Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism



Abstract
:1. Introduction: Apoptosis
2. Acute Myeloid Leukemia (AML)
3. Emerging Therapies for Patients with AML Targeting the BCL-2 Pathway
4. Targeting the Energetic Metabolism in AML cells
4.1. Abnormalities of the Glycolytic Pathway
4.2. Abnormalities of the Mitochondrial Energetic Metabolism
5. Metabolomics and BH3-Profiling Studies: Two Precious Tools for the Discovery and Prediction of Drug Sensitivity of Leukemic Cells
6. The Extrinsic Apoptotic Pathway: TRAIL-Rs, Caspase-8 and Their Abnormalities in AMLs
7. ONC201: A TRAIL—Inducing Small Chemical Compound
8. Autophagy Pathway: Abnormalities a Therapeutic Opportunities in AML
9. TP53-Mutated AMLs
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; Annichiarfico-Petruzzelli, M.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar]
- Nagata, S. Apoptosis and clearance of apoptotic cells. Ann. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Humphreis, L.; Espona-Fiedler, M.; Lorigley, D.B. FLIP as a therapeutic target in cancer. FEBS J. 2018, 285, 4104–4123. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamas-Din, A.; Kale, J.; Lewber, B.; Andrews, D.B. Mechanisms of action of BCL-2 family proteins. Cold Spring Harb. Perspect. 2013, 5, a008714. [Google Scholar] [CrossRef]
- Letai, A. Apoptosis and cancer. Ann. Rev. Cancer Biol. 2017, 1, 275–294. [Google Scholar] [CrossRef]
- Dohner, H.; Wiesdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Shlush, L.I. Age-related clonal hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef]
- Bowman, R.L.; Busque, L.; Levine, R.L. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 2018, 22, 157–170. [Google Scholar] [CrossRef]
- Acuna-Hildago, R.; Sengul, H.; Steehouwer, M.; van der Vorst, M.; Vermeulen, S.H.; Kiemeney, L.; Veltman, J.; Gliussen, C.; Hoischen, A. Ultra-sensitive sequencing identifies high prevalence of clonal hematopoiesis-associated mutations throughout adult life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef]
- Desai, P.; Mencia-Trinchant, M.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Abelson, S.; Collord, G.; Ng, S.; Cohen, N.M.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; Luben, R.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell 2018, 23, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Meisel, M.; Hinterleitner, R.; Pacis, A.S.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Wong, T.N.; Miller, C.A.; Jotte, M.R.M.; Bagegni, N.; Baty, J.D.; Schmidt, A.P.; Cashen, A.F.; Duncavage, E.J.; Helton, N.M.; Fiala, M.; et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat. Commun. 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 2017, 21, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.J.; Lindsley, R.C.; Tchekmedyian, V.; Mar, B.G.; Shi, J.; Jaiwswal, S.; Bosworth, A.; Francisco, L.; Le, H.; et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J. Clin. Oncol. 2017, 35, 1598–1605. [Google Scholar] [CrossRef]
- Hirsch, C.M.; Nazha, A.; Kneen, K.; Abazeed, M.E.; Meggendorfer, M.; Przychodzen, B.P.; Nadarajah, N.; Adema, V.; Nagata, Y.; Goyal, A.; et al. Consequences of mutant TET2 on clonality and subclonal hierarchy. Leukemia 2018, 32, 1751–1761. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.C.; Buccisano, F.; Cloos, J.; Grimmwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable disease in AML: A consensus document from the European Leukemia Net MRD working party. Blood 2019, 131, 1275–1291. [Google Scholar] [CrossRef]
- Jangen-Lavrencicx, M.; Grob, T.; Hauekaup, D.; Kavelaars, T.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular minimal residual disease in acute myeloid leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Rothenberg-Thurley, M.; Amler, S.; Goerlich, D.; Kohnke, T.; Konstandin, N.P.; Schneider, S.; Sauerland, M.C.; Herold, T.; Hubmann, M.; Ksiezyk, B.; et al. Persistence of pre-leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia 2018, 32, 1598–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzbecker, U.; Middeke, J.M.; Sockel, K.; Herbst, R.; Wold, D.; Baldus, C.D.; Oelschlagel, U.; Mutherig, A.; Fransecky, L.; Noppeney, R.; et al. Measurable residual disease-guided treatment with azacytidine to prevent hematological, relapse in patients with myelodysplastic syndrome and acute myeloid leukemia (RELAZA2): An open-label, multicenter, phase 2 trial. Lancet Oncol. 2018, 19, 1668–1679. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Jacoby, M.A.; Chang, G.S.; Miller, C.A.; Edwin, N.; Shao, J.; Elliott, K.; Robinson, J.; Abel, H.; Fulton, R.S.; et al. Mutation clearance after transplantation for myelodysplastic syndrome. N. Engl. J. Med. 2018, 379, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.; Radivoyevitch, T.; Suzuki, H.; Prsychoden, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Tyndel, M.S.; Kim, H.J.; Ahn, J.S.; Choi, S.H.; Park, H.J.; Kim, Y.K.; Yang, D.H.; Lee, J.J.; Jung, S.H.; et al. The clonal origins of leukemic progression of myelodysplasia. Leukemia 2017, 31, 1928–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stasch, J.M.; Heumuller, C.; Bleul, S.; Rothenberg-Thurley, M.; Riba, J.; Renz, N.; vel Szic, K.S.; Pfeifer, D.; Follo, M.; Pahl, H.L.; et al. Gene mutations and clonal architecture in myelodysplastic syndromes and changes upon progression to acute myeloid leukemia and under treatment. Br. J. Haematol. 2018, 182, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kao, Y.R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.R.; Montagna, C.; Will, B.; Verma, A.; Steidl, U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2019, in press. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Pellagatti, A.; Karimi, M.; Sato-Otsubo, A.; Sato, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Gene expression and risk of leukemic transformation in myelodysplasia. Blood 2017, 130, 2642–2653. [Google Scholar] [CrossRef]
- Yokoyama, K.; Shimizu, E.; Yokoyama, N.; Nakamura, S.; Kasajima, R.; Ogawa, M.; Takei, T.; Ito, M.; Kobayashi, A.; Yamaguchi, R.; et al. Cell-lineage level-targeted sequencing to identify acute myeloid leukemia with myelodysplasia-related changes. Blood Adv. 2018, 2, 2513–2521. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Herol, T.; Turley, M.R.; Amler, S.; Saueland, M.C.; Garlich, D.; Schneider, S.; Konstandin, N.P.; Dufour, A.; Braundi, K.; et al. Spectrum and prognostic relevance of driver gene mutation in acute myeloid leukemias. Blood 2016, 128, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomic of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Medinger, M.; Passweg, J.R. Acute myeloid leukaemia genomics. Br. J. Haematol. 2017, 179, 530–542. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Assi, S.A.; Imperato, M.R.; Coleman, D.J.; Pichin, A.; Polturi, S.; Ptasinska, A.; Chin, P.S.; Blair, H.; Cauchy, P.; James, S.R.; et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat. Genet. 2019, 51, 151–162. [Google Scholar] [CrossRef]
- Prossek, V.V.; Rotherberg-Turley, M.; Sauerland, M.C.; Herold, T.; Ksienzyk, B.; Konstandin, N.P.; Gerlich, D.; King, U.; Faldum, A.; et al. Genetics of acute myeloid leukemia in the elderly: Mutation spectrum and clinical impact in intensively treated patients aged ≥75 years. Haematologica 2018, in press. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Mor, B.G.; Mazzola, E.; Grammon, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1379. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; Huether, R.; et al. Pan-cancer genome and transcriptome analyses of 1,699 pediatric leukaemias and solid tumors. Nature 2018, 555, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; O’Connor, C.; Canete, A.; Bittencourt-Silvestre, J.; Sarrou, E.; Prendergast, A.; Choi, J.; Johnston, P.; Wells, C.; Gibson, B.; et al. Age-specific biological and molecular profiling distinguishes pediatric from adult acute myeloid leukaemias. Nat. Commun. 2018, 9, 5280. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Richtey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Mg, S.; Trotman-Grant, A.; Medeiros, J.J.; Rao-Bhatia, A.; Jaciw-Zurakowski, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Mitchell, A.; Kennedy, J.A.; Chen, W.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Marceau-Renaut, A.; Villenet, C.; Petit, A.; Russeau, A.; Ng, S.; Paquet, A.; Gonzales, F.; Barthelemy, A.; Lepretre, F.; et al. The stem cell-associated gene expression signature allows risk stratification in pediatric acute myeloid leukemia. Leukemia 2019, in press. [Google Scholar] [CrossRef]
- Zeijemaker, W.; Grob, T.; Meijer, R.; Hanekamp, D.; Kelder, A.; Carbaat-Ham, J.C.; Oussoren-Brockoff, Y.; Snel, A.N.; Veldhuizen, D.; Scholten, W.J.; et al. 34+/CD38− leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia 2019, in press. [Google Scholar]
- Alexander, T.B.; Gu, Z.; Iacobubbi, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The genetic basis and cell of origin of mixed phenotype acute leukemia. Nature 2018, 562, 373–377. [Google Scholar] [CrossRef]
- Miller, C.A.; Wilson, R.K.; Ley, T.J. Genomic landscapes and clonality of de novo AML. N. Engl. J. Med. 2013, 369, 1472–1473. [Google Scholar]
- Paguirigan, A.L.; Smith, J.; Meshinchi, S.; Carroll, M.; Maley, C.; Radich, J.P. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci. Transl. Med. 2016, 7, 281re2. [Google Scholar] [CrossRef]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Kco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Margini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar]
- Quek, L.; Otto, G.W.; Garnett, C.; Lhermitte, L.; Karamitros, D.; Stoilova, B.; Lau, I.J.; Doondea, J.; Usukhbayar, B.; Kennedy, A.; et al. Genetically distinct leukemic stem cells in human CD34-acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J. Exp. Med. 2016, 213, 1513–1535. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; Sciambi, A.; Treusch, S.; Durruthy-Durruthy, R.; Gohale, K.; Jacob, J.; Chen, T.X.; Geis, J.A.; Oldham, W.; Matthews, J.; et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018, 28, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Ebrahem, Q.; Mahfour, R.Z.; Hasipek, M.; Enane, F.; Radivovevitch, T.; Rapin, N.; Pezychodzen, B.; Hu, Z.; Balusu, R.; et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub regulating granulo-monocytic fates. J. Clin. Investig. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabradan, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018, 34, 499–512. [Google Scholar] [CrossRef]
- Liu, Y.; He, P.; Liu, F.; Shi, L.; Zhu, H.; Zhao, J.; Wang, Y.; Cheng, X.; Zhang, M. Prognostic significance of NPM1 mutations in acute myeloid leukemia: A meta-analysis. Mol. Clin. Oncol. 2014, 2, 275–281. [Google Scholar] [CrossRef]
- Boddu, P.; Kantarjian, H.; Borthakur, G.; Kadia, T.; Daver, N.; Pierce, S.; Andreef, M.; Ravandi, F.; Cortes, J.; Kornblau, S.M. Co-occurrence of FLT3-TKD and NPM1 mutations defines a highly favorable prognostic AML group. Blood Adv. 2017, 1, 1546–1550. [Google Scholar] [CrossRef]
- Patel, S.S.; Kuo, F.C.; Gibson, C.J.; Steensma, P.P.; Soiffer, R.J.; Alyea, E.P.; Chen, Y.A.; Fathi, A.T.; Graubert, T.A.; Brusmer, A.M.; et al. High NPM1-mutant allele burden at diagnosis predicts unfavorable outcomes in de novo AML. Blood 2018, 31, 2616–2625. [Google Scholar]
- Shludh, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Parkin, B.; Londono-Joshi, A.; Kang, Q.; Kang, Q.; Tewari, M.; Rhim, A.D.; Malek, S.N. Ultrasensitive mutation detection identifies rare residual cells causing acute myelogenous leukemia relapse. J. Clin. Investig. 2017, 127, 3484–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corces-Zimmerman, M.R.; Hong, W.J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, N.; Miraki-Moud, F.; Ermini, L.; Titley, L.; Vijayaraghavan, G.; Papaemmanuil, E.; Campbell, P.; Gribben, J.; Taussing, D.; Greaves, M. Single cell analysis of clonal architecture in acute myeloid leukemia. Leukemia 2019, in press. [Google Scholar]
- Eisfeld, A.K.; Kohlschmidt, J.; Mrozek, K.; Blechly, J.S.; Walker, C.J.; Nicolet, D.; Orwick, S.; Maharry, S.E.; Carroll, A.J.; Stone, R.M.; et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years or older who respond favorably to standard chemotherapy: An analysis of Alliance studies. Leukemia 2018, 32, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Hollein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 mutated AML can relapse with wild-type NPM1: Persistent clonal hematopoiesis. Blood Adv. 2018, 2, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovi, J.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of minimal residual disease in standard risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef]
- Van der Lee, D.; Reijmers, R.M.; Honders, M.W.; Hagedoorn, R.S.; de Jong, R.; Kester, M.; van der Steen, D.; de Ru, A.; Kweekel, C.; Bijen, H.M.; et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Investig. 2019, in press. [Google Scholar] [CrossRef]
- Vasu, S.; Kohlschmidt, J.; Mrozek, K.; Eisfeld, A.K.; Nicolet, D.; Sterling, L.J.; Becker, H.; Mezler, K.H.; Papaioannou, D.; Powell, B.L.; et al. Ten-year outcome of patients with acute myeloid leukemia not treated with allogeneic transplantation in first complete remission. Blood Adv. 2018, 2, 1645–1650. [Google Scholar] [CrossRef]
- Sood, R.; Hansen, N.F.; Donovan, F.X.; Carrington, B.; Bucci, D.; Maskeri, B.; Young, A.; Trivedi, N.S.; Kohlschmidt, J.; Stone, R.M.; et al. Somatic mutational landscape of AML with inv(16) or t(8;21) identifies patterns of clonal evolution in relapse. Leukemia 2016, 30, 501–504. [Google Scholar] [CrossRef]
- Faber, Z.J.; Chen, X.; Gedman, A.L.; Boggs, K.; Cheng, J.; Ma, J.; Radtke, I.; Chao, J.R.; Walsh, M.P.; Song, G.; et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 2016, 48, 1551–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisfeld, A.K.; Kohlschmidt, J.; Schwind, S.; Nicolet, D.; Blashly, J.S.; Orwick, S.; Shah, C.; Bainazar, M.; Kroll, K.W.; Walker, C.J.; et al. Mutations in the CCND1 and CCND2 genes are frequent events in adult patients with t(8;21)(q22;q22) acute myeloid leukemia. Leukemia 2017, 31, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Soria, N.; McKenzie, L.; Draper, J.; Ptasinska, A.; Issa, H.; Potturi, S.; Blair, H.J.; Pickin, A.; Isa, A.; Chin, P.S.; et al. The oncogenic transcription factor RUNX1/ETO corrupts cell cycle regulation to drive leukemic transformation. Cancer Cell 2018, 34, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Christen, F.; Hoyer, K.; Yoshida, K.; Hou, H.A.; Waldmeter, N.; Heuser, M.; Hills, R.K.; Chan, W.; Hablesteiter, R.; Blau, O.; et al. Genomic landscape and clonal evolution of acute myeloid leukemia with (8;21): An international study on 331 patients. Blood 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, S.; Murphy, T.; Law, A.; Yehudal, D.; Ho, J.M.; Chan, S.; Schimmer, A.D. Emerging therapies for acute myeloid leukemia: Translating biology into the clinic. JCI Insight 2017, 2, e95679. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell. Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Schenk, R.L.; Strasser, A.; Dewson, G. BCL-2: Long and winding path from discovery to therapeutic target. Biochem. Biophys. Res. Commun. 2017, 482, 459–469. [Google Scholar] [CrossRef]
- Porwit-MacDonald, A.; Ivory, K.; Wilkinson, S.; Wheatley, K.; Wong, L.; Janossy, G. Bcl-2 protein expression in normal human bone marrow precursors and in acute myelogenous leukemia. Leukemia 1995, 9, 1191–1198. [Google Scholar]
- Andreef, M.; Jiang, S.; Zhang, X.; Konopleva, M.; Estrov, Z.; Snell, V.E.; Xie, Z.; Okcu, M.F.; Sanchez-Williams, G.; M Dong, J.; et al. Expression of Bcl-2-related genes in normal and AML progenitors: Changes induced by chemotherapy and retinoic acid. Leukemia 1999, 13, 1881–1892. [Google Scholar] [CrossRef]
- Vo, T.T.; Ryan, J.; Carrasco, R.; Neuberg, D.; Rossi, D.J.; Stone, R.M.; De Angelo, D.J.; Frattini, M.G.; Letai, A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012, 151, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Bhola, P.D.; Mar, B.G.; Lindsley, R.C.; Ryan, J.A.; Hogdal, L.J.; Vo, T.T.; De Angelo, D.J.; Galinsky, I.; Ebert, B.L.; Letai, A. Functionally identifiable apoptosis-insensitive subpopulations determine chemoresistance in acute myeloid leukemia. J. Clin. Investig. 2016, 126, 3827–3836. [Google Scholar] [CrossRef] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belili, B.A.; Bruncko, M.; Deowerth, T.L.; Dinges, J.; Hadjuk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumors. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Perini, G.F.; Ribeiro, G.N.; Pinto Neto, J.V.; Campos, L.T.; Hammerschalk, N. BCL-2 as therapeutic target for hematological malignancies. J. Hematol. Oncol. 2018, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; Blum, W.; et al. Efficacy and biological correlates of response in a phase II study of Venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rostogi, S.; Hong, W.J.; Zhao, F.; Medeiros, B.C.; Tycoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chyla, B.; Daver, N.; Doyle, K.; McKeegan, E.; Huang, X.; Ruvolo, V.; Wang, Z.; Chen, K.; Souers, A.; Leverson, J.; et al. Genetic biomarkers of sensitivity and resistance to Venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am. J. Hematol. 2018, in press. [Google Scholar] [CrossRef]
- Kontro, M.; Kumar, A.; Majumder, M.M.; Eldfors, S.; Parsons, A.; Pemovska, T.; Saarela, J.; Yadav, B.; Malani, D.; Floisand, Y.; et al. HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia. Leukemia 2017, 31, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.; Shi, Y.; Kornblau, S.; Lu, H.; Konoplev, S.; Antony, A.; Ruvolo, V.; Wiu, Y.H.; Zhang, N.; Coomber, K.R.; et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann. Hematol. 2012, 91, 1861–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogenberger, J.M.; Kornblau, S.M.; Pierceall, W.E.; Lena, R.; Chow, D.; Shi, C.X.; Mantei, J.; Ahmann, G.; Gonzalez, I.M.; Choudhary, A.; et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 2014, 28, 1657–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teh, T.C.; Nguyen, N.Y.; Moujalled, D.M.; Segal, D.; Pomilio, G.; Rijal, S.; Jabbour, A.; Cummins, K.; Lackovic, K.; Blombery, P.; et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia 2018, 32, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminarMy efficacy of venetoclax with decitabine or azacytidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schh, A.C.; Candoni, A.; et al. International phase 3 study of azacytidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; Yang, D.; Aribi, A.; Ali, H.; Sandhu, K.; Al Maliki, M.M.; Mei, M.; Salhotra, A.; Khaled, S.; Nakamura, R.; et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica 2018, 103, e404–e407. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.; Strickland, S.A.; Roboz, G.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Martinelli, G.; Walter, R.B.; Enjeti, A.; Fakouhi, K.; et al. Safety and efficacy of Venetoclax plus low-dose Cytarabine in treatment-naïve patients aged ≥65 years with acute myeloid leukemia. Blood 2016, 128, 102. [Google Scholar]
- Wei, A.; Strickland, S.A.; Roboz, G.J.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Chyla, B.; Popovic, R.; et al. Phase 1-2 study of Venetoclax with low-dose cytarabine in treatment-naïve, elderly patients with acute myeloid leukemia unfit for intensive chemotherapy: 1-year outcomes. Blood 2017, 130, 890. [Google Scholar]
- Daver, N.; Pollyea, D.A.; Yee, K.; Fenaux, P.; Brandwein, J.; Vey, N.; Martinelli, G.; Kelly, K.R.; Roboz, G.; Garcia, J.S.; et al. Preliminary results from a phase 1b study evaluating BCL-2 inhibitor Venetoclax in combination with the MEK inhibitor Cobimetinib or MDM2 inhibitor Idasanutlin in patients with relapsed or refractory (R/) AML. Blood 2017, 130, 813. [Google Scholar]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreef, M. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: Mechanisms and superior antileukemic efficacy. Cancer Cell 2017, 32, 748–760. [Google Scholar] [CrossRef]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting BCL-2 and PI3K induces BAX-dependent mitochondrial apoptosis in AML cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef]
- Reyna, D.E.; Garner, T.P.; Lopez, A.; Kopp, F.; Choudhary, G.S.; Sridharan, A.; Narayanagari, S.R.; Mitchell, K.; Dong, B.; Bartholdy, B.A.; et al. Direct activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell 2017, 32, 490–505. [Google Scholar] [CrossRef]
- Reichenbach, F.; Wiedenmann, C.; Schalk, E.; Becker, D.; Funk, K.; Scholz-Kreisel, P.; Todt, F.; Wolleschak, D.; Dohner, K.; Marquardt, J.U.; et al. Mitochondrial BAX determines the predisposition to apoptosis in human AML. Clin. Cancer Res. 2017, 23, 4805–4816. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.E.; Eide, C.A.; Kaempf, A.; Khanna, V.; Savage, S.L.; Rofelty, A.; English, I.; Ho, H.; Pandva, R.; Bolosky, W.J.; et al. Molecularly targeted drug combinations demonstrate selective effectiveness for myeloid- and lymphoid-derived hematologic malignancies. Proc. Natl. Acad. Sci. USA 2017, 114, E7554–E7563. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Luo, H.; Payton, J.E.; Cain, J.; Ley, T.J.; Opfenman, J.T.; Tomasson, M.H. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin. Investig. 2010, 120, 2109–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, S.H.; Karp, J.E.; Svingen, P.A.; Krajewski, S.; Burke, P.J.; Gore, S.D.; Reed, J.C. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998, 91, 991–1000. [Google Scholar] [PubMed]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Lino, T.; Rocnik, J.L.; Kikushige, Y.; Mosi, Y.; Shima, T.; Iwasaki, H.; Takemaka, K.; et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, K.; Liu, Q.; Gowda, K.; Barth, B.M.; Claxton, D.; Amin, S.; Loughran, T.P.; Wang, H.G. Maritoclax induces apoptosis in acute myeloid leukemia cells with elevated Mcl-1 expression. Cancer Biol. Ther. 2014, 15, 1077–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Dai, N.; Tang, Z.; Fang, H. Small-molecule Mcl-1 inhibitors: Emerging anti-tumor agents. Eur. J. Med. Chem. 2018, 146, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Kotschy, A.; Szlavik, Z.; Murray, J. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Abulwerdi, F.; Liao, C.; Liu, M.; Azmi, A.S.; Aboukameel, A.; Mady, A.S.; Gulappa, T.; Cierpicki, T.; Owens, S.; Zhang, T.; et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2014, 13, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Zhang, H.; Chen, J.; Tahir, S.K.; Phillips, D.C.; Xue, T.; Nimmer, P.; Jin, S.; Smith, M.; Xiao, Y.; et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015, 6, e1590. [Google Scholar] [CrossRef] [PubMed]
- Phase I Study of S64315 Administered Intravenously in Patients with Acute Myeloid Leukaemia or Myelodysplastic Syndrome; NCT02979366; National Cancer Institute: Bethesda, MD, USA, 2017.
- Caenepeel, S.R.; Belmontes, B.; Sun, J.; Coxon, A.; Moody, G.; Hughes, P.E. Preclinical evaluation of AMG176, a novel, potent and selective Mcl-1 inhibitor with robust anti-tumor activity in Mcl-1 dependent cancer models. Cancer Res. 2017, 77. [Google Scholar] [CrossRef]
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar] [PubMed]
- Hird, A.W.; Secrist, J.P.; Adam, A.; Belmonte, M.A.; Gangl, E.; Gibbons, F.; Hargreaves, D.; Johannes, J.W.; Kazmirski, S.L.; Kettle, J.G.; et al. AZD5991: A potent and selective macrocyclic inhibitor of Mcl-1 for treatment of hematologic cancers. Cancer Res. 2017, 77. [Google Scholar] [CrossRef]
- Moujalled, D.M.; Pomilio, G.; Ghiurau, C.; Ivey, A.; Salmon, J.; Rijal, S.; Macraild, S.; Zhang, L.; Teh, T.C.; Tiong, I.S.; et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensitaffar, J.; Hogdal, L.J.; et al. A novel MCL-1 inhibitor combined with Venetoclax restores Venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [PubMed]
- Luedtke, D.A.; Niu, X.; Pan, X.; Zhao, J.; Liu, S.; Edwards, H.; Chen, K.; Lin, H.; Taub, J.W.; Ge, Y. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal. Transduct. Target Ther. 2017, 2, 17012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, N.S.; Bilardi, R.A.; Ng, A.P.; Xu, Z.; Robati, M.; Vandenberg, C.J.; Cory, S. Impact of elevated anti-apoptotic MCL-1 and BCL-2 on the development and treatment of MLL-AF9 in mice. Cell Death Differ. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Araghi, R.R.; Bird, G.H.; Ryan, J.A.; Jenson, J.M.; Godes, M.; Pritz, J.R.; Grant, R.A.; Letai, A.; Walensky, L.D.; Keating, A.E. Iterative optimization yelds Mcl-1-targeting stapled peptides with selective cytotoxicity to Mcl-1-dependent cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E886–E895. [Google Scholar] [CrossRef]
- Brennan, M.S.; Chang, C.; Tai, L.; Lessene, G.; Strasser, A.; Dewson, G.; Kelly, G.L.; Herold, M.J. Humanized Mcl-1 mice enable accurate pre-clinical evaluation of MCL-1 inhibitors destined for clinic al use. Blood 2018, in press. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Foster, M.C.; Blackford, A.L.; Litzow, M.R.; Morris, L.E.; Strickland, S.A.; Lancet, J.E.; Bose, P.; Levy, M.Y.; Tibes, R.; et al. Randomized multicenter phase II study of flavopiridol (alvocidib), cytarabine, and mitoxantrone (FLAM) versus cytarabine/daunorubicin (7+3) in newly diagnosed acute myeloid leukemia. Haematologica 2015, 100, 1172–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidner, J.F.; Foster, M.C.; Blackford, A.L.; Litzow, M.R.; Morris, L.E.; Strickland, S.A.; Lancet, J.E.; Bose, P.; Levy, M.Y.; Tibes, R.; et al. Final results of randomized multicenter phase II study of flavopiridol (alvocidib), cytarabine, and mitoxantrone (FLAM) versus cytarabine/daunorubicin (7+3) in newly diagnosed high-risk acute myeloid leukemia. Leuk. Res. 2018, 72, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Vigil, C.E.; Lin, T. Phase II study incorporating a novel BH3-profiling biomarker approach of alvocidib followed by cytarabine and mitoxantrone in relapsed/refractory acute myeloid leukemia (AML). In Proceedings of the 23rd Annual Congress of the European Hematology Association, Stockolm, Sweden, 14–17 June 2018. [Google Scholar]
- Zeidner, J.F.; Lin, T.L.; Vigil, C.E.; Dalovisio, A.; Wang, E.S.; Levy, M.Y.; Frattini, M.G.; Lee, D.J.; Fernandez, P.; Miguel, J.M.; et al. Zella 201: A biomarker-guided phase II study of alvocidib followed by cytarabine and mitoxantrone in MCL-1 dependent relapsed/refractory acute myeloid leukemia (AML). Blood 2018. [Google Scholar] [CrossRef]
- Dey, J.; Deckwerth, T.L.; Kerwin, W.S.; Casalini, J.R.; Merrell, A.J.; Grenley, M.O.; Burns, C.; Ditzler, S.H.; Dixon, C.P.; Beirne, E.; et al. Voriciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk diffuse large B-cell lymphoma to BCL2 inhibition. Sci. Rep. 2017, 7, 18007. [Google Scholar] [CrossRef] [PubMed]
- Boffo, S.; Damato, A.; Alfano, L.; Giordano, A. CDK9 inhibitors in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 36. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.; Gregory, G.P.; Verbrugge, I.; Kats, L.; Hilton, J.J.; Vidacs, E.; Lee, E.M.; Lock, R.B.; Zuber, J.; Shortt, J.; et al. The CDK9 inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of MLL-rearranged acute myeloid leukemia. Cancer Res. 2016, 76, 1158–1169. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–376. [Google Scholar] [CrossRef]
- Konopleva, M.; Konoplev, S.; Hu, W.; Konitskey, A.V.; Afamesic, B.V.; Andreef, M. Stromal cells prevent apoptosis of AMl cells by up-regulation of anti-apoptotic proteins. Leukemia 2002, 16, 1713–1724. [Google Scholar] [CrossRef]
- O’Reilly, E.; Dhami, S.; Baev, D.; Ortutay, C.; Halpin-McCormick, A.H.; Morell, R.; Santocanale, C.; Samali, A.; Quinn, J.; O’Dwyer, M.; et al. Repression of Mcl-1 expression by the CDC7/CDK9 inhibitor PHA-767491 overcomes bone marrow stroma-mediated drug resistance in AML. Sci. Rep. 2018, 8, 15752. [Google Scholar] [CrossRef]
- Fiskus, W.; Cai, T.; Di Nardo, C.; Kornblau, S.; Borthakur, G.; Kadia, T.; Pammaraju, N.; Bose, P.; Masorova, L.; Rajapakshe, K.; et al. Supeiror efficacy of cotreatment with BET protein inhibitor and BCL2 or MCL1 inhibitor agains AML bast progenitor cells. Blood Cancer J. 2019, 9, 4. [Google Scholar] [CrossRef]
- Kronke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minzel, W.; Venkatachalem, A.; Fink, A.; Hung, E.; Brachya, G.; Burstain, I.; Shaham, M.; Rivlin, A.; Omer, I.; Zinger, A.; et al. Small molecule co-targeting CKIα and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell 2018, 175, 171–185. [Google Scholar] [CrossRef]
- Powell, J.A.; Lewis, A.C.; Zhu, W.; Toubia, J.; Pitman, M.R.; Wallington-Beddoe, C.T.; Moretti, P.; Iarossi, D.; Samaraweera, S.E.; Cummings, N.; et al. Targeting sphingosine kinase 1 induces MCL1-dependent cell death in acute myeloid leukemia. Blood 2017, 129, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Toman, R.E.; Gorapaju, S.K.; Maceyka, M.; Nava, V.E.; Sankala, H.; Payne, S.G.; Bektas, M.; Ishii, I.; Chun, J.; et al. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J. Biol. Chem. 2003, 278, 40330–40336. [Google Scholar] [CrossRef]
- Powell, J.A.; Wallington-Beddoe, C.T.; Pitson, S.M. Targeting sphingosine kinase 1 in acute myeloid leukemia: Translation to clinic. Int. J. Hematol. Oncol. 2017, 6, 31–34. [Google Scholar] [CrossRef]
- Hengst, J.A.; Dick, T.E.; Sharma, A.; Doi, K.; Hegde, S.; Tan, S.F.; Geffert, L.M.; Fox, T.E.; Sharma, A.K.; Desai, D.; et al. SKI-178: A multitargeted inhibitor of sphingosine kinase and microtubule dynamics demonstrating therapeutic efficacy in acute myeloid leukemia models. Cancer Transl. Med. 2017, 3, 109–121. [Google Scholar]
- Testa, U.; Labbaye, C.; Castelli, G.; Pelosi, E. Oxidative stress and hypoxia in normal and leukemia stem cells. Exp. Hematol. 2016, 44, 540–560. [Google Scholar] [CrossRef]
- Chen, W.L.; Wang, J.H.; Zhao, A.H.; Xu, X.; Wang, Y.H.; Chen, T.L.; Li, J.M.; Mi, J.Q.; Zhu, Y.M.; Liu, Y.F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.L.; Wang, Y.Y.; Zhao, A.; Xia, L.; Xie, G.; Su, M.; Zhao, L.; Liu, J.; Qu, C.; Wei, R.; et al. Enhanced fructose utilization mediated by SLCA5 is a unique metabolic feature of acute myeloid leukemia with therapeutic potential. Cancer Cell 2016, 30, 779–791. [Google Scholar] [CrossRef]
- Braun, M.; Qorraj, M.; Buttner, M.; Klein, F.A.; Saul, D.; Aigner, M.; Huber, W.; Mackensen, A.; Jitschin, R.; Mougiakos, D. CXCL2 promotes glycolytic reprogramming in acute myeloid leukemia via the CXCR4/mTOR axis. Leukemia 2016, 30, 1740–1792. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Pak, S.; Hall, M.N. mTOR signaling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Poulain, L.; Sujobert, P.; Zylberstejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.L.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemic cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.; Dutta, S.; Opatz, S.; Vosberg, S.; Reiter, K.; Leubolt, G.; Metzeler, K.H.; Herold, T.; Bamopoulos, S.; Braundl, K.; et al. ZBTB7A mutations in acute myeloid leukemia with t(8;21) translocation. Nat. Commun. 2016, 7, 11733. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Israelsen, W.J.; Lee, D.; Yu, V.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Heiden, M.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Coelho, F.; Nunes, C.; Gouveia-Fernandes, S.; Rosas, R.; Silva, F.; Gameiro, P.; Cravalho, T.; Gomes da Silva, M.; Cadecadas, J.; Dias, S.; et al. Monocarboxylate transporter 1 (MCT1), a tool to stratify acute myeloid leukemia (AML) patients and a vehicle to kill cancer cells. Oncotarget 2017, 8, 82803–82823. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, A.I.; MacGregor, G.G. Glucose-dependent growth arrest of leukemia cells by MCT1 inhibition: Feeding Warburg’s sweet tooth and blocking acid export as anticancer strategy. Biomed. Pharmacother. 2018, 98, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 2018, 25, 3047–3058. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwy, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Jigl, S.; Reidel, V.; Muller-Thomas, C.; Konig, J.; Schanaecker, J.; Hockendorf, U.; Huberle, C.; Gorka, O.; Schmidt, B.; Burghart, R.; et al. Blockade of BCL-2 proteins efficiently induces apoptosis in progenitor cells of high-risk myelodysplastic syndromes patients. Leukemia 2016, 30, 112–123. [Google Scholar]
- Stevens, B.M.; Khan, N.; D’Alessandro, A.; Nemkov, T.; Winters, A.; Jones, C.L.; Zhang, W.; Pollyea, D.A.; Jordan, C.T. Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes. Nat. Commun. 2018, 9, 3694. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mendoza, S.E.; Rego, E.M. Targeting the mitochondria in acute myeloid leukemia. Appl. Cancer Res. 2017, 37, 22. [Google Scholar] [CrossRef]
- Amaya, M.L.; Pollyea, D.A. Targeting the IDH2 pathway in acute myeloid leukemia. Clin. Cancer Res. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Akhtari, M.; Alachkar, H. Characterization of mutations in mitochondrial encoded electron transport chain complexes in acute myeloid leukemia. Sci. Rep. 2018, 8, 13301. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; De Dios, I.; Funari, V.; Sudarsanam, S.; Jiang, S.P.; Agersborg, S.; Hummel, J.; Blocker, F.; Albitar, M. IDH1 and IDH2: Founding or progressor mutations in myeloid neoplasms. Blood 2017, 130, 405. [Google Scholar]
- Falini, B.; Spinelli, O.; Meggendorfer, M.; Martelli, M.P.; Bigerna, B.; Ascani, S.; Stein, H.; Rambaldi, A.; Haferlach, A. IDH1-R132 changes vary according to NPM1 and othe mutations status in AML. Leukemia 2019, in press. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Cappelli, L.V.; Walter, W.; Haferlach, C.; Kern, W.; Falini, B.; Haferlach, T. IDH1R132, IDH2R140 and IDH2R172 in AML: Different genetic landscapes correlate with outcome and may influence targeted treatment strategies. Leukemia 2018, 32, 1249–1253. [Google Scholar] [CrossRef]
- Xu, Q.; Li, Y.; Lv, N.; Xu, Y.; Li, Y.; Li, W.; Yao, Z.; Chen, X.; Huang, S.; et al. Correlation between isocitrate dehydrogenase gene aberrations and prognosis of patients with acute myeloid leukemia: A systematic review and meta-analysis. Clin. Cancer Res. 2017, 23, 4511–4522. [Google Scholar] [CrossRef]
- Di Nardo, C.D.; Rausch, C.R.; Benton, C.; Kadia, T.; Jain, N.; Pemmaraju, N. Clinical experience with the BCL-2 inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am. J. Hematol. 2018, 93, 401–407. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable remissions with Ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Roboz, G.J.; DiNardo, C.; Stein, E.M.; deBotton, S.; Mims, A.; Prince, G.T.; Altman, J.; Arellano, M.; Donnellan, W.; Erba, H.; et al. Ivosidenib (AG-220) induced durable remissions and transfusion independence in patients with IDH1-mutant untreated AML: Results from a phase 1 dose-escalation and expansion study. Blood 2018, 132, 561. [Google Scholar]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2018, in press. [Google Scholar] [CrossRef]
- Quek, L.; David, M.D.; Kennedy, A.; Metzner, M.; Amatangelo, M.; Shih, A.; Stoilova, B.; Quivoron, C.; Heiblig, M.; Willekens, C.; et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 2018, 24, 1167–1177. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenbug, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schwartzman, J.M.; Hou, S.; Famulare, C.; Patel, M.; Roshal, M.; Do, R.K.; Zehir, A.; et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov. 2018, in press. [Google Scholar] [CrossRef]
- Norsoworthy, K.; Mulkey, F.; Word, A.; Przepiorka, D.; Deisseroth, A.; Farrell, A.; Farrell, A.; Pazdur, R. Incidence of differentiation syndrome with Ivosidenib (IVO) and Enasidenib (ENA) for treatment of patients with relapsed or refractory (R/R) isocitrate dehydrogenase (IDH)1- or IDH2-mutated acute myeloid leukemia (AML): A systematic analysis by the U.S. Food and Drug Administration (FDA). Blood 2018, 132, 288. [Google Scholar]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–390. [Google Scholar] [CrossRef]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukemia. Nature 2017, 545, 500–505. [Google Scholar] [CrossRef]
- Green, A.S.; Chapuis, N.; Maciel, T.T.; Willems, L.; Lambert, M.; Arnoult, C.; Boyer, O.; Bardet, V.; Park, S.; Foretz, M.; et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood 2010, 116, 4262–4273. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, I.; Mitsumori, T.; Nozaki, Y.; Yamamoto, T.; Shobu-Sueki, Y.; Nakajima, K.; Kirito, K. Negative regulation of the LKB1/AMPK pathway by ERK in human acute myeloid leukemia cells. Exp. Hematol. 2015, 43, 524–533. [Google Scholar] [CrossRef]
- Sujobert, P.; Poulain, L.; Paubelle, E.; Zylberstein, F.; Grenier, A.; Lambert, M.; Twosend, E.C.; Brusq, J.M.; Nicodeme, E.; Decrooqc, J.; et al. Co-activation of AMPK and mTORC1 induces cytotoxicity in acute myeloid leukemia. Cell Rep. 2015, 11, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell 2015, 17, 585–596. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell. Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FISI1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell 2018, 23, 86–100. [Google Scholar] [CrossRef]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Pratz, K.; Pullarkat, K.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Ventoclax combined with decitabine or azacytidine in treatment-naïve, elderly patients with acute myeloid leukemia. Blood 2018, in press. [Google Scholar]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacytidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; LeMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyver, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liyanage, S.U.; Hurren, R.; Voisin, V.; Bridon, G.; Wang, X.; Xu, C.J.; MacLean, N.; Siriwardena, T.P.; Gronda, M.; Yehudai, D.; et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood 2017, 129, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Acroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phopshorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Baran, N.; Lodi, A.; Sweeney, S.R.; Renu, P.; Kuruvilla, V.M.; Cavazos, A.; Herranz, D.; Skwarska, A.; Warmoes, M.; Davis, E.; et al. Mitochondrial Complex I inhibitor IACS-010759 reverses the NOTCH1-driven metabolic reprogramming in T-ALL via blockade of oxidative phosphorylation: Synergy with chemotherapy and glutaminase inhibition. Blood 2018, 132, 4020. [Google Scholar]
- Di Francesco, M.E.; Marszalek, J.R.; McAfoos, T.; Carroll, C.L.; Cang, Z.; Liu, G.; Theroff, J.; Bardenhager, J.P.; Bandi, M.L.; Molina, J.R.; et al. Discovery and development of IACS-010759, a novel inhibitor of Complex I currently in phase I studies to exploit oxidative phosphorylation dependency in acute myeloid leukemia and solid tumors. Cancer Res. 2018, 78, 1655. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseine, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.L.; Trinh, D.L.; Ries, R.E.; Wang, J.; Gerbing, R.B.; Ma, Y.; Topham, J.; Hughes, M.; Pleasance, E.; Mungall, A.J.; et al. MicroRNA expression-based model indicates event-free survival in pediatric acute myeloid leukemia. J. Clin. Oncol. 2017, 35, 3964–3977. [Google Scholar] [CrossRef] [PubMed]
- Sperb, N.; Krowiorz, K.; Grasiedeck, S.; Rueb, C.; Pochert, N.; Schneider, E.; Johansson, P.; Malmberg, E.D.; Arabanian, L.; Maetzig, T.; et al. Members of the microRNA-106a-363 cluster associate with unfavorable outcome in adult acute myeloid leukemia patients and promote leukemogenesis in vivo through increased metabolic activity. Blood 2018, 132, 3924. [Google Scholar]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, M.; Sun, Y.; Hellmich, C.; Marlein, C.R.; Mistry, J.; Forde, E.; Piddock, R.E.; Shafat, M.S.; Morfakis, A.; Mehta, T.; et al. Acute myeloid leukemia induces pro-tumoral p16INK4a driven senescence in the bone marrow microenvironment. Blood 2018, in press. [Google Scholar]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [Green Version]
- Fenouille, N.; Bassil, C.F.; Ben-Sahra, I.; Banajiba, L.; Alexa, G.; Ramos, A.; Pikman, Y.; Conway, A.S.; Burgess, M.R.; Li, Q.; et al. The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat. Med. 2017, 23, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benajiba, L.; Alexe, G.; Su, A.; Raffoux, E.; Soulier, J.; Hemann, M.T.; Hermien, O.; Itzykson, R.; Stegmaier, K.; Puisant, A. Creatine kinase pathway inhibition alters GSK and WNT signaling in EVI1-positive AML. Leukemia 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Meatabol. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardee, T.S.; Lee, K.; Luddy, J.; Maturo, C.; Rodriguez, R.; Isom, S.; Miller, L.D.; Stadelman, K.M.; Levitan, D.; Hurd, D.; et al. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2014, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghirardeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A phase I study of CPI-613 in combination with high-dose cytarabine and mitoxantrone for relapsed or refractory acute myeloid leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef]
- Pardee, T.S.; Isom, S.; Ellis, L.R.; Howard, D.S.; Bhave, R.; Manuel, M.; Dralle, S.; Lyerly, D.S.; Powell, B.L. Therapeutic manipulation of cancer cell metabolism with the mitochondrial inhibitor CPI-613 in addition to chemotherapy abrogates the adverse prognostic effect of age in relapsed and refractory AML. Blood 2018, 132, 1355. [Google Scholar]
- Ju, H.Q.; Zhan, G.; Huang, A.; Sun, Y.; Wen, S.; Yang, J.; Lu, W.; Xu, R.; Li, J.; Li, Y.; et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia 2017, 31, 2143–2150. [Google Scholar] [CrossRef] [Green Version]
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.; Vohra, S.; Medina-Perez, P.; Basheer, F.; Marando, L.; Di Lisio, L.; Dias, J.; et al. Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018, 131, 1639–1653. [Google Scholar] [CrossRef]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, M.A.; Nemkov, T.; Reisz, J.A.; Zaberezhnyy, V.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp. Hematol. 2018, 58, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; D’Alessandro, A.; Alvarez-Calderon, F.; Kim, J.; Nemkov, T.; Rozhok, A.I.; Kumar, A.; Kumar, V.; Pollyea, D.A.; Wempe, M.F.; et al. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, E6669–E6678. [Google Scholar] [CrossRef] [PubMed]
- Stockard, B.; Garrett, T.; Guingab-Cagmat, J.; Meshinchi, S.; Lamba, J. Distinct metabolic features differentiating FLT3-ITD AML from FLT3-WT childhood acute myeloid leukemia. Sci. Rep. 2018, 8, 5534. [Google Scholar] [CrossRef] [PubMed]
- Hospital, M.A.; Jacquel, A.; Mazed, F.; Saland, E.; Larrue, C.; Mondesir, J.; Birsen, R.; Green, A.S.; Lambert, M.; Sujobert, P.; et al. RSK2 is a new Pim2 target with pro-survival functions in FLT3-ITD-positive acute myeloid leukemia. Leukemia 2018, 32, 597–605. [Google Scholar] [CrossRef]
- Nelson, J.; Qurashi, A.; Ghole, V.S.; Deshmukh, D.R. Regulation of orotic acid biosynthesis and excretion induced by oral glutamine administration in mice. Biochem. Med. Met. Biol. 1993, 49, 338–350. [Google Scholar] [CrossRef]
- Sykes, D.B.; Kfoury, Y.S.; Mercier, F.E.; Wawer, M.J.; Law, J.M.; Haynes, M.K.; Lewis, T.A.; Schnajnovitz, A.; Jain, E.; Lee, D.; et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell 2016, 167, 171–186. [Google Scholar] [CrossRef]
- Sykes, D.B. The emergence of dihydrooratate dehydrogenase (DHODH) is a therapeutic target in acute myeloid leukemia. Exp. Opin. Ther. Targets 2018, 22, 893–898. [Google Scholar] [CrossRef]
- Lewis, T.A.; Sykes, D.B.; Law, J.M.; Munoz, B.; Rustiquel, J.K.; Scadden, D.T.; Schreiber, S.L. Development of ML390: A human DHODH inhibitor that induces differentiation in acute myeloid leukemia. ACS Med. Chem. Lett. 2016, 7, 1112–1117. [Google Scholar] [CrossRef]
- Sainas, S.; Pippione, A.C.; Lupino, E.; Giorgis, M.; Circosta, P.; Gaidano, V.; Goyal, P.; Bonanni, D.; Rolando, B.; Cignetti, A.; et al. Targeting myeloid differentiation using potent 2-hydroxypyrazolo (1,5-a) pyridine scaffold-based human dihydroorotate dehydrogenase inhibitors. J. Med. Chem. 2018, 61, 6034–6055. [Google Scholar] [CrossRef]
- Janzer, A.; Gradi, S.; Christian, S.; Zimmermann, K.; Merz, C.; Meyer, H.; Stellfeld, T.; Guenther, J.; Stoeckigt, D.; Seidel, H.; et al. BAY 2402234: A novel, selective dihydroorotate dehydrogenase (DHODH) inhibitor for the treatment of myeloid malignancies. Cancer Res. 2018, 78. [Google Scholar] [CrossRef]
- Zhou, J.; Quah, J.Y.; Chooi, J.Y.; Toh, S.; Ng, Y.; Lin, B.; Osato, M.; Seet, Q.; Ooi, L.; Lindmark, B.; et al. ASLAN003, a novel and potent dihyroorotate dehydrogenase (DHODH) inhibitor, induces differentiation of acute myeloid leukemia. Blood 2018, 132, 4047. [Google Scholar]
- Ting, S.B.; Ng, C.H.; Bajel, A.; Hwang, W.; Ho, S.J.; Chng, W.J.; McHale, M.; Hsieh, C.Y.; Shih, H.J.; McIntyre, N.; et al. Preliminary results of a phase 2a dose optimization study of ASLAN003 (DHODH inhibitor) in acute myeloid leukemia (AML) patients who are ineligible for standard therapy: Early signs of activity. Blood 2018, 132, 2676. [Google Scholar]
- Wu, D.; Wang, W.; Chen, W.; Lian, F.; Lang, L.; Huang, Y.; Xu, Y.; Zhang, N.; Chen, Y.; Liu, M.; et al. Pharmacological inhibition of dihydroorotate dehydrogenase induces apoptosis and differentiation in acute myeloid leukemia cells. Haematologica 2018, 103, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Khurtonenko, A.A.; Roudko, V.V.; Chernyak, B.V.; Vartapetian, A.B.; Chumakov, P.M.; Evstafieva, A.G. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12828–12833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladds, M.; van Leuwen, I.; Drummond, C.; Chu, S.; Haely, A.; Popova, G.; Pastor Fernandez, A.; Mollick, T.; Darekar, S.; Sedimbi, S.K.; et al. A DHODH inhibitor increases p53 synthesis and enhances tumor cell killing by p53 degradation blockage. Nat. Commun. 2018, 9, 1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Weetall, M.; Trotta, C.; Cintron, C.; Ma, J.; Kim, M.J.; Furia, B.; Romfo, C.; Graci, J.D.; Li, W.; et al. Targeting of hematologic malignancies with PTC299, a novel potent inhibitor of dihydroorotate dehydrogenase with favorable pharmaceutical properties. Mol. Cancer Ther. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Kfoury, Y.; Aziz, N.; Elkhoury, J.; Hallgren, B.; Scadden, D.T.; Heiden, M.V.; Sykes, D.B. DHODH inhibitors in the treatment of acute myeloid leukemia: Defining the mechanism of action and the basis of the metabolic therapeutic window. Blood 2018, 132, 2716. [Google Scholar]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G.; Godley, L.A.; Koivunen, P. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Goncalves, E.; Zecchni, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; Rajeeve, V.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [Green Version]
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubachova, S.; Endaya, B.; Judasova, K.; et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab. 2019, 29, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Campos-Sandoval, J.A.; Marquez, J. Glutaminase isoenzymes in the metabolic therapy of cancer. Biochem. Biophys. Acta Rev. Cancer 2018, 1870, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Meng, Y.; Li, L.; Xu, P.; Wang, J.; Li, Z.; Bian, J. Overview of the development of glutaminase inhibitors: Achievements and future directions. J. Med. Chem. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Jong, M.; Kim, S.H.; Im, C.Y.; Hwang, H.J. Recent development of small molecule glutaminase inhibitors. Curr. Top. Med. Chem. 2018, 18, 432–443. [Google Scholar]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, M.; Miwa, H.; Shikami, M.; Tsunekawa-Imai, N.; Suganuma, K.; Mizuno, S.; Takahashi, M.; Mizutani, M.; Hanamura, I.; Nitta, M. Importance of glutamine metabolism in leukemia cells by energy production through TCA cycle and by redox homeostasis. Cancer Investig. 2014, 326, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennet, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 7022, 8981–8987. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 465, 966–970. [Google Scholar] [CrossRef]
- Emadi, A.; Jun, S.A.; Tsukamoto, T.; Fathi, A.T.; Minden, M.D.; Dang, C.V. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp. Hematol. 2014, 424, 247–251. [Google Scholar] [CrossRef]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79734. [Google Scholar] [CrossRef]
- Wang, E.S.; Frankfurt, O.; Orford, K.W.; Bennett, M.; Flinn, I.W.; Maris, M.; Konopleva, M. Phase 1 study of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase in patients with relapsed/refractory leukemia. Blood 2015, 126, 2566. [Google Scholar]
- Cai, T.; Lorenzi, P.L.; Pontikos, M.A.; Lodi, A.; Han, L.; Zhang, Q.I.; Ma, H.; Rahmani, M.; Bhagat, T.D.; Horvath, T.D.; et al. Gls inhibitor CB-839 modulates cellular metabolism in AML and potently suppresses AML cell growth when combined with 5-Azacitine. Blood 2016, 128, 4064. [Google Scholar]
- Gregory, M.A.; Nemkov, T.; Zaberezhnyy, V.; Park, H.J.; Gehrke, S.; Hansen, K.C.; D’Alessandro, A.D.; DeGregori, J. Targeting glutamine metabolism and redox state for leukemia therapy. bioRxiv 2018. [Google Scholar] [CrossRef]
- Emadi, A.; Law, J.Y.; Strovel, E.T.; Lapidus, R.G.; Jeng, L.J.B.; Lee, M.; Blitzer, M.G.; Carter-Cooper, B.A.; Sewell, D.; et al. Asparaginase Erwinia chrysanthemi effectively depletes plasma glutamine in adult patients with relapsed/refractory acute myeloid leukemia. Cancer Chemother. Pharmacol. 2018, 81, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Lorenzi, P.L.; Anishkin, A.; Purwalha, P.; Sukharev, S.; Rempe, S.B.; Weinstein, J.N. The glutaminase activity of L-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood 2014, 123, 3596–3606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.A.; Su, Y.; Zhang, J.Y.; Antanasijevic, A.; Caffrey, M.; Schalk, A.M.; Liu, L.; Rondelli, D.; Oh, A.; Mahmud, D.L.; et al. A novel l-Asparaginase with low l-Glutaminase coactivity is highly efficacious against both T- and B-cell acute lymphoblastic leukemias in vivo. Cancer Res. 2018, 78, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- CXhan, W.K.; Tan, L.; Harutyunyan, K.; Du, D.; Marin, L.; Yin, E.; Konopleva, M.; Weinstein, J.; Lorenzi, P.L. The glutaminase activity of L-Asparaginase mediates suppression of Asns upregulation. Blood 2018, 132, 3959. [Google Scholar]
- Bertuccio, S.N.; Serravalle, S.; Astolkfi, A.; Lonetti, A.; Indio, V.; Leszl, A.; Pession, A.; Melchionda, F. Identification of a cytogenetic and molecular subgroup of acute myeloid leukemias showing sensitivity to L-Asparaginase. Oncotarget 2017, 8, 109915–109923. [Google Scholar] [CrossRef]
- Nakamura, A.; Nambu, T.; Ebara, S.; Hasegawa, Y.; Toyoshima, K.; Tsuchiya, Y.; Tomita, D.; Fujimoto, J.; Kurasawa, O.; Takahara, C.; et al. Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc. Natl Acad. Sci. USA 2018, 115, E7776–E7785. [Google Scholar] [CrossRef]
- Miroki-Moud, F.; Ghazaly, Z.; Ariza-McNaughton, L.; Hody, K.A.; Clear, A.; Anjos-Afonso, F.; Liapis, K.; Granthan, M.; Sahzabi, F.; Cavenagh, J.; et al. Arginine deprivation using pegylated arginine deaminase has activity against acute myeloid leukemia cells in vivo. Blood 2015, 125, 4060–4068. [Google Scholar] [CrossRef]
- Mussai, F.; Egan, S.; Higginbutham-Jones, J.; Perry, T.; Beggy, A.; Odintsova, E.; Loke, J.; Pong, K.; Lo, A.; Ng, M.; et al. Arginine dependence of acute myeloid leukemia blast proliferation: A novel therapeutic target. Blood 2015, 125, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.T.; Qi, Y.; Wang, Y.C.; Chi, K.K.; Chung, Y.; Ouyang, C.; Chen, Y.R.; Oh, M.E.; Sheng, X.; Tang, Y.; et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun. Biol. 2018, 1, 178. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.J.; Jiang, S.S.; Hung, W.C.; Borthakur, G.; Lin, S.F.; Pemmaraju, N.; Jabbour, E.; Bomalaski, J.S.; Chen, Y.P.; Hsiao, H.H.; et al. A phase II study of arginine deaminase (ADI-PEG20) in relapsed/refractory or poor risk acute myeloid leukemia patients. Sci. Rep. 2018, 7, 11253. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Queck, L.; McEwen-Smith, R.; Qurweshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, F.; Zhang, Y.; Li, X.; Chen, C.; Zhou, M.; Yu, Z.; Liu, Y.; Zhao, Y.; Hao, X.; et al. PPM1K regulates hematopoiesis and leukemogenesis through CDC20-mediated ubiquitination of Meis1 and p21. Cell Rep. 2018, 23, 1461–1475. [Google Scholar] [CrossRef]
- Jeyaraju, D.V.; Voisin, V.; Xu, C.; Barghout, S.H.; Khan, D.H.; Hurren, R.; Gronda, M.; Wang, X.; Jitkova, Y.; Sharon, D.; et al. Inhibiting the mitochondrial sulfhyryl oxidase ALR reduces Cox17 and alters mitochondrial crista structure leading to the differentiation of AML and stem cells. Blood 2017, 130, 881. [Google Scholar]
- Singh, R.P.; Jeyaraju, D.V.; Voisin, V.; Xu, C.; Barghout, S.H.; Khan, D.H.; Hurren, R.; Gronda, M.; Wang, X.; Jitkova, Y.; et al. Targeting the mitochondrial metallochaperone Cox17 reduces DNA methylation and promotes AML differentiation through copper dependent mechanism. Blood 2018, 132, 1339. [Google Scholar]
- Shi, J.; Fu, H.; Jia, Z.; He, K.; Fu, L.; Wang, W. High expression of CPT1A predicts adverse outcomes: A potential therapeutic target for acute myeloid leukemia. EBiomedicine 2016, 14, 55–64. [Google Scholar] [CrossRef]
- Samudio, I.; Hamoncey, R.; Fiegl, M.; Kantarjan, H.; Konopleva, M.; Korchin, B.; Kaluicrachchi, K.; Bormann, W.; Duvvuri, S.; Tagetmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, M.R.; Mirabilli, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef] [PubMed]
- German, N.J.; Yoon, H.; Yusuf, R.Z.; Murphy, J.P.; Finley, L.; Laurent, G.; Haas, W.; Statterstrom, K.; Guarnerio, J.; Zagonjor, E.; et al. PHD3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2. Mol. Cell 2016, 63, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sakihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Saitoh, K.; Yang, H.; Sekihara, K.; Yamatani, K.; Ruvolo, V.; Taka, H.; Kaga, N.; Kikkawa, M.; Arai, H.; et al. Inhibition of FAO in AML co-cultured with BM adipocytes: Mechanisms of survival and chemosensitization to cytarabine. Sci. Rep. 2018, 8, 16837. [Google Scholar] [CrossRef] [PubMed]
- Piragyte, I.; Clapes, T.; Polyzou, A.; Geltink, R.K.; Lefkopoulos, S.; Yin, N.; Cauchy, P.; Curtis, J.D.; Klaeylé, L.; Lang, X.; et al. A metabolic interplay coordinated by HLX regulates myeloid differentiation and AML through partly overlapping pathways. Nat. Commun. 2018, 9, 3090. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, D.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.H.; et al. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Limboussaki, A.; Gemelli, C.; Testa, A.; Facchni, G.; Ferrari, F.; Mavilio, F.; Grande, A. PPAR-deltas is a ligand-dependent negative regulator of vitamin D3-induced monocyte differentiation. Carcinogenesis 2009, 30, 230–237. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Alexeev, E.; Nusbacher, N.; Minhajuddin, M.; Stevens, B.M.; Winters, A.C.; Lin, X.; Ashton, J.M.; et al. Subversion of systemic glucose metabolism as a mechanism to support the growth of leukemia cells. Cancer Cell 2018, 34, 659–673. [Google Scholar] [CrossRef]
- Spratlin, J.L.; Serkova, N.J.; Eckhardt, S.G. Clinical applications of metabolomics in oncology. Clin. Cancer Res. 2009, 15, 431–440. [Google Scholar] [CrossRef]
- Nagrath, D.; Caneba, C.; Karedath, T.; Bellance, N. Metabolomics for mitochondrial and cancer studies. Biochim. Biophys. Acta 2011, 1807, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.K.; DeBerardinis, R.J. Applications of metabolomics to study cancer metabolism. BBA Rev. Cancer 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Stuani, L.; Riols, F.; Millard, P.; Sabatier, M.; Batut, A.; Saland, E.; Viars, F.; Tonini, L.; Zaghdoudi, S.; Linares, L.K.; et al. Stable isotope labeling highlights enhanced fatty acid and lipid metabolism in human acute myeloid leukemia. Int. J. Mol. Sci. 2018, 19, 3325. [Google Scholar] [CrossRef]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Koirsmeyer, S.; Armstrong, S.A.; Leatai, A. Mitochondrial primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.; Montero, J.; Rocco, J.; Letai, A. iBH3: Simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol. Chem. 2016, 397, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.A.; Brunelle, J.K.; Letai, A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 12895–12900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, J.; Sarosiek, K.A.; De Angelo, J.D.; Maertens, O.; Ryan, J.; Ercan, D.; Piao, H.; Horowitz, N.S.; Berkowitz, R.S.; Matulonis, O.; et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 2015, 160, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Stephausky, J.; Cai, T.; Griffin, J.K.; Cabal-Hierre, L.; Togani, K.; Hegdal, L.G.; Galinsky, I.; Morgan, E.A.; Aster, J.C.; et al. Blastic plasmocytoid dendritic cell neoplasm is dependent on BCL2 and sensitive to Venetoclax. Cancer Discov. 2017, 7, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Letai, A. Dynamic BH3 profiling-poking cancer cells with a stick. Mol. Cell Oncol. 2016, 3, e1040144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Mochizuki, Y.; Ogahara, I.; Watanabe, T.; Hogdal, L.; Takagi, S.; Sato, K.; Keneko, A.; Kajita, H.; Uchida, N.; et al. Overcoming mutational complexity in acute myeloid leukemia by inhibition of critical pathways. Sci. Transl. Med. 2017, 9, 1214. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.; Laumann, K.; Geyer, S.; Bloomfield, C.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Pallis, M.; Burrows, F.; Ryan, J.; Grundy, M.; Abdul-Aziz, A. Complementary dynamic BH3 profiles predict co-operativity between the multi-kinase inhibitor TG0” and the BH3 mimetic ABT-199 in acute myeloid leukemia cells. Oncotarget 2017, 8, 16220–16232. [Google Scholar] [CrossRef] [PubMed]
- Grundy, M.; Seedhouse, C.; Jones, T.; Elmi, L.; Hasll, M.; Graham, A.; Russell, N.; Pallis, M. Predicting effective pro-apoptotic anti-leukaemic drug combinations using co-operative dynamic BH3 profiling. PLoS ONE 2018, 13, e0190682. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, J.; Kojima, K.; McQueen, T.; Ruvolo, V.; Chachad, D.; Nogueras-Gonzalez, G.M.; Huang, X.; Pierceall, W.E.; Dettman, E.J.; Cardone, M.H.; et al. Mitochondrial profiling of acute myeloid leukemia in the assessment of response to apoptosis modulating drugs. PLoS ONE 2015, 10, e0138377. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Roberts, A.; Juarez, D.; Vo, T.T.; Bhatt, S.; Herzog, L.O.; Mallya, S.; Bellin, R.J.; Agarwal, S.K.; Salem, A.H.; et al. Statins enhance efficacy of venetoclax in blood cancer. Sci. Transl. Med. 2018, 10, eaaq1240. [Google Scholar] [CrossRef] [PubMed]
- Von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; Von Goetz, M.; Yee, S.F.; Toptal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the apoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar] [CrossRef]
- Dufour, F.; Rattier, T.; Shirley, S.; Picarda, G.; Constantinescu, A.A.; Morle, A.; Zakaria, A.B.; Marcion, G.; Causse, S.; Szegezdi, E.; et al. N-glycosylation of mouse TRAIL-R and human TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ. 2017, 24, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Micheau, O. Regulation of TNF-regulated apoptosis-inducing ligand signaling by glycosylation. Int. J. Mol. Sci. 2018, 19, 715. [Google Scholar] [CrossRef] [PubMed]
- Wuchter, C.; Krappmann, D.; Cai, Z.; Ruppert, V.; Scheidereit, C.; Dorken, B. In vitro susceptibility to TRAIL-induced apoptosis of acute leukemia cells in the context of TRAIL receptor gene expression and constitutive NF-kB activity. Leukemia 2001, 15, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Riccioni, R.; Pasquini, L.; Mariani, G.; Saulle, E.; Rossini, A.; Diverio, D.; Pelosi, E.; Vitale, A.; Chierichini, A.; Cedrone, M.; et al. TRAIL decoy receptors mediate resistance of acute myeloid leukemia cells to TRAIL. Haematologica 2005, 90, 612–624. [Google Scholar] [PubMed]
- Cappellini, A.; Mantovani, I.; Tazzari, P.L.; Grafone, T.; Martinelli, G.; Cocco, L.; Martelli, A.M. Application of flow cytometry to molecular medicine: Detection of tumor necrosis factor-related apoptosis-inducing ligand receptors in acute myeloid leukemia blasts. Int. J. Mol. Med. 2005, 16, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Riccioni, R.; Pelosi, E.; Riti, V.; Castelli, G.; Lo-Coco, F.; Testa, U. Immunophenotypic features of acute myeloid leukemia patients exhibiting high FLT3 expression not associated with mutations. Br. J. Haematol. 2011, 153, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Nuebling, T.; Wild, J.; Jung, J.; Kroell, T.; Kanz, L.; Salih, H.R.; Schmetzer, H. Death receptor expression in blasts in AML is associated with unfavorable prognosis. Anticancer Res. 2015, 35, 4043–4052. [Google Scholar]
- Chalumeau, M.E.; Ossenkoppele, G.J.; van Rhenhen, A.; van Dreunen, L.; Jirka, S.M.; Zevenbergen, A.; Schuurhuis, G.J.; Loosdrecht, A.A. High TRAIL-R3 expression on leukemic blasts is associated with poor outcome and induces apoptosis-resistance which can be overcome by targeting TRAIL-R2. Leuk. Res. 2011, 35, 741–749. [Google Scholar] [Green Version]
- Suh, W.S.; Kim, Y.S.; Schimmer, A.D.; Kitada, S.; Minden, M.; Andreef, M.; et al. Synthetic triterpenoids activate a pathway for apoptosis in AML cells involving downregulation of FLIP and sensitization to TRAIL. Leukemia 2003, 17, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Carter, B.Z.; Mak, D.H.; Schober, W.D.; Dietrich, M.F.; Pinilla, C.; Vassilev, L.T.; Reed, J.C.; Andreef, M. Triptolide sensitizes AML cells to TRAIL-induced apoptosis via decrease of XIAP and p53-mediated increase of DR5. Blood 2008, 111, 3742–3750. [Google Scholar] [CrossRef] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Coe, S.; Grant, S. The multikinase inhibitor sorafenib potentiates TRAIKL lethality in human leukemia cells in association wiith Mcl-1 and cFLIPL down-regulation. Cancer Res. 2007, 67, 9490–9500. [Google Scholar] [CrossRef]
- Tazzari, P.L.; Tabellini, G.; Ricci, F.; Papa, V.; Bortul, R.; Chiarini, F.; Evangelisti, C.; Martinelli, G.; Bontadini, A.; Cocco, L.; et al. Synergistic proapoptotic activity of recombinant TRAIL plus the AKT inhibitor perifosine in acute myelogenous leukemia cells. Cancer Res. 2008, 68, 9394–9403. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Aloisio, F.; De Gabrieli, A.; Consalvo, M.I.; Lavorgna, S.; Voso, M.T.; Lo-Coco, F.; Graziani, G. The poly(ADP-ribose) polymerase inhibitor olaparib induces up-regulation of death receptors in primary acute myeloid leukemia blasts by NF-kB activation. Cancer Lett. 2018, 423, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, G.; Mirandola, P.; Carubbi, C.; Micheloni, C.; Malinverno, C.; Lunghi, P.; Bonati, A.; Vitale, M. Phorbol ester-induced PKC epsilon down-modulation sensitizes AML cells to TRAIL-induced apoptosis and cell differentiation. Blood 2009, 113, 3080–3087. [Google Scholar] [CrossRef] [PubMed]
- DiMarcantonio, D.; Martinez, E.; Sidoli, S.; Vadaketh, J.; Nieborowska-Skorska, M.; Gupta, A.; Meadoes, J.M.; Ferraro, F.; Masselli, E.; Challen, G.A.; et al. Protein kinase C epsilon is a key regulator of mitochondrial redox homeostasis in acute myeloid leukemia. Clin. Cancer Res. 2018, 24, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Haimovichi, A.; Humbert, M.; Federzoni, E.A.; Shan-Krauer, D.; Brunner, T.; Frese, S.; Kaufmann, T.; Torbett, B.E.; Tschan, M.P. PU.1 supports TRAIL-induced cell death by inhibiting NF-kB-mediated cell survival and inducing DR5 expression. Cell Death Differ. 2017, 24, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, D.; Ryoo, H.D. Regulation of cell death by IAPs and their antagonists. Curr. Top. Dev. Biol. 2015, 114, 185–208. [Google Scholar] [PubMed]
- Ibrahim, A.M.; Mansour, I.M.; Wilson, M.M.; Mokhtar, D.A.; Helai, A.M.; Al Wakeel, H.M. Study of surviving and X-linked inhibitor of apoptosis protein (XIAP) genes in acute myeloid leukemia (AML). Lab. Hematol. 2012, 18, 1–10. [Google Scholar] [CrossRef]
- Tamm, I.; Richter, S.; Oltersdorf, D.; Creutzig, U.; Harbott, J.; Scholz, F.; Karawajew, L.; Ludwig, W.D.; Wuchter, C. High expression levels of x-linked inhibitor of apoptosis protein and surviving correlate with poor overall survival in childhood de novo acute myeloid leukemia. Clin. Cancer Res. 2004, 10, 3737–3744. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Hasegawa, M.; Kurata, M.; Yamamoto, K.; Abe, S.; Inoue, M.; Takemura, T.; Hirokawa, K.; Suzuki, K.; Kitagawa, M. Expression of IAP-family proteins in adult acute mixed lineage leukemia (AMLL). Am. J. Hematol. 2005, 78, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Saraei, R.; Soleimani, M.; Akbari, A.A.M.; Hagh, M.F.; Hassanzadeh, A.; Solali, S. The role of XIAP in resistance to TNF-related apoptosis-inducing ligand (TRAIL) in leukemia. Biomed. Pharmacother. 2018, 107, 1010–1019. [Google Scholar] [CrossRef]
- Moreno-Martinez, D.; Nomdedu, M.; Lara-Castillo, M.C.; Etxabe, A.; Pratcorona, M.; Tesi, N.; Diaz-Beyà, M.; Rozman, M.; Montesserrat, E.; Urbano-Ispizua, A.; et al. XIAP inhibitors induce differentiation and impair clonogenic capacity of acute myeloid stem cells. Oncotarget 2014, 5, 4337–4346. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Lan, J.; Huang, Q.; Chen, X.; Sun, X.; Liu, X.; Yang, P.; Jin, T.; Wang, S.; Mou, X. Embelin sensitizes acute myeloid leukemia cells to TRAIL through XIAP inhibition and NF-kB inactivation. Cell Biochem. Biophys. 2015, 71, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D.; Estey, E.H.; Borthakur, G.; Carter, B.Z.; Schiller, G.J.; Tallman, M.S.; Altman, J.K.; Karp, J.E.; Kassis, J.; Hedley, D.W.; et al. Phase I/II trial of AEG35156 X-linked apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J. Clin. Oncol. 2009, 27, 4741–4746. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D.; Herr, W.; Hanel, M.; Borthakur, G.; Frankel, A.; Horst, H.A.; Martin, S.; Kassis, J.; Desjardins, P.; Seiter, K.; et al. Addition of AEG35156 XIAP antisense oligonucleotide in reinduction chemotherapy does not improve remission rates in patients with primary refractory acute myeloid leukemia in randomized phase II study. Clin. Lymphoma Myeloma Leuk. 2011, 11, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lu, X.; Tan, T.Z.; Chng, W.J. X-linked inhibitor of apoptosis inhibition sensitizes acute myeloid leukemia response to TRAIL and chemotherapy through potentiated induction of proapoptotic machinery. Mol. Oncol. 2018, 12, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.H.; Yu, Y.; Zhong, L.; Adams, J.; Lam, F.; Li, P.; Noll, B.; Milne, R.; Pang, J.; Wang, S. CDKI-73: An orally bioavailable and highly efficacious CDK9 inhibitor against acute myeloid leukemia. Investig. New Drugs 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, A.; Chen, Z.; Van Waes, C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin. Cancer Res. 2016, 23, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Lueck, S.C.; Russ, A.C.; Botzenhardt, U.; Schlenk, R.F.; Zobel, K.; Deshayes, K.; Vucic, D.; Dohner, H.; Dohner, K.; Fulda, S.; et al. Smac mimetic induces cell death in a large proportion of primary acute myeloid leukemia samples, which correlates with defined molecular markers. Oncotarget 2016, 7, 49539–49551. [Google Scholar] [CrossRef] [Green Version]
- Safferhal, C.; Rohde, K.; Fulda, S. Therapeutic targeting of necroptosis by Smac mimetic bypasses apoptosis resistance in acute myeloid leukemia cells. Oncogene 2017, 36, 1487–1502. [Google Scholar] [CrossRef]
- Volk, A.; Li, J.; Xin, J.; You, D.; Zhang, J.; Liu, X.; Xiao, Y.; Breslin, P.; Li, Z.; Wei, W.; et al. Co-inhibition of NF-kB and JNK is synergistic in TNF-expressing human AML. J. Exp. Med. 2014, 211, 1093–1108. [Google Scholar] [CrossRef]
- Li, J.; Volk, A.; Zhang, J.; Cannova, J.; Dai, S.; Hao, C.; Hu, C.; Sun, J.; Xu, Y.; Wei, W.; et al. Sensitizing leukemia stem cells to NF-kB inhibitor treatment in vivo by inactivation of both TNF and IL-1 signaling. Oncotarget 2017, 8, 8420–8435. [Google Scholar] [PubMed]
- Bertoli, S.; Picard, M.; Bérard, E.; Griessinger, E.; Larrue, C.; Mouchel, P.L.; Vergez, F.; Tavitian, S.; Yon, E.; Ruiz, J.; et al. Dexamethasone in hyperleukocytic acute myeloid leukemia. Haematologica 2018, 103, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Singh, S.; Hag, W. IAP proteins antagonist: An introduction and chemistry of Smac mimetics under clinical development. Curr. Med. Chem. 2018, 25, 3768–3795. [Google Scholar] [CrossRef]
- Heaton, W.L.; Senina, A.V.; Pomicter, A.D.; Salama, M.E.; Clair, P.M.; Yan, D.; Bell, R.N.; Gilliland, J.M.; Prchal, J.T.; O’Hare, T.; et al. Autocrine Tnf signaling favors malignant cells in myelofibrosis in a Tnfr2-dependent fashion. Leukemia 2018, 32, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Carter, B.Z.; Kantarjian, H.M.; Cortes, J.E.; Kadia, T.M.; Garcia-Manero, G.; DiNardo, C.D.; Bose, P.; Daver, N.G.; Konopleva, M.; et al. LCL161, an oral Smac mimetic/IAP antagonist for patients with myelofibrosis (MF): Novel translational findings among long-term responders in a phase 2 clinical trial. Blood 2018, 132, 687. [Google Scholar]
- Thorburn, J.; Andrysik, Z.; Staskiewicz, L.; Gummp, J.; Maycotte, P.; Oberst, A.; Green, D.R.; Espinosa, J.M.; Thorburn, A. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014, 7, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Goodall, M.L.; Fitwalter, B.; Zhaedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.M.; Rabinowich, H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef]
- Morgan-Lappe, S.E. ABBV-621: A best-in-class TRAIL-receptor agonist fusion protein that enhances optimal clustering for the treatment of solid and hematologic tumors. Cancer Res. 2017, 77. [Google Scholar] [CrossRef]
- Tahir, S.-K.; Smith, M.L.; Solomon, L.R.; Zhang, H.; Xue, J.C.; Xiao, Y.; Cheng, D.; Buchanan, G.; Morgan-Lappe, S.; Phillips, D.C. ABBV-621 is a novel and potent TRAIL receptor agonist fusion protein that induces apoptosis alone and in combination with navitoclax and venetoclax in hematological tumors. Blood 2017, 130, 2812. [Google Scholar]
- Li, M.; Wu, X.M.; Gao, J.; Zhang, C.L.; Ke, K.; Wang, Y.C.; Zheng, Y.S.; Yao, J.F.; Guan, Y.Y.; et al. Mutations in the P10 region of procaspase-8 lead to chemotherapy resistance in acute myeloid leukemia by impairing procaspase-8 dimerization. Cell Death Dis. 2018, 9, 516. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yao, J.; Zhang, X.; Chen, X.; Chen, J.; Guan, Y.; Yang, X. Q482H mutation of procaspase-8 in acute myeloid leukemia abolishes caspase-8-mediated apoptosis by impairing procaspase-8. Biochem. Biophys. Res. Commun. 2018, 495, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and hierarchical binding of c-FLIP and Caspase-8: A unified model defines how c-FLIP isoforms differentially control cell fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.M.; Li, Y.; Li, Z.; Vajihala, P.R.; Cruz, A.C.; Srivastava, D.B.; Di Maio, F.; Penczek, P.A.; Siegel, R.M.; et al. Cryo-EM structure of caspase-8 tandem DED filament reveals assembly and regulation mechanisms of the death-inducing signaling complex. Mol. Cell 2016, 64, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Brumatti, G.; Ma, C.; Lalaoui, N.; Nguyen, N.Y.; Navarro, M.; Tanzer, M.C.; Richmond, J.; Ghisi, M.; Salmon, J.M.; Silke, N.; et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med. 2016, 8, 339ra69. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Krigsfeld, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S.; Dolof, N.G.; Messaris, E.; Scata, K.A.; Wang, W.; et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci. Transl. 2013, 5, 171ra17. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Krinsfeld, G.; Patel, L.; Mayes, P.A.; Dicker, D.T.; Wu, G.S.; El-Deiry, W.S. Identification of TRAIL-inducing compounds highlights small molecule ONC201/TIC10 as a unique anti-cancer agent that activates the TRAIL pathway. Mol. Cancer 2015, 14, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci. Signal. 2016, 9, ra17. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.L.; Van den Heuvel, A.P.; Allen, J.E.; Prabhu, V.V.; Dicker, D.T.; El-Deiry, W.S. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci. Signal. 2016, 9, ra18. [Google Scholar] [CrossRef]
- Kline, C.L.; Ralff, M.D.; Lulla, A.R.; Wagner, J.M.; Abbash, P.H.; Dicker, D.T.; Allen, J.E.; El-Deiry, D.S. Role of dopamine receptors in the anticancer activity of ONC201. Neoplasia 2018, 20, 80–91. [Google Scholar] [CrossRef]
- Allen, J.E.; Kline, C.L.; Probhu, V.V.; Wagner, J.; Ishizawa, J.; Madhukae, N.; Lev, A.; Baumeister, M.; Zhou, L.; Lulla, A.; et al. Discovery and clinical introduction of first-in-class imipridone ONC 201. Oncotarget 2016, 7, 74380–74392. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.N.; Bertino, J.R.; Kaufman, M.L.; Mayer, T.; Mon, R.; Silk, A.; Silk, A.; Chan, N.; Malhotra, J.; Rodriguez, C.; et al. First-in-human clinical trial of oral ONC 201 in patients with refractory solid tumors. Cancer Res. 2017, 23, 4163–4169. [Google Scholar]
- Prabhu, V.V.; Talekar, M.K.; Lulla, A.R.; Kline, C.L.; Zhou, L.; Hall, J.; Van den Heuvel, A.P.J.; Dicker, D.T.; Bobar, J.; Grupp, S.A.; et al. Single agent and synergistic combinatorial efficacy of first-in-class small molecule imipridone ONC 201 in hematological malignancies. Cell Cycle 2018, 17, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Kline, C.L.; Zhou, L.; Campbell, K.S.; MacFarlane, A.; Olsranski, A.J.; Cai, K.Q.; Hensley, H.H.; Ross, E.A.; Rolff, M.D.; et al. Dose intensification of TRAIL-inducing ONC 201 inhibits metastasis and promotes intratumoral NK cell recruitment. J. Clin. Investig. 2018, 128, 2325–2338. [Google Scholar] [CrossRef] [PubMed]
- Auberger, P.; Puisant, A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 2017, 129, 547–552. [Google Scholar] [CrossRef]
- Ianniciello, A.; Rattigan, K.M.; Helgason, G.V. The ins and outs of autophagy and metabolism in hematopoietic and leukemic stem cells: Food for thought. Front. Cell Dev. Biol. 2018, 6, 120. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.-E.W.; et al. The autophagy protein ATG7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Kawai, Y.; Fergusson, M.M.; Rovira, I.I.; Bishop, A.J.R.; Montoyama, N.; Cao, L.; Finkel, T. ATG7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012, 336, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lasinger, O.M.; Flach, J.; Kerovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lee, J.Y.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, G.L. FIP 200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Xu, C.; Zhang, X.; Long, J.; Rezaeian, A.H.; Liu, C.; Furth, M.E.; Kridel, S.; Pasche, B.; Bian, X.W.; et al. ATAD3A suppresses PINK1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat. Immunol. 2018, 19, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kenler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuring, J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Diff. 2017, 8, e2927. [Google Scholar] [CrossRef] [PubMed]
- Visconte, V.; Przychodzen, B.; Han, Y.; Nawrochi, S.T.; Thota, S.; Kelly, K.R.; Patel, B.J.; Hirsch, C.; Advani, A.S.; Carraway, H.E.; et al. Complete mutational spectrum of the autophagy interactome: A novel class of tumor suppressor genes in myeloid neoplasms. Leukemia 2016, 31, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Marconi, G.; Fontana, M.C.; Papayannidis, C.; Padella, A.; Lo Monaco, S.; Abbenante, M.C.; Sartor, C.; Bertamini, L.; Nanni, J.; di Rorà, A.G.L.; et al. Prognostic significance of alterations of pathways regulating autophagy in acute myeloid leukemia. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Bosnjak, M.; Ristic, B.; Arsikin, K.; Mircic, A.; Suzin-Zivkovic, V.; Perovic, V.; Bogdanovic, A.; Paunovic, V.; Markovic, I.; Bumbarisevic, V.; et al. Inhibition of mTOR-dependent autophagy sensitizes leukemic cells to cytarabine-induced apoptotic death. PLoS ONE 2014, 9, e94374. [Google Scholar] [CrossRef]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; McQueen, T.; Davis, R.E.; Hail, N.; Kautarjian, H.; Andreef, M.; Porthakur, G. ATG7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.E.; Besson, A.; Manenti, S.; Joffre, C.; Mansta-De Mas, V. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2018, 37, 787–797. [Google Scholar] [CrossRef]
- Rudat, S.; Pfaus, A.; Cheng, Y.Y.; Holtmann, J.J.; Ellegast, J.M.; Buhler, C.; Marcantonio, D.D.; Martinez, E.; Gollner, S.; Wickenhauser, C.; et al. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia 2018, 32, 2189–2202. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Li, T.S. Dual role of mitophagy in cancer drug resistance. Anticancer Res. 2018, 38, 617–621. [Google Scholar] [PubMed]
- Tian, Y.; Huang, Z.; Wang, Z.; Yin, C.; Zhou, L.; Zhang, L.; Huang, K.; Zhou, H.; Jiang, X.; Li, J.; et al. Identification of novel molecular markers for prognosis estimation of acute myeloid leukemia: Overexpression of PDCD7, FIS1 and Ang2 may indicate poor prognosis in pretreatment patients with acute myeloid leukemia. PLoS ONE 2014, 9, e84150. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Shaid, S.; Vakhurushava, O.; Koschade, S.E.; Klann, K.; Thoken, M.; Baker, F.; Zhang, J.; Oellerich, T.; Surun, D.; et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood 2018, in press. [Google Scholar] [CrossRef]
- Dany, M.; Gencer, S.; Nganga, R.; Thomas, R.J.; Oleinik, N.; Baron, K.D.; Szulc, Z.M.; Ruvolo, P.; Kornblau, S.; Andreef, M.; et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 2016, 128, 1944–1958. [Google Scholar] [CrossRef]
- Segala, G.; David, M.; de Medina, P.; Porot, M.C.; Serhan, N.; Vergez, F.; Mougel, A.; Saland, E.; Carayon, K.; Leignadier, J.; et al. Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat. Commun. 2017, 8, 1903. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Amaral, J.D.; Xavier, J.M.; Steer, C.J.; Rodrigues, C.M. The role of p53 in apoptosis. Discov. Med. 2010, 9, 145–152. [Google Scholar]
- Denisenko, T.V.; Pivnyuk, A.D.; Zhitonsky, B. p53-autophagy-metastasis. Cancers 2018, 10, 148. [Google Scholar] [CrossRef]
- Tandermit, E.; Maiuri, C.; Galluzzi, L.; Vitali, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplsmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin 2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Diff. 2018, in press. [Google Scholar] [CrossRef]
- Chee, J.L.; Saidin, S.; Lane, D.P.; Leong, S.M.; Noll, J.E.; Neilsen, P.M.; Phua, Y.T.; Gabra, H.; Lim, T.M. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle 2013, 12, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Frank, A.K.; Pietsch, A.C.; Dumont, P.; Tao, J.; Murphy, M.E. Wild-type and mutant p53 proteins interact with mitochondrial caspase-3. Cancer Biol. Ther. 2011, 11, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Stengel, A.; Kern, W.; Haferlach, H.; Meggendorfer, M.; Fasan, A.; Haferlach, C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: An analysis of 3307 cases. Leukemia 2017, 31, 705–711. [Google Scholar] [CrossRef]
- Rose, D.; Haferlach, T.; Schnittger, S.; Perglerova, K.; Kern, W.; Haferlach, C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia 2017, 31, 11–17. [Google Scholar] [CrossRef]
- Iacobucci, I.; Wen, J.; Meggendorfer, M.; Carmichael, C.; Choi, J.K.; Maasih, K.; Li, Y.; Payne, D.; Tomizawa, D.; Kiyokawa, N.; et al. The genomic landscape of childhood and adult erythroid leukemia. Blood 2016, 128, 39. [Google Scholar]
- Hou, H.A.; Chou, W.C.; Kuo, Y.; Liu, C.Y.; Lin, L.I.; Tseng, M.H.; Chiang, Y.C.; Liu, M.C.; Tang, J.L.; Yao, M.; et al. TP53 mutations in de novo acute myeloid leukemia patients: Longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015, 5, e331. [Google Scholar] [CrossRef]
- Bochtler, T.; Granzow, M.; Stolzel, F.; Kunz, C.; Mohr, B.; Kartal-Kaess, M.; Hinderhofer, K.; Heilig, C.E.; Kramer, M.; Thiede, C.; et al. Marker chromosomes can arise from chromothripsis and predict adverse prognosis in acute myeloid leukemia. Blood 2017, 129, 133–1342. [Google Scholar] [CrossRef]
- Fontana, M.C.; Marconi, G.; Feenstra, J.D.M.; Fonzi, E.; Papayannidis, C.; Ghelli Luserna di Rorà, A.; Padella, A.; Solli, V.; Franchini, E.; Ottaviani, E.; et al. Chromotrhripsis in acute myeloid leukemia: Biological features and impact on survival. Leukemia 2018, 32, 1609–1620. [Google Scholar] [CrossRef]
- Kuykendall, A.; Duployez, N.; Boissel, N.; Lancet, J.E.; Welch, J.S. Acute myeloid leukemia: The good, the bad, and the ugly. Am. Soc. Clin. Oncol. Educ. Book 2018, 23, 556–573. [Google Scholar] [CrossRef]
- Assi, R.; Gur, H.D.; Loghavi, S.; Konoplev, S.N.; Konopleva, M.; Daver, N.; Tashakori, M.; Kadia, T.; Routbort, M.; Salem, A.; et al. P53 protein overexpression in de novo acute myeloid leukemia patients with normal diploid karyotype correlates with FLT3 internal tandem duplication and worse relapse-free survival. Am. J. Hematol. 2018, 93, 1376–1383. [Google Scholar] [CrossRef]
- Huang, R.; Liao, X.; Li, Q. Identification of key pathways and genes in TP53 mutation acute myeloid leukemia: Evidence from bioinformatics analysis. Oncol Targets Ther. 2018, 11, 163–173. [Google Scholar] [CrossRef]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.; Shen, D.; Hundsal, J.; Fulton, R.S.; et al. Tole of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.B.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Chikulwar, A.; Saliba, R.M.; Chen, J.; Rondon, G.; Patel, K.P.; Khogeer, H.; Shah, A.R.; Shah, A.R.; Randoplh, B.V.; et al. Prognostic factors influencing survival after allogeneic transplantation for AML/MDS patients with TP53 mutations. Blood 2018, 131, 2989–2992. [Google Scholar] [CrossRef]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Loughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinic-molecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef]
- Kadia, T.M.; Cortes, J.; Ravandi, F.; Jabbour, E.; Konopleva, M.; Benton, C.B.; Burger, J.; Sasaki, K.; Borthakar, C.; Di Nardo, C.; et al. Cladribine and low-dose cytarabine alternating with Decitabine as front-line therapy for elderly patients with acute myeloid leukemia: A phase 2 single-arm trial. Lancet Hematol 2018, 5, e411–e421. [Google Scholar] [CrossRef]
- Uy, G.L.; Duncavage, E.J.; Cheng, G.S.; Jacoby, M.A.; Miller, C.A.; Shao, J.; Health, S.; Elliott, K.; Reineck, T.; Fulton, R.S.; et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. Leukemia 2017, 31, 872–881. [Google Scholar] [CrossRef]
- Daver, N.; Kantarjian, H.; Garcia-Manero, G.; Jabbour, E.; Borthakur, G.; Brandt, M.; Pierce, S.; Vaughan, K.; Ning, J.; Nogueras Gonzalez, G.M.; et al. Vosaroxin in combination with decitabine in newly diagnosed older patients with acute myeloid leukemia or high-risk myelodysplastic syndrome. Haematologica 2017, 102, 1709–1717. [Google Scholar] [CrossRef] [Green Version]
- Montalban-Bravo, G.; Takahashi, K.; Garcia-Maneiro, G. Decitabine in TP53-mutated AML. N. Engl. J. Med. 2017, 376, 796–797. [Google Scholar]
- Short, N.J.; Kantarjian, H.M.; Loghavi, S.; Huang, X.; Qiao, W.; Borthakur, G.; Kadia, T.M.; Daver, N.; Ohaniona, M.; DiNardo, C.D.; et al. Treatment with 5-day versus a 10-day schedule of decitabine in older patients with newly diagnosed acute myeloid leukemia: A randomized phase 2 trial. Lancet Hematol. 2018, in press. [Google Scholar]
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.; Herba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S.; et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 2018, 131, 1415–1424. [Google Scholar] [CrossRef]
- Prokocimer, M.; Moichadsky, A.; Rotter, V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: Projections on diagnostic workup and therapy. Blood 2017, 130, 699–712. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Komblau, S.M. p53 pathway dysfunction is highly prevalent in acute myeloid leukemic independent of TP53 mutational status. Leukemia 2017, 31, 1296–1305. [Google Scholar] [CrossRef]
- Wei, A.H.; Chua, C.C.; Tiong, I.S.; Fong, C.Y.; Ting, S.B.; Macraild, S.; Salmon, J.M.; Ivey, A.; Nguyen, J.; Yuen, F.; et al. Molecular patterns of response and outcome in the chemotherapy and venetoclax in elderly AML trial (CAVETA study). Blood 2018, 132, 333. [Google Scholar]
- Wei, A.; Strickland, S.A.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.K.; Hong, W.J.; Chyla, B.; Popovic, R.; et al. Venetoclax with low-dose cytarabine induces rapid, deep, and durable responses in previously untreated older adults with AML ineligible for intensive chemotherapy. Blood 2018, 132, 284. [Google Scholar]
- Alfayez, M.; Patel, K.; Cortes, J.E.; Kadia, T.M.; Ravandi, F.; DiNardo, C.D.; Daver, N.G.; Pemmaraju, N.; Kantarjian, H.M.; Barthakur, G. Impact of variant allele frequency of mutant PTPN11 in AML: Single institution experience of 122 patients. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Stevens, B.M.; Ljones, C.L.; Winters, A.; Dugan, J.; Abbott, D.; Savona, M.R.; Fesik, S.W.; Pollyea, D.A.; Jordan, C.T. PTPN11 11 mutations confer unique metabolic properties and increase resistance to Venetoclax and Azacitidine in acute myeloid leukemia. Blood 2018, 132, 909. [Google Scholar]
- Salmon, J.M.; Pomilio, G.; Moujalled, D.M.; Macraild, S.; The, C.E.; Riajl, S.; Ivey, A.; The, T.C.; Ekert, P.G.; Kraus-Berthier, L.; et al. Combined BCL-2 and HDAC has potent and TP53 independent activity in AML. Blood 2018, 132, 1426. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; Seet, C.S.; et al. Ivosidenib or enasidenib combined with induction and consolidation chemotherapy in patients with newlty diagnosed AML with an IDH1 or IDH2 mutation is safe, effective, and leads to MRD-negative complete remissions. Blood 2018, 132, 560. [Google Scholar]
- Cortes, J.E.; Watts, J.; Prebet, T.; Schiller, G.J.; Lee, S.; Yang, J.; Wang, E.S.; Dinner, S.; Ferrell, P.B.; Donnellann, W.; et al. FT-1202, and IDH1m inhibitor, in combination with azacytidine in patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS): Results from a phase 1 study. Blood 2018, 132, 1452. [Google Scholar]
- Cathelin, S.; Sharon, D.; Subedi, A.; Cojocari, D.; Phillips, D.C.; Leverson, J.D.; MacBeth, K.; Nicolay, B.; Narayanaswamy, R.; Ronseaux, S.; et al. Combination of enasidenib and venetoclax shows superior anti-leukemic activity against IDH2 mutated AML in patient-derived xenograft models. Blood 2018, 132, 562. [Google Scholar]
- Middeke, J.M.; Rollig, C.; Kramer, M.; Kramer, A.; Bochtler, T.; Scholl, S.; Hochaus, A.; Jost, E.; Brummendord, T.; Naumann, R.; et al. Clinical characteristics and outcome in IDH1/2 mutant AML patients-analysis of 3898 newly diagnosed patients with acute myeloid leukemia. Blood 2018, 132, 1461. [Google Scholar]
- Ahr, K.; Famulare, C.; Koche, R.; Spitzer, B.; Levine, R.L.; Tallman, M.S.; Goldberg, A.D.; Stein, E.M.; Glass, J.L. AML with mutations in IDH1 and DNMT3A exhibits a distinct epigenetic signature with poorer overall survival. Blood 2018, 132, 1471. [Google Scholar]
- Thota, S.; Patel, B.J.; Carraway, H.E.; Griffiths, E.A.; Przespolewski, A.; Thompson, J.E.; Kuzmanovic, T.; Nazha, A.; Vipul, S.; Hirsch, C.M.; et al. Clinical outcomes for patients with myeloid malignancies harboring IDH1/23 mutations after intensive chemotherapy. Blood 2018, 132, 1389. [Google Scholar]
- Wang, F.; Morita, K.; DiNardo, C.D.; Patel, K.; MacBeth, K.; Tosolini, A.; Frattini, M.G.; Matthews, J.; Little, L.; Gumbs, C.; et al. Comprehensive genomic analysis of IDH inhibitor-treated AML samples delineates molecular mechanisms of differentiation and heterogeneous patterns of acquired resistance. Blood 2018, 132, 441. [Google Scholar]
- Bhola, P.D.; Letai, A. Mitochondria-judges and executioners of cell death sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef]
- Thol, F.; Klesse, S.; Kohler, L.; Gabdoulline, R.; Kloos, A.; Liebich, A.; Wichmann, M.; Chaturvedi, A.; Fabisch, J.; Gaidzik, V.I.; et al. Acute myeloid leukemia derived from lympho-myeloid clonal hematopoiesis. Leukemia 2017, 31, 1286–1295. [Google Scholar] [CrossRef]
- Boyd, A.L.; Aslostovar, L.; Reid, J.; Ye, W.; Tanasijevic, B.; Porras, D.P.; Shapovalova, Z.; Almakadi, M.; Foley, R.; Leber, B.; et al. identification of chemotherapy-induced leukemic-regenerating cells reveals a transient vulnerability of human AML recurrence. Cancer Cell 2018, 24, 483–498. [Google Scholar] [CrossRef]
- Etxabe, A.; Lara-Castillo, M.C.; Cornet-Masana, J.M.; Banus-Mulet, A.; Nomdedeu, M.; Torrente, M.A.; Pratcorona, M.; Diaz-Beya, M.; Esteve, J.; Risueno, R.M. Inhibition of serotonin receptor type 1 in acute myeloid leukemia impairs leukemia stem cell functionality: A promising novel therapeutic target. Leukemia 2017, 31, 2288–2302. [Google Scholar] [CrossRef]
- Slostovar, L.; Boyd, A.L.; Almakadi, M.; Collins, T.J.; Leong, D.P.; Tirona, R.G.; Kim, R.B.; Julian, J.A.; Xenocostas, A.; Leber, B.; et al. A phase 1 trial evaluating thioridazine in combination with cytarabine in patients with acute myeloid leukemia. Blood Adv. 2018, 2, 1935–1945. [Google Scholar] [CrossRef]









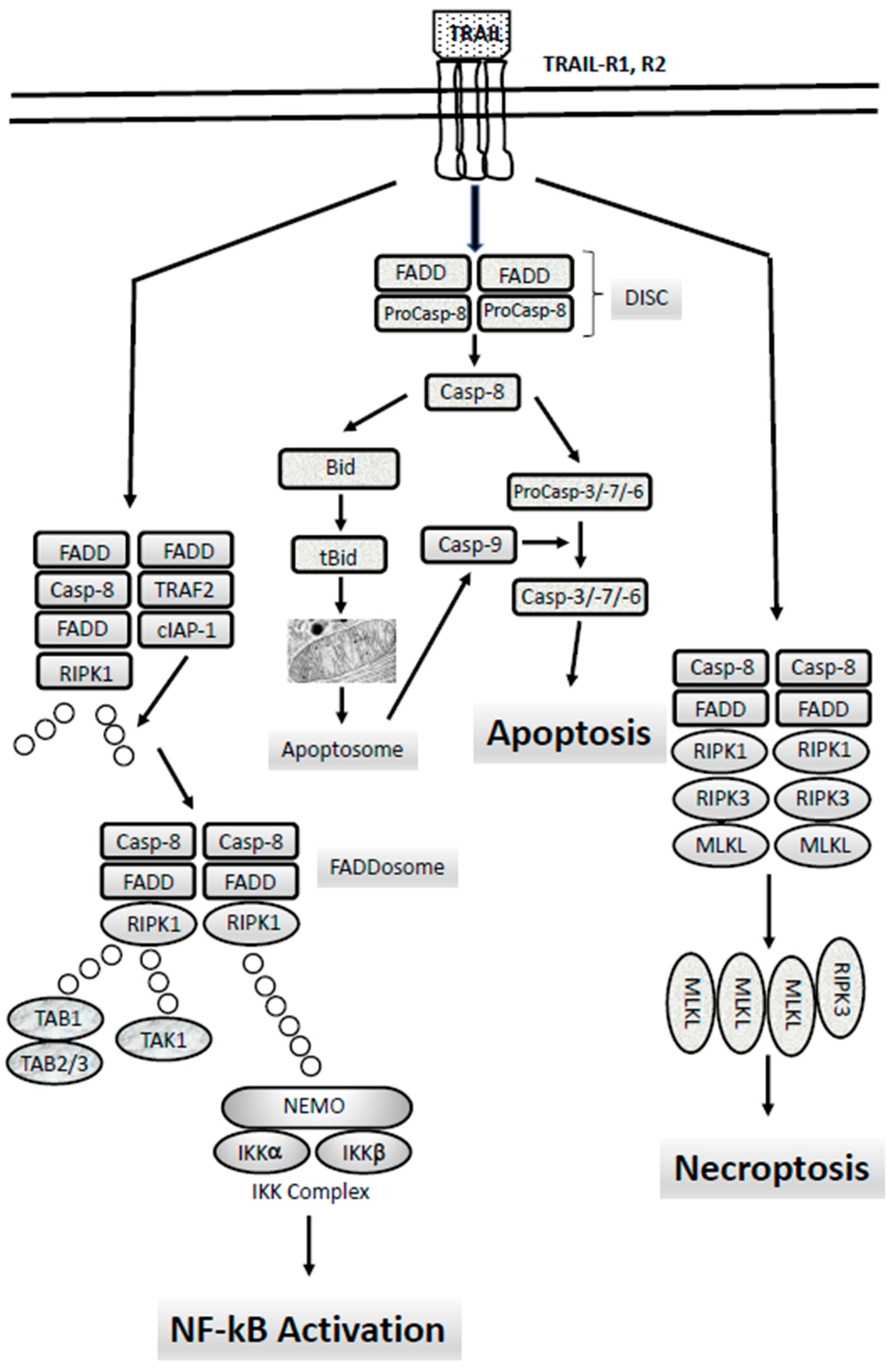{kind=link}
{kind=link}
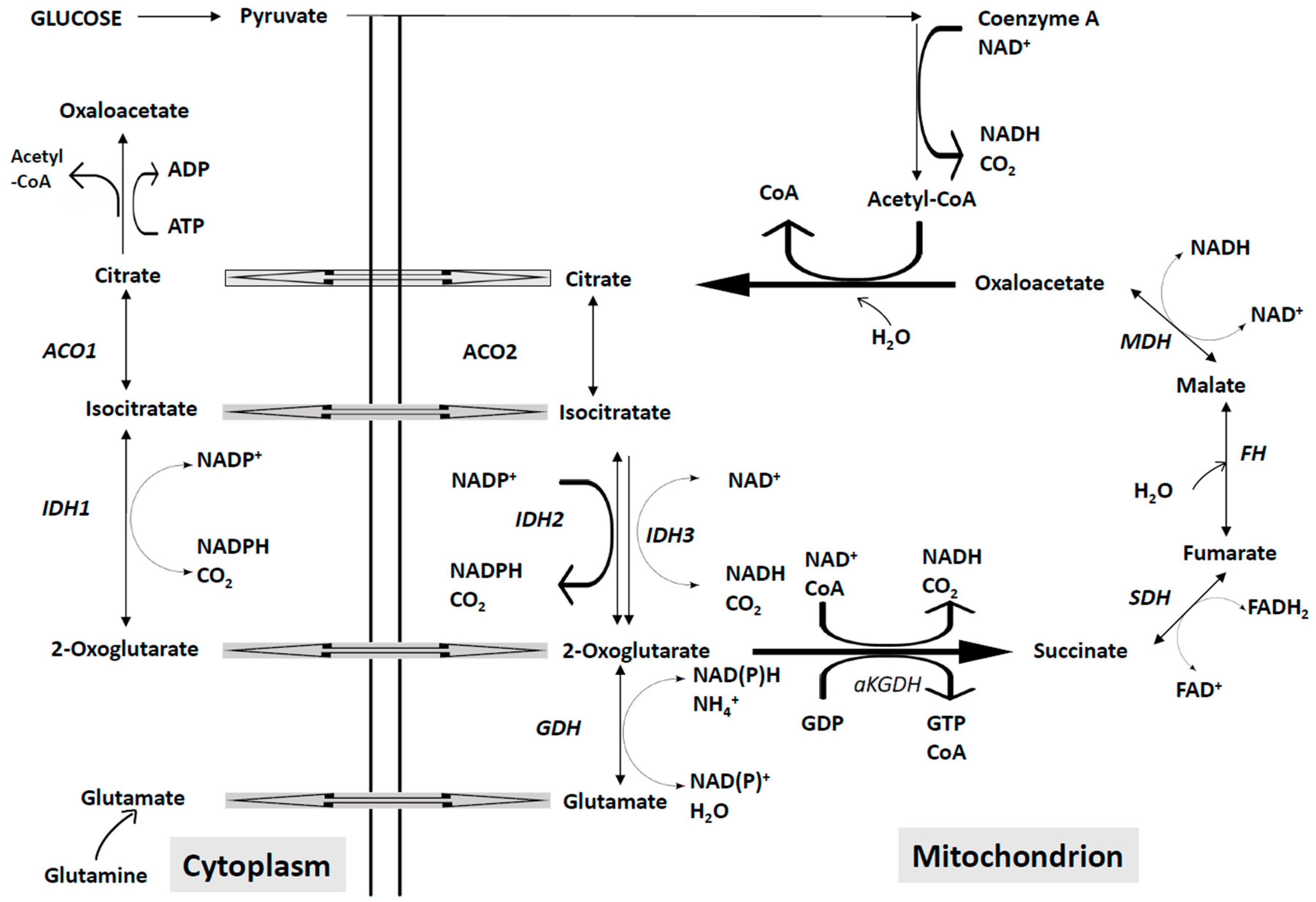{kind=link}
{kind=link}
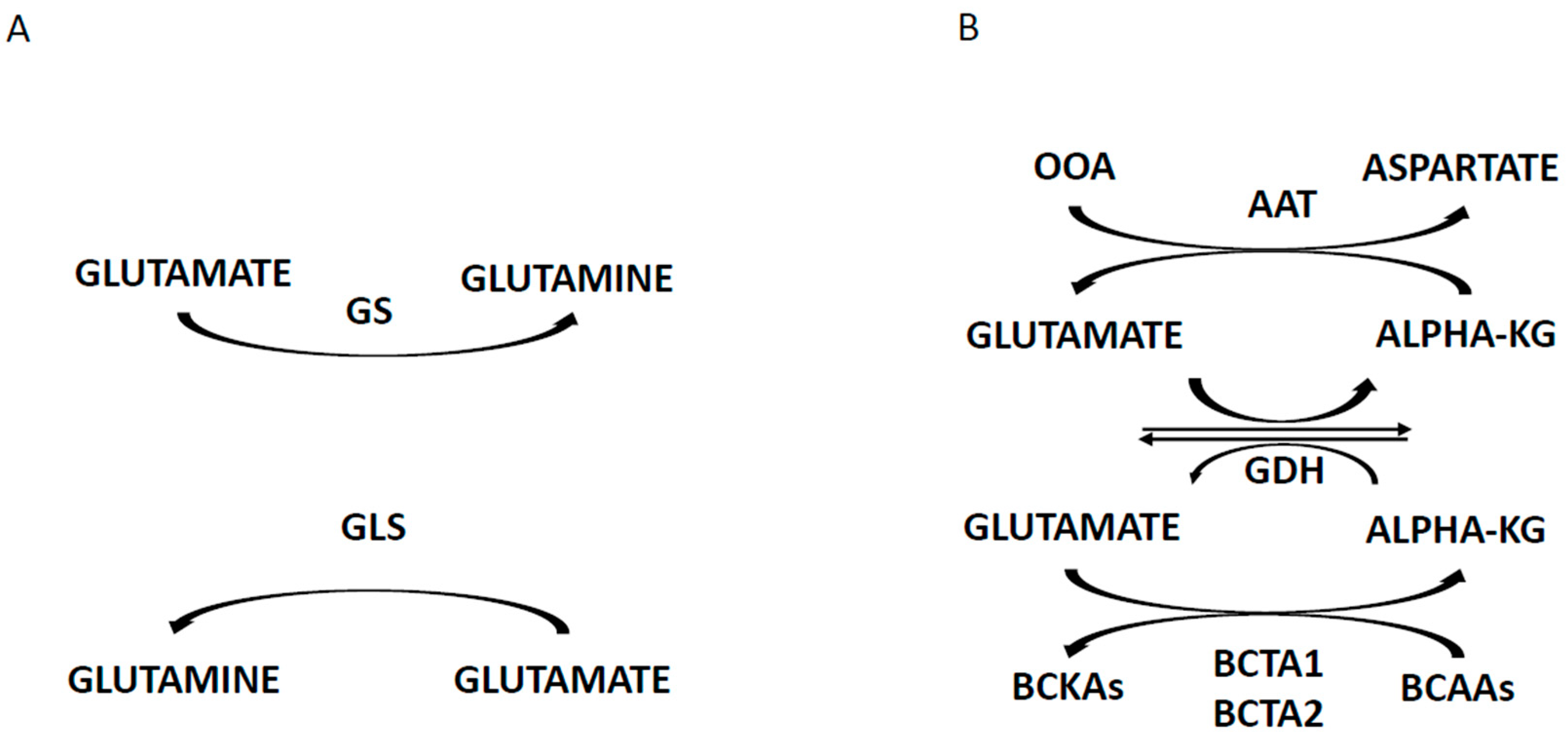{kind=link}
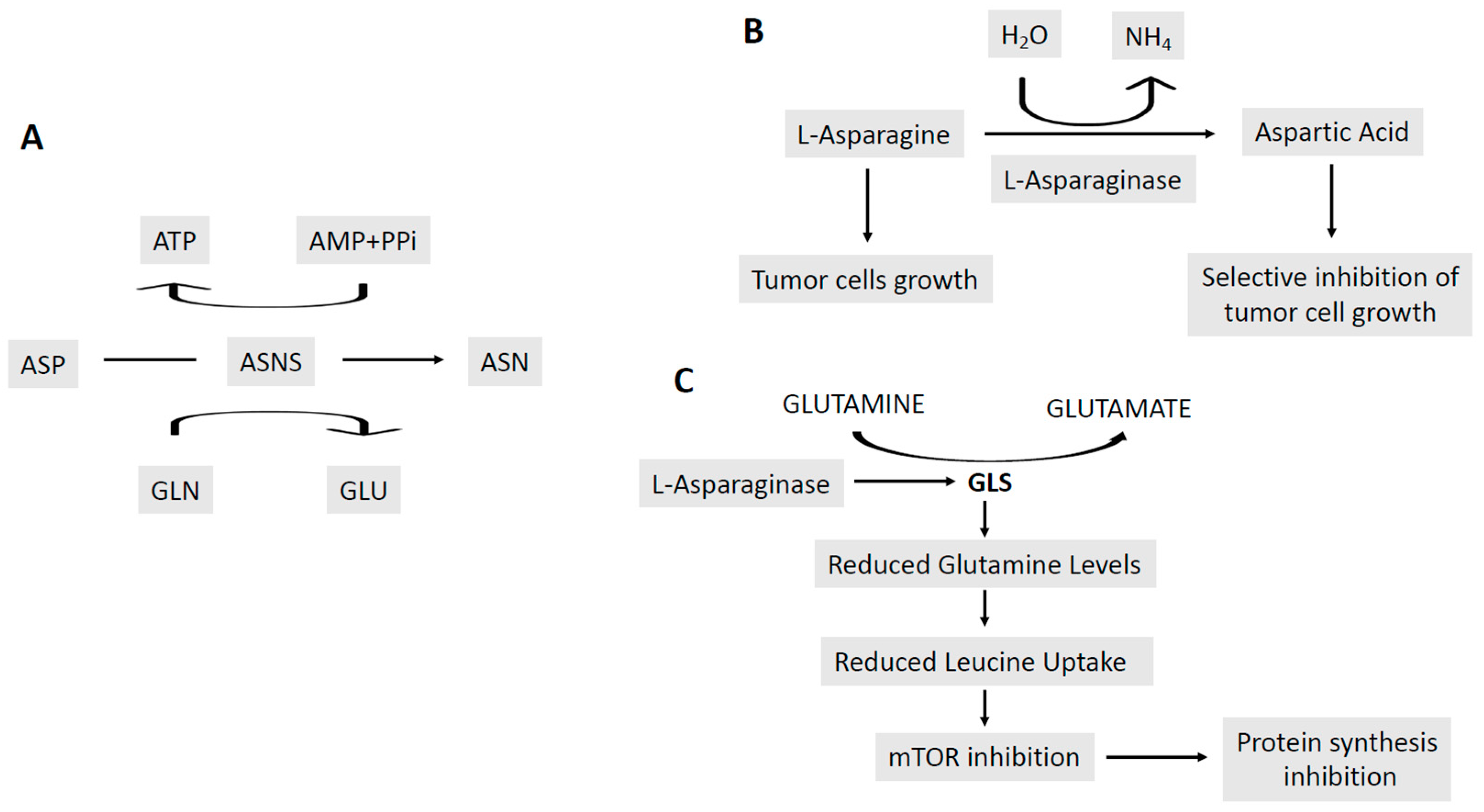{kind=link}
{kind=link}
{kind=link}
| Prognostic Group | AML Subtypes (Concurrent Mutation Frequency) | Probability of CR (%) | Probability of Relapse (%) |
|---|---|---|---|
| Low Risk | t(8;21)(q22;q22); RUNX1-RUNX1T1 1–6% (KIT 25%, NRAS 20%, Cohesin 20%, ZBTB7A 20%, ASXL1 10%) inv(16)(p13.1q22)/ t(16;16)(p13.1;q22) CBFB-MYH11 1–6% (NRAS 40%, KIT 35%, FLT3-TKD 20%, KRAS 15%) t(15;17)(q22;q12); PML-RARA 5–13% (FLT3-ITD 35%, FLT3-TKD 15%, WT1 15%) NK and NPM1+/FLT3-ITD−/WT (DNMT3A 50%, FLT3-ITD 40%, cohesion 20%, NRSA 20%, IDH1 15%, IDH2 R140 15%, TET2 15%) NK and CEBPA+/+ (biallelic) 1–5% (GATA2 30%, NRAS 30%, WT1 20%, CSF3R 20%, 9q− 15%) | 80–95 | 5–40 |
| Intermediate Risk | NK and NPM1−/FLT3-ITD−/low (without adverse-risk genetic lesions) NK and NPM1+/FLT3-ITDhigh t(9;11)(p21.3;q23.3); MLLT3-KMT2A 1% (NRAS 20%, FLT3-TKD 10%, FLT3-ITD 5%, +8/+8q 30%) Cytogenetic abnormalities not in the low or high risk groups t(8;21), inv(16) and t(16;16) with KIT mutations;BCFB-MYH11 1–6% (NRAS 40%, KIT 35%, FLT3-TKD 20%, KRAS 15%, +8/+8q 15%) | 50–80 | 50–80 |
| High Risk | t(6;9)(p23;q34.1); DEK-NUP214 1% (FLT3-ITD 70%, KRAS 20%) t(3;3)(q21.3q26.2), inv(3)(q21.3q26.2); GATA2, MECOM (EVI1) 1% (NRAS 30%, KRAS 15%, PTPN11 20%, GATA2 15%, SF3B1 20%, ETV6 15%, PHF6 15%, RUNX1 10%, ASXL1 10%) ASXL1mut 5–15% (RUNX1 20%, IDH2-R140 13%, +8 15%) t(9;22)(q34.1;q11.2); BCR-ABL1 monosomy 7 (-7) monosomy 5 (-5) deletion of long arm (q) of chromosome 7 (−7q) abnormalities of 3q, 17p, 11q multiple cytogenetic abnormalities (≥3–5) NK and NPM1−/FLT3-ITDhigh RUNXmut 5–20% (SRSF2 25%, ASXL1 20%, KMT2A 20%, IDH2-R140 12%, −7/7q− 10% TP53mut (complex karyotype 68%, −5/5q 47%, −7/7q 44%, −12/12p 17%, +8/8q 16%, c-kit 15%, CEBPA 15%) | <50 | >90 |
| Clinical Trial. Gov Identifier | Clinical Trial | Molecular Target | Phase | Relevant Preliminary Results |
|---|---|---|---|---|
| NCT01994837 | A phase 2 study of ABT-199 in subjects with AML | BCL-2 | II | ORR 38% CR 19% |
| NCT02287233 | A phase 1/2 study of venetoclax in combination with low-dose cytarabine in treatment-naïve subjects with acute myelogenous leukemia who are ≥ 60 years of age and who are not eligible for standard anthracycline-based induction therapy | BCL-2 | I/II | CR 62% |
| NCT03069352 | A study of venetoclax in combination with low dose cytarabine versus low dose cytarabine alone in treatment naïve patients with AML who are ineligible for intensive chemotherapy | BCL-2 | III | Phase II: ORR 64% |
| NCT02993523 | A study of venetoclax in combination with azacytidine versus azacytidine in treatment naïve subjects with AML who are ineligible for standard induction therapy | BCL-2 | III | Phase II: ORR 67% |
| NCT03404193 | Venetoclax in combination with 10-day decitabine in newly diagnosed elderly or relapsed/refractory AML and relapsed high-risk myelodysplastic syndrome | BCL-2 | II | |
| NCT03214562 | Study of the BCL-2 inhibitor venetoclax in combination with standard intensive AML induction/consolidation therapy with FLAG-IDA in patients with newly diagnosed or relapsed/refractory AML | BCL-2 | IB/II | |
| NCT0267044 | A study of venetoclax in combination with cobimetinib and venetoclax in combination with Idasanutlin in patients aged ≥ 60 years with relapsed or refractory AML who are not eligible for cytotoxic therapy | BCL-2 MEK MDM2 | IB/II | ORR (Ven + Cobi) 18% ORR (Ven + Ida) 20% CR (Ven + Cobi) 18% CR (Ven + Ida) 15% |
| NCT02203773 | Study of ABT-199 in combination with azacytidine or decitabine (chemo combo) in subjects with AML Group A: venetoclax + decitabine Group B: venetoclax + azacitidine Group C:venetoclax + decitabine and posaconazole (CYP3A inhibitor) | BCL-2 | IB | CR or CRi 61% (overall) CR or CRi 60% (group A 65%; group B 59%; group C 67%) |
| NCT03194932 | Study of venetoclax in combination with chemotherapy in pediatric patients with refractory or relapsed AML or acute leukemia of ambiguous lineage | BCL-2 | I/II | |
| ACTRN12616000445471 | A phase IB clinical evaluation of venetoclax in combination with chemotherapy (cytarabine+idarubicin) in older patients with acute myeloid leukemia | BCL-2 | IB | |
| NCT02391480 | A phase 1 study evaluating the safety and pharmacokinetics of ABBV-075 in subjects with advanced cancer (expansion combination treatment cohort of ABBV-075 and venetoclax) | BET BCL-2 | I/II | |
| NCT03537482 | A phase i study of safety, tolerability, pharmacokinetic and pharmacodynamics property of orally administered APG-2575 in patients with hematologic malignancies | BCL-2 | I | |
| CT03082209 | TRAIL receptor agonist ABBV-621 in subjects with previously treated solid tumors and hematologic malignancies (group II: ABBV-621 and venetoclax) | BCL-2 TRAIL-R | I |
| Clinical Trial. Gov Identifier | Clinical Trial | Molecular Target | Phase and Patients |
|---|---|---|---|
| NCT 02979366 | Phase I study of S64315 administered intravenously in patients with acute myeloid leukemia and myelodysplastic syndrome | MCL-1 (BH3-groove) | I, relapsed/refractory AMLs or secondary MDSs |
| NCT o2675452 | AMG 176 first in human trial in subjects with relapsed or refractory multiple myeloma and subjects with relapsed or refractory AML | MCL-1 (BH3-groove) | I, relapsed/refractory multiple myeloma and AML |
| NCT 03218683 | Study of AZD 5991 in relapsed or refractory hematologic malignancies | MCL-1 | I, relapsed/refractory hematologic malignancies |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castelli, G.; Pelosi, E.; Testa, U. Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism. Cancers 2019, 11, 260. https://doi.org/10.3390/cancers11020260
Castelli G, Pelosi E, Testa U. Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism. Cancers. 2019; 11(2):260. https://doi.org/10.3390/cancers11020260
Chicago/Turabian StyleCastelli, Germana, Elvira Pelosi, and Ugo Testa. 2019. "Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism" Cancers 11, no. 2: 260. https://doi.org/10.3390/cancers11020260
APA StyleCastelli, G., Pelosi, E., & Testa, U. (2019). Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism. Cancers, 11(2), 260. https://doi.org/10.3390/cancers11020260