J.; Winikoff, S.; Landsittel, D.P.; Gorelik, E.; Marrangoni, A.M.; Velikokhatnaya, L.; Winans, M.T.; Lee, K.; Moser, A.; Bartlett, D.; et al. Multianalyte profiling of serum cytokines for detection of pancreatic cancer. Cancer Biomark. Sect. A Dis. Markers 2005, 1, 259–269. [Google Scholar] [CrossRef]
- Brand, R.E.; Nolen, B.M.; Zeh, H.J.; Allen, P.J.; Eloubeidi, M.A.; Goldberg, M.; Elton, E.; Arnoletti, J.P.; Christein, J.D.; Vickers, S.M.; et al. Serum biomarker panels for the detection of pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Goymer, P. Early detection for pancreatic cancer. Nat. Rev. Cancer 2008, 8, 408. [Google Scholar] [CrossRef]
- Sener, S.F.; Fremgen, A.; Menck, H.R.; Winchester, D.P. Pancreatic cancer: A report of treatment and survival trends for 100,313 patients diagnosed from 1985–1995, using the National Cancer Database. J. Am. Coll. Surg. 1999, 189, 1–7. [Google Scholar] [CrossRef]
- Hu, J.; Locasale, J.W.; Bielas, J.H.; O’Sullivan, J.; Sheahan, K.; Cantley, L.C.; Heiden, M.G.V.; Vitkup, D. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat. Biotechnol. 2013, 31, 522. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Chen, Z.; Farshidfar, F.; Xu, X.; Lorenzi, P.L.; Wang, Y.; Cheng, F.; Tan, L.; Mojumdar, K.; Du, D.; et al. Molecular Characterization and Clinical Relevance of Metabolic Expression Subtypes in Human Cancers. Cell Rep. 2018, 23, 255–269.e4. [Google Scholar] [CrossRef] [PubMed]
- Harsha, H.C.; Kandasamy, K.; Ranganathan, P.; Rani, S.; Ramabadran, S.; Gollapudi, S.; Balakrishnan, L.; Dwivedi, S.B.; Telikicherla, D.; Selvan, L.D.N.; et al. A Compendium of Potential Biomarkers of Pancreatic Cancer. PLoS Med. 2009, 6, e1000046. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, S.; Zhang, B.; Ni, Q.; Yu, X.; Xu, J. Circulating biomarkers for early diagnosis of pancreatic cancer: Facts and hopes. Am. J. Cancer Res. 2018, 8, 332–353. [Google Scholar] [PubMed]
- Oldfield, L.E.; Connor, A.A.; Gallinger, S. Molecular Events in the Natural History of Pancreatic Cancer. Trends Cancer 2017, 3, 336–346. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114. [Google Scholar] [CrossRef]
- Diamandis, E.P. The failure of protein cancer biomarkers to reach the clinic: Why, and what can be done to address the problem? BMC Med. 2012, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wendl, M.C.; McMichael, J.F.; Raphael, B.J. Expanding the computational toolbox for mining cancer genomes. Nat. Rev. Genet. 2014, 15, 556. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Braunstein, G.; Koeffler, H.P. LAMC2 as a therapeutic target for cancers. Expert Opin. Ther. Targets 2014, 18, 979–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauri, P.; Scarpa, A.; Nascimbeni, A.C.; Benazzi, L.; Parmagnani, E.; Mafficini, A.; Della Peruta, M.; Bassi, C.; Miyazaki, K.; Sorio, C. Identification of proteins released by pancreatic cancer cells by multidimensional protein identification technology: A strategy for identification of novel cancer markers. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1125–1127. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Jaffee, E.M. Annexin A2 is a new antigenic target for pancreatic cancer immunotherapy. Oncoimmunology 2012, 1, 112–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grützmann, R.; Lüttges, J.; Sipos, B.; Ammerpohl, O.; Dobrowolski, F.; Alldinger, I.; Kersting, S.; Ockert, D.; Koch, R.; Kalthoff, H.; et al. ADAM9 expression in pancreatic cancer is associated with tumour type and is a prognostic factor in ductal adenocarcinoma. Br. J. Cancer 2004, 90, 1053. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Sliker, B.; Peters, H.L.; Tuli, A.; Herskovitz, J.; Smits, K.; Purohit, A.; Singh, R.K.; Dong, J.; Batra, S.K.; et al. Amyloid precursor protein and amyloid precursor-like protein 2 in cancer. Oncotarget 2016, 7, 19430–19444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Hasebe, T.; Oda, T.; Sasaki, S.; Kinoshita, T.; Konishi, M.; Ochiai, T.; Ochiai, A. Cytoplasmic expression of laminin γ2 chain correlates with postoperative hepatic metastasis and poor prognosis in patients with pancreatic ductal adenocarcinoma. Cancer 2002, 94, 1894–1901. [Google Scholar] [CrossRef] [PubMed]
- Kosanam, H.; Prassas, I.; Chrystoja, C.C.; Soleas, I.; Chan, A.; Dimitromanolakis, A.; Blasutig, I.M.; Ruckert, F.; Gruetzmann, R.; Pilarsky, C.; et al. Laminin, gamma 2 (LAMC2): A promising new putative pancreatic cancer biomarker identified by proteomic analysis of pancreatic adenocarcinoma tissues. Mol. Cell. Proteom. 2013, 12, 2820–2832. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.-H.; Lee, S.K. Long noncoding RNAs in cancer cells. Cancer Lett. 2018, 419, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, F.; De Giovanni, C.; Nanni, P.; Forni, G.; Lollini, P.L. 2011: The immune hallmarks of cancer. Cancer Immunol. Immunother. CII 2011, 60, 319–326. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Mak, T.W. The current state of cancer metabolism. Nat. Rev. Cancer 2016, 16, 613. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Muñoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.-H.; Hong, J.-Y.; Park, H.J.; Lee, S.K. The role of exosomes and miRNAs in drug-resistance of cancer cells. Int. J. Cancer 2017, 141, 220–230. [Google Scholar] [CrossRef]
- Bach, D.-H.; Park, H.J.; Lee, S.K. The Dual Role of Bone Morphogenetic Proteins in Cancer. Mol. Ther. Oncol. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Hamada, S.; Satoh, K.; Fujibuchi, W.; Hirota, M.; Kanno, A.; Unno, J.; Masamune, A.; Kikuta, K.; Kume, K.; Shimosegawa, T. MiR-126 acts as a tumor suppressor in pancreatic cancer cells via the regulation of ADAM9. Mol. Cancer Res. MCR 2012, 10, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Keklikoglou, I.; Hosaka, K.; Bender, C.; Bott, A.; Koerner, C.; Mitra, D.; Will, R.; Woerner, A.; Muenstermann, E.; Wilhelm, H.; et al. MicroRNA-206 functions as a pleiotropic modulator of cell proliferation, invasion and lymphangiogenesis in pancreatic adenocarcinoma by targeting ANXA2 and KRAS genes. Oncogene 2015, 34, 4867–4878. [Google Scholar] [CrossRef] [PubMed]
- Malgerud, L.; Lindberg, J.; Wirta, V.; Gustafsson-Liljefors, M.; Karimi, M.; Moro, C.F.; Stecker, K.; Picker, A.; Huelsewig, C.; Stein, M.; et al. Bioinformatory-assisted analysis of next-generation sequencing data for precision medicine in pancreatic cancer. Mol. Oncol. 2017, 11, 1413–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Gill, E.E.; Hancock, R.E.W. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823. [Google Scholar] [CrossRef]
- Long, N.P.; Jung, K.H.; Yoon, S.J.; Anh, N.H.; Nghi, T.D.; Kang, Y.P.; Yan, H.H.; Min, J.E.; Hong, S.S.; Kwon, S.W. Systematic assessment of cervical cancer initiation and progression uncovers genetic panels for deep learning-based early diagnosis and proposes novel diagnostic and prognostic biomarkers. Oncotarget 2017, 8, 109436–109456. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 18. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 26. [Google Scholar] [CrossRef]
- Pedersen, T.L.; Benesty, M. Lime: Local Interpretable Model-Agnostic Explanations. R Package Version 0.4.1. 2018. Available online: https://CRAN.R-project.org/package=lime (accessed on 27 January 2019).
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef]
- Tan, Y.; Tan, Y.; Lu, L.; Zhang, H.; Sun, C.; Liang, Y.; Zeng, J.; Yang, X.; Li, D.; Zou, H. HPCDb: An integrated database of pancreatic cancer. bioRxiv 2017. [Google Scholar] [CrossRef]
- Dayem Ullah, A.Z.; Cutts, R.J.; Ghetia, M.; Gadaleta, E.; Hahn, S.A.; Crnogorac-Jurcevic, T.; Lemoine, N.R.; Chelala, C. The pancreatic expression database: Recent extensions and updates. Nucleic Acids Res. 2014, 42, D944–D949. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.; Ali, I.; Silins, I.; Pyysalo, S.; Guo, Y.; Hogberg, J.; Stenius, U.; Korhonen, A. Cancer Hallmarks Analytics Tool (CHAT): A text mining approach to organize and evaluate scientific literature on cancer. Bioinformatics 2017, 33, 3973–3981. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.-Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A redesign and expansion of the drug–gene interaction database. Nucleic Acids Res. 2018, 46, D1068–D1073. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-K.; Long, N.P.; Jung, J.; Kim, T.J.; Na, E.; Kang, Y.P.; Kwon, S.W.; Jang, J. Integrative lipidomic and transcriptomic analysis of X-linked adrenoleukodystrophy reveals distinct lipidome signatures between adrenomyeloneuropathy and childhood cerebral adrenoleukodystrophy. Biochem. Biophys. Res. Commun. 2018. [Google Scholar] [CrossRef]
- Long, N.P.; Park, S.; Anh, N.H.; Min, J.E.; Yoon, S.J.; Kim, H.M.; Nghi, T.D.; Lim, D.K.; Park, J.H.; Lim, J.; et al. Efficacy of Integrating a Novel 16-Gene Biomarker Panel and Intelligence Classifiers for Differential Diagnosis of Rheumatoid Arthritis and Osteoarthritis. J. Clin. Med. 2019, 8, 50. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Entrez ID | RNA Alteration | Protein Alteration | NGS Results | Meta-Analysis | Neoplasm # vs. Normalcy | PDAC vs. Neoplasm # | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Log FC | p-Value | FDR | cES | p-Value | ||||||
| AGR2 | 10551 | ↑RT-PCR 1,2, ↑SAGE 1,2, ↑DM 1 | ↑IHC 1, ↑LFQ-MS 1, ↑WB 2 | 3.67 | 1.55 × 10−15 | 7.37 × 10−13 | 1.93 | 0 | ↑ | X |
| GAPDH | 2597 | ↑DM 1, ↑SAGE 1, NB 2 | ↑ICAT 1, ↑WB 1,2, ↓SILAC-TMS 2 | 1.48 | 6.08 × 10−4 | 1.27 × 10−2 | 1.68 | 2.02 × 10−13 | X | X |
| LAMC2 | 3918 | ↑DM 1,2 | ↑IHC 1 | 3.91 | 1.15 × 10−7 | 1.17 × 10−5 | 2.37 | 0 | ↑ | X |
| MMP11 | 4320 | ↑DM 1, ↑ISH 1, ↑SAGE 1,2, ↑NB 1,2 | ↑IHC 1, ↑WB 2 | NA | NA | NA | 1.62 | 7.90 × 10−14 | X | X |
| TAGLN2 | 8407 | NA | ↑SILAC-TMS 1, ↑ICAT 1 | NA | NA | NA | 1.73 | 1.64 × 10−5 | ↑ | X |
| ADAM9 | 8754 | ↑DM 1,2, ↑RT-PCR 1,2 | ↑IHC 1 | NA | NA | NA | 1.71 | 1.41 × 10−5 | X | X |
| ANXA2 | 302 | ↑DM 1 | ↑IHC 1, ↑ICAT 1, ↑WB 1,2, ↑MS 1, ↑TDE 1, ↑LFQ-MS 1 | 1.16 | 2.26 × 10−3 | 3.35 × 10−2 | 1.63 | 8.33 × 10−9 | X | X |
| APLP2 | 334 | ↑DM 1,2 | ↑SILAC-TMS 1,2, ↑SILAC 2 | NA | NA | NA | 1.53 | 1.11 × 10−5 | X | X |
| CDH3 | 1001 | ↑DM 1,2, ↑RT-PCR 1,2 | ↑IHC 1 | 2.72 | 8.70 × 10−6 | 4.50 × 10−4 | 2.02 | 0 | X | ↑ |
| MSLN | 10232 | ↓DM 1, ↑DM 1,2, ↑RT-PCR 1,2, ↑SAGE 1,2, ↑ISH 1 | ↑IHC 1 | 5.21 | 1.89 × 10−15 | 8.77 × 10−13 | 2.03 | 4.88 × 10−4 | ↑ | X |
| SERPINB5 | 5268 | ↑DM 1,2, ↑NB 2 | ↑IHC 1,2, ↑SILAC-TMS 1 | 8.35 | 6.54 × 10−30 | 2.96 × 10−26 | 1.96 | 0 | ↑ | X |
| CD82 | 3732 | ↑NB 1, ↑ISH 1, ↑DM 1 | ↑IHC 1 | 1.39 | 4.55 × 10−4 | 1.03 × 10−2 | 1.62 | 7.90 × 10−14 | X | X |
| CLDN18 | 51208 | ↑DM 1 | ↑IHC 1 | 2.57 | 9.43 × 10−5 | 2.97 × 10−3 | 1.64 | 7.60 × 10−9 | ↑ | X |
| EPHA2 | 1969 | ↑DM 1,2 | ↑SILAC-TMS 1, ↑WB 2 | 1.69 | 4.95 × 10−5 | 1.78 × 10−3 | 1.56 | 2.99 × 10−13 | X | ↑ |
| EZR | 7430 | NA | ↑IHC 1, SILAC-TMS 1 | NA | NA | NA | 1.68 | 0 | ↑ | X |
| FXYD3 | 5349 | ↑DM 1,2, ↑SAGE 1, ↑NB 1, ↑ISH 1 | NA | 4.71 | 4.03 × 10−21 | 5.21 × 10−18 | 1.91 | 0 | ↑ | X |
| GPRC5A | 9052 | ↑DM 1,2, ↑SAGE 1,2 | ↑IHC 1 | 3.98 | 2.08 × 10−8 | 2.61 × 10−6 | 1.91 | 1.25 × 10−8 | ↑ | ↑ |
| ITGA2 | 3673 | ↑DM 1,2 | ↑IHC 1, ↑WB 1 | 2.94 | 1.20 × 10−6 | 8.72 × 10−5 | 2.04 | 2.02 × 10−13 | ↑ | X |
| ITGB6 | 3694 | ↑DM 1 | ↑IHC 1 | 2.56 | 4.55 × 10−6 | 2.71 × 10−4 | 1.79 | 2.78 × 10−8 | X | X |
| MET | 4233 | ↑DM 1, ↑RT-PCR 1,2 | NA | NA | NA | NA | 1.90 | 1.97 × 10−7 | X | X |
| MST1R | 4486 | ↑DM 1,2, ↑RT-PCR 2 | ↑IHC 1, ↑WB 2 | 2.84 | 9.03 × 10−7 | 6.81 × 10−5 | 2.06 | 0 | ↑ | X |
| NQO1 | 1728 | ↑DM 1,2 | ↑IHC 1, ↑WB 2 | 3.71 | 1.88 × 10−9 | 3.04 × 10−7 | 2.15 | 1.63 × 10−5 | ↑ | X |
| SLC2A1 | 6513 | ↑DM 1,2 | ↑IHC 1 | 3.28 | 6.34 × 10−10 | 1.16 × 10−7 | 2.03 | 0 | ↑ | ↑ |
| Gene Symbol | Entrez ID | Cox Regression | |
|---|---|---|---|
| Cox Coefficient | FDR | ||
| AGR2 | 10551 | 0.05 | 0.76 |
| GAPDH | 2597 | 0.21 | 0.19 |
| LAMC2 | 3918 | 0.42 | 0.02 |
| MMP11 | 4320 | 0.17 | 0.28 |
| TAGLN2 | 8407 | 0.19 | 0.22 |
| ADAM9 | 8754 | 0.41 | 0.02 |
| ANXA2 | 302 | 0.32 | 0.04 |
| APLP2 | 334 | 0.23 | 0.14 |
| CDH3 | 1001 | 0.37 | 0.03 |
| MSLN | 10232 | 0.26 | 0.10 |
| SERPINB5 | 5268 | 0.38 | 0.02 |
| CD82 | 3732 | 0.11 | 0.51 |
| CLDN18 | 51208 | 0.09 | 0.63 |
| EPHA2 | 1969 | 0.33 | 0.05 |
| EZR | 7430 | 0.28 | 0.08 |
| FXYD3 | 5349 | 0.17 | 0.30 |
| GPRC5A | 9052 | 0.37 | 0.03 |
| ITGA2 | 3673 | 0.41 | 0.02 |
| ITGB6 | 3694 | 0.49 | 0.01 |
| MET | 4233 | 0.66 | 0.00 |
| MST1R | 4486 | 0.22 | 0.17 |
| NQO1 | 1728 | 0.14 | 0.43 |
| SLC2A1 | 6513 | 0.27 | 0.08 |
| Gene Symbol | miRNA–Gene Interaction | Druggable Genome | Drug–Gene Interaction 3 | |||
|---|---|---|---|---|---|---|
| miRNA 1 That Targets the Gene | Validation Methods | Expression Profile in PC (Correlation Coefficient 2) | ||||
| Strong Evidence | Less Strong Evidence | |||||
| ADAM9 | hsa-miR-126-3p | RA, WB, qPCR | MA | 0.315 | Yes | Ilomastat |
| hsa-miR-33a-5p | RA, WB, qPCR | 0.373 | ||||
| hsa-miR-125a-5p | qPCR | 0.320 | ||||
| ANXA2 | hsa-miR-155-5p | RA, WB, qPCR | MA | −0.599 | Yes | NA |
| hsa-miR-206 | RA, WB, qPCR | −0.312 | ||||
| APLP2 | NA | Yes | NA | |||
| CDH3 | NA | Yes | NA | |||
| MSLN | hsa-miR-21-5p | RA | 0.715 | Yes | Amatuximab | |
| SERPINB5 | hsa-miR-21-5p | RA, WB, qPCR | MA, NGS | 0.709 | Yes | NA |
| hsa-miR-103a-3p | RA, WB, qPCR | MA, NGS | 0.501 | |||
| CD82 | NA | Yes | NA | |||
| CLDN18 | NA | Yes | Claudiximab | |||
| EPHA2 | NA | Yes | Dasatinib, Dorsomorphin, Regorafenib, Vandetanib | |||
| EZR | hsa-miR-183-5p | RA, WB, qPCR | 0.424 | NA | NA | |
| hsa-miR-204-5p | RA, WB, qPCR | MA | −0.542 | |||
| hsa-miR-205-5p | RA | MA, NGS | 0.358 | |||
| FXYD3 | NA | No | NA | |||
| GPRC5A | hsa-miR-103a-3p | RA, WB, qPCR | NGS | 0.514 | Yes | NA |
| ITGA2 | hsa-miR-16-5p | qPCR | NGS, pSILAC | 0.509 | Yes | Abciximab, CHEMBL36326, Eptifibatide, Tirofiban, Vatelizumab |
| ITGB6 | NA | Yes | DI17E6, Intetumumab, STX-100 | |||
| MET | hsa-miR-34c-5p | RA, WB, qPCR | MA, NGS | 0.337 | Yes | ARRY-300, ABT-700, AMG-337, AMG-208, Amuvatinib, Altiratinib, Amoxicillin, Alectinib, BMS-698769, BMS-777607, BMS-794833, BMS-817378, BPI-9016, Crizotinib, Clofibrate, Cabozantinib, Cabozantinib S-Malate, Capmatinib, Crenolanib, Emibetuzumab, EMD-1204831, Foretinib, Golvatinib, JNJ-38877605, LY-2875358, MK-8033, MGCD-265, Merestinib, MK-2461, Onartuzumab, PF-04217903, PHA-665752, Pyrazinamide, Rilonacept, SGX-523, Savolitinib, Tepotinib, Tivantinib, Tanespimycin, SAR-125844, TAS-115 |
| hsa-miR-199a-3p | RA, WB, qPCR | MA | 0.365 | |||
| hsa-miR-34a-5p | RA, WB, qPCR | 0.478 | ||||
| hsa-miR-23b-3p | RA, WB, qPCR | 0.447 | ||||
| hsa-miR-27a-3p | RA, WB, qPCR | 0.444 | ||||
| hsa-miR-27b-3p | RA, WB, qPCR | 0.458 | ||||
| hsa-miR-31-5p | RA, qPCR | 0.594 | ||||
| hsa-miR-34a-3p | WB | 0.380 | ||||
| MST1R | NA | Yes | BMS-777607, Foretinib, MGCD-265, MK-2461, MK-8033 | |||
| NQO1 | NA | Yes | Apaziquone, Dicumarol, Vatiquinone | |||
| SLC2A1 | hsa-miR-22-3p | RA, WB, qPCR | NGSs | 0.331 | Yes | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, N.P.; Jung, K.H.; Anh, N.H.; Yan, H.H.; Nghi, T.D.; Park, S.; Yoon, S.J.; Min, J.E.; Kim, H.M.; Lim, J.H.; et al. An Integrative Data Mining and Omics-Based Translational Model for the Identification and Validation of Oncogenic Biomarkers of Pancreatic Cancer. Cancers 2019, 11, 155. https://doi.org/10.3390/cancers11020155
Long NP, Jung KH, Anh NH, Yan HH, Nghi TD, Park S, Yoon SJ, Min JE, Kim HM, Lim JH, et al. An Integrative Data Mining and Omics-Based Translational Model for the Identification and Validation of Oncogenic Biomarkers of Pancreatic Cancer. Cancers. 2019; 11(2):155. https://doi.org/10.3390/cancers11020155
Chicago/Turabian StyleLong, Nguyen Phuoc, Kyung Hee Jung, Nguyen Hoang Anh, Hong Hua Yan, Tran Diem Nghi, Seongoh Park, Sang Jun Yoon, Jung Eun Min, Hyung Min Kim, Joo Han Lim, and et al. 2019. "An Integrative Data Mining and Omics-Based Translational Model for the Identification and Validation of Oncogenic Biomarkers of Pancreatic Cancer" Cancers 11, no. 2: 155. https://doi.org/10.3390/cancers11020155
APA StyleLong, N. P., Jung, K. H., Anh, N. H., Yan, H. H., Nghi, T. D., Park, S., Yoon, S. J., Min, J. E., Kim, H. M., Lim, J. H., Kim, J. M., Lim, J., Lee, S., Hong, S. -S., & Kwon, S. W. (2019). An Integrative Data Mining and Omics-Based Translational Model for the Identification and Validation of Oncogenic Biomarkers of Pancreatic Cancer. Cancers, 11(2), 155. https://doi.org/10.3390/cancers11020155