Modelling of Protein Kinase Signaling Pathways in Melanoma and Other Cancers


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Principles of Mitogen-Activated Protein Kinase (MAPK) and Cellular Homolog of v-Kit Hardy-Zuckerman 4 Feline Sarcoma Viral Oncogene (c-KIT) Signaling in Melanoma
2.1. BRAF Signaling
2.2. RAS Signaling
2.3. Signalling through Receptor Tyrosine Kinases
3. Signaling through Other Mutated Pathways
4. Treatment Resistance to MAPK Inhibition
5. The ‘Omics’ Approach in Melanoma and Treatment Sensitivity
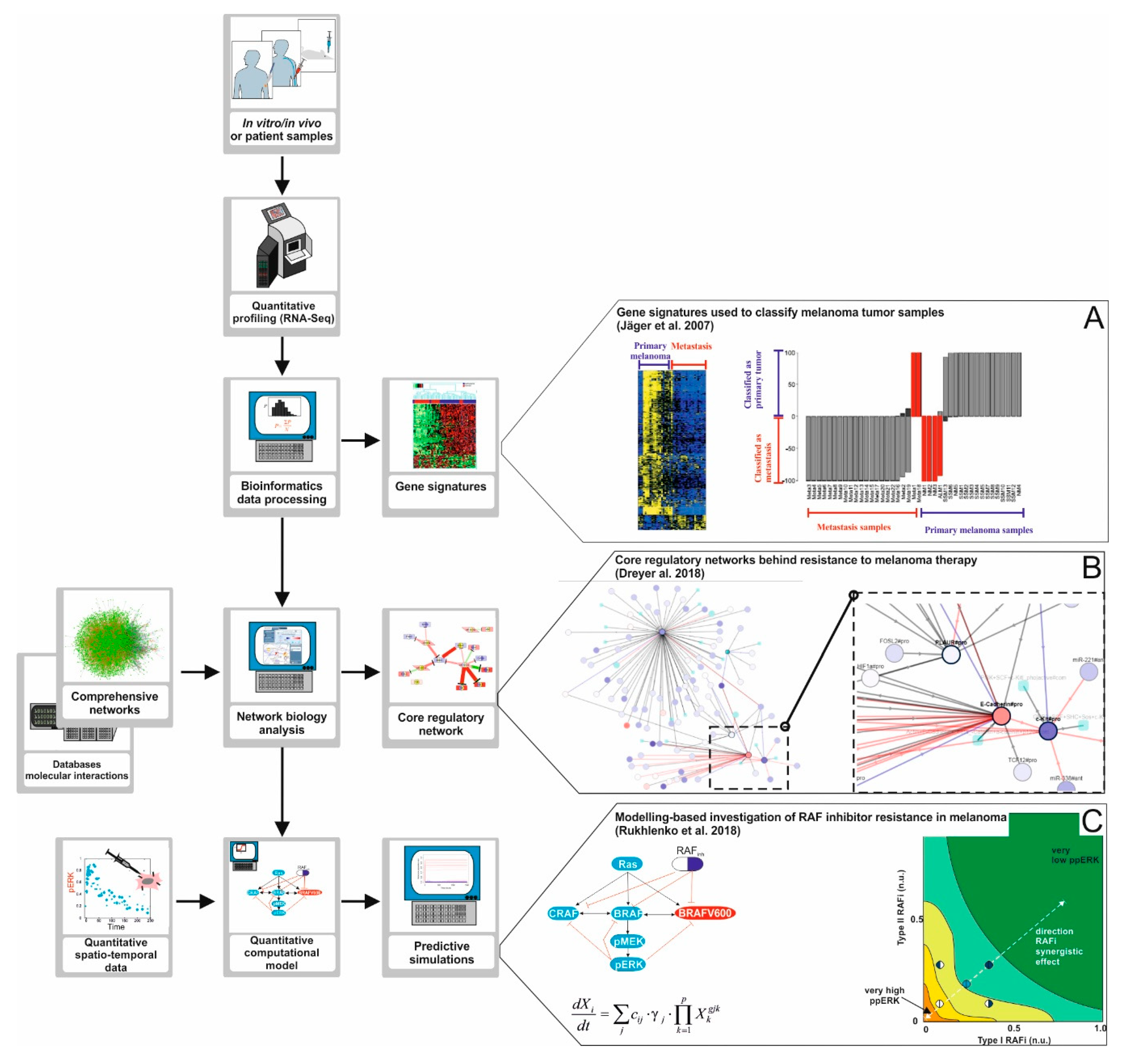
6. Systems Biology Approaches Used to Understand Kinase Inhibitor Activity and Resistance: from Networks to Computational Models
7. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumor types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Schadendorf, D.; van Akkooi, A.C.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, X.; Tamgüney, T.M.; Collisson, E.A.; Lin, L.-J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, N.; Rukhlenko, O.S.; Kolch, W.; Kholodenko, B.N. MAPK kinase signalling dynamics regulate cell fate decisions and drug resistance. Curr. Opin. Struct. Biol. 2016, 41, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Garbe, C.; Abusaif, S.; Eigentler, T.K. Vemurafenib. Recent Results Cancer Res. 2014, 201, 215–225. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Flaherty, K.T. New strategies in melanoma: Entering the era of combinatorial therapy. Clin. Cancer Res. 2015, 21, 2424–2435. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Vanella, V.; Festino, L.; Strudel, M.; Simeone, E.; Grimaldi, A.M.; Ascierto, P.A. PD-L1 inhibitors in the pipeline: Promise and progress. Oncoimmunology 2017, 7, e1365209. [Google Scholar] [CrossRef]
- Wellbrock, C.; Arozarena, I. The Complexity of the ERK/MAP-Kinase Pathway and the Treatment of Melanoma Skin Cancer. Front. Cell Dev. Biol. 2016, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.; Tam, A.; Pomerantz, J.; Wong, M.; Holash, J.; Bardeesy, N.; Shen, Q.; O’Hagan, R.; Pantginis, J.; Zhou, H.; et al. Essential role for oncogenic Ras in tumour maintenance. Nature 1999, 400, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef]
- McNeal, A.S.; Liu, K.; Nakhate, V.; Natale, C.A.; Duperret, E.K.; Capell, B.C.; Dentchev, T.; Berger, S.L.; Herlyn, M.; Seykora, J.T.; et al. CDKN2B Loss Promotes Progression from Benign Melanocytic Nevus to Melanoma. Cancer Discov. 2015, 5, 1072–1085. [Google Scholar] [CrossRef]
- Raaijmakers, M.I.G.; Widmer, D.S.; Narechania, A.; Eichhoff, O.; Freiberger, S.N.; Wenzina, J.; Cheng, P.F.; Mihic-Probst, D.; Desalle, R.; Dummer, R.; et al. Co-existence of BRAF and NRAS driver mutations in the same melanoma cells results in heterogeneity of targeted therapy resistance. Oncotarget 2016, 7, 77163–77174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandalà, M.; Merelli, B.; Massi, D. Nras in melanoma: Targeting the undruggable target. Crit. Rev. Oncol. Hematol. 2014, 92, 107–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.K.; Reynoird, N.; Khatri, P.; Jansen, P.W.; Wilkinson, A.W.; Liu, S.; Barbash, O.; Van Aller, G.S.; Huddleston, M.; Dhanak, D.; et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Postow, M.A.; Carvajal, R.D. Therapeutic implications of KIT in melanoma. Cancer J. 2012, 18, 137–141. [Google Scholar] [CrossRef]
- Kalinsky, K.; Lee, S.; Rubin, K.M.; Lawrence, D.P.; Iafrarte, A.J.; Borger, D.R.; Margolin, K.A.; Leitao, M.M.; Tarhini, A.A.; Koon, H.B.; et al. A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: A trial of the ECOG-ACRIN Cancer Research Group (E2607). Cancer 2017, 123, 2688–2697. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed]
- Gottesdiener, L.S.; O’Connor, S.; Busam, K.J.; Won, H.; Solit, D.B.; Hyman, D.M.; Shoushtari, A.N. Rates of ERBB2 Alterations across Melanoma Subtypes and a Complete Response to Trastuzumab Emtansine in an ERBB2-Amplified Acral Melanoma. Clin. Cancer Res. 2018, 24, 5815–5819. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, S.; Preziosi, M.; Rothberg, P.G.; Zeitouni, N.; Corson, N.; Xu, L. ERBB3 is required for metastasis formation of melanoma cells. Oncogenesis 2014, 3, e110. [Google Scholar] [CrossRef] [Green Version]
- Birkeland, E.; Zhang, S.; Poduval, D.; Geisler, J.; Nakken, S.; Vodak, D.; Meza-Zepeda, L.A.; Hovig, E.; Myklebost, O.; Knappskog, S.; et al. Patterns of genomic evolution in advanced melanoma. Nat. Commun. 2018, 9, 2665. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Agrawal, N.S.; Wei, X.; Yates, K.E.; Lin, J.C.; Wunderlich, J.R.; Cronin, J.C.; Cruz, P.; Rosenberg, S.A.; Samuels, Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat. Genet. 2009, 41, 1127–1132. [Google Scholar] [CrossRef]
- Prickett, T.D.; Wei, X.; Cardenas-Navia, I.; Teer, J.K.; Lin, J.C.; Walia, V.; Gartner, J.; Jiang, J.; Cherukuri, P.F.; Molinolo, A.; et al. Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nat. Genet. 2011, 43, 1119–1126. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Walia, V.; Lin, J.C.; Teer, J.K.; Prickett, T.D.; Gartner, J.; Davis, S.; Stemke-Hale, K.; Davies, M.A.; Gershenwald, J.E.; et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat. Genet. 2011, 43, 442–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosero, R.A.; Villares, G.J.; Bar-Eli, M. Protease-Activated Receptors and other G-Protein-Coupled Receptors: The Melanoma Connection. Front. Genet. 2016, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Manca, A.; Lissia, A.; Cossu, A.; Rubino, C.; Ascierto, P.A.; Stanganelli, I.; Palmieri, G. Mutations in ERBB4 may have a minor role in melanoma pathogenesis. J. Investig. Dermatol. 2013, 133, 1685–1687. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Sanchez-Laorden, B.; O’Brien, K.; Brunton, H.; Ferguson, J.; Young, H.; Dhomen, N.; Flaherty, K.T.; Frederick, D.T.; Cooper, Z.A.; et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov. 2014, 4, 1214–1229. [Google Scholar] [CrossRef]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wölfel, T.; Hölzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat. Commun. 2015, 6, 8755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hölzel, M.; Tüting, T. Inflammation-Induced Plasticity in Melanoma Therapy and Metastasis. Trends Immunol. 2016, 37, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.-K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [Green Version]
- Riveiro-Falkenbach, E.; Villanueva, C.A.; Garrido, M.C.; Ruano, Y.; García-Martín, R.M.; Godoy, E.; Ortiz-Romero, P.L.; Ríos-Martín, J.J.; Santos-Briz, A.; Rodríguez-Peralto, J.L. Intra- and Inter-Tumoral Homogeneity of BRAF(V600E) Mutations in Melanoma Tumors. J. Investig. Dermatol. 2015, 135, 3078–3085. [Google Scholar] [CrossRef]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.A.; Golub, T.R.; Lander, E.S.; Hynes, R.O. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000, 406, 532–535. [Google Scholar] [CrossRef] [Green Version]
- Bittner, M.; Meltzer, P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, J.; Koczan, D.; Thiesen, H.-J.; Ibrahim, S.M.; Gross, G.; Spang, R.; Kunz, M. Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser-microdissected melanoma tissues. Clin. Cancer Res. 2007, 13, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Mauerer, A.; Roesch, A.; Hafner, C.; Stempfl, T.; Wild, P.; Meyer, S.; Landthaler, M.; Vogt, T. Identification of new genes associated with melanoma. Exp. Dermatol. 2011, 20, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Kunz, M.; Löffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 2018. [Google Scholar] [CrossRef]
- Kuźbicki, L.; Aładowicz, E.; Chwirot, B.W. Cyclin-dependent kinase 2 expression in human melanomas and benign melanocytic skin lesions. Melanoma Res. 2006, 16, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, F.S.; Cantone, M.; Eberhardt, M.; Jaitly, T.; Walter, L.; Wittmann, J.; Gupta, S.K.; Khan, F.M.; Wolkenhauer, O.; Pützer, B.M.; et al. A web platform for the network analysis of high-throughput data in melanoma and its use to investigate mechanisms of resistance to anti-PD1 immunotherapy. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2315–2328. [Google Scholar] [CrossRef] [PubMed]
- Rukhlenko, O.S.; Khorsand, F.; Krstic, A.; Rozanc, J.; Alexopoulos, L.G.; Rauch, N.; Erickson, K.E.; Hlavacek, W.S.; Posner, R.G.; Gómez-Coca, S.; et al. Dissecting RAF Inhibitor Resistance by Structure-based Modeling Reveals Ways to Overcome Oncogenic RAS Signaling. Cell Syst. 2018, 7, 161–179. [Google Scholar] [CrossRef]
- Khan, F.M.; Marquardt, S.; Gupta, S.K.; Knoll, S.; Schmitz, U.; Spitschak, A.; Engelmann, D.; Vera, J.; Wolkenhauer, O.; Pützer, B.M. Unraveling a tumor type-specific regulatory core underlying E2F1-mediated epithelial-mesenchymal transition to predict receptor protein signatures. Nat. Commun. 2017, 8, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zecena, H.; Tveit, D.; Wang, Z.; Farhat, A.; Panchal, P.; Liu, J.; Singh, S.J.; Sanghera, A.; Bainiwal, A.; Teo, S.Y.; et al. Systems biology analysis of mitogen activated protein kinase inhibitor resistance in malignant melanoma. BMC Syst. Biol. 2018, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.; Schmitz, U.; Lai, X.; Engelmann, D.; Khan, F.M.; Wolkenhauer, O.; Pützer, B.M. Kinetic modeling-based detection of genetic signatures that provide chemoresistance via the E2F1-p73/DNp73-miR-205 network. Cancer Res. 2013, 73, 3511–3524. [Google Scholar] [CrossRef]
- Lai, X.; Gupta, S.K.; Schmitz, U.; Marquardt, S.; Knoll, S.; Spitschak, A.; Wolkenhauer, O.; Pützer, B.M.; Vera, J. MiR-205-5p and miR-342-3p cooperate in the repression of the E2F1 transcription factor in the context of anticancer chemotherapy resistance. Theranostics 2018, 8, 1106–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Muller, M.; Smith, D.; Dutta, B.; Komurov, K.; Iadevaia, S.; Ruths, D.; Tseng, J.T.; Yu, S.; Yu, Q.; et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene 2011, 30, 4567–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, R.; Clifton-Bligh, R.; Molloy, M.P. Phosphoproteomics of MAPK inhibition in BRAF-mutated cells and a role for the lethal synergism of dual BRAF and CK2 inhibition. Mol. Cancer Ther. 2014, 13, 1894–1906. [Google Scholar] [CrossRef]
- Blüthgen, N.; Legewie, S.; Kielbasa, S.M.; Schramme, A.; Tchernitsa, O.; Keil, J.; Solf, A.; Vingron, M.; Schäfer, R.; Herzel, H.; et al. A systems biological approach suggests that transcriptional feedback regulation by dual-specificity phosphatase 6 shapes extracellular signal-related kinase activity in RAS-transformed fibroblasts. FEBS J. 2009, 276, 1024–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vera, J.; Rath, O.; Balsa-Canto, E.; Banga, J.R.; Kolch, W.; Wolkenhauer, O. Investigating dynamics of inhibitory and feedback loops in ERK signalling using power-law models. Mol. Biosyst. 2010, 6, 2174–2191. [Google Scholar] [CrossRef] [Green Version]
- Korkut, A.; Wang, W.; Demir, E.; Aksoy, B.A.; Jing, X.; Molinelli, E.J.; Babur, Ö.; Bemis, D.L.; Onur Sumer, S.; Solit, D.B.; et al. Perturbation biology nominates upstream-downstream drug combinations in RAF inhibitor resistant melanoma cells. eLife 2015, 4. [Google Scholar] [CrossRef]
- Klinger, B.; Sieber, A.; Fritsche-Guenther, R.; Witzel, F.; Berry, L.; Schumacher, D.; Yan, Y.; Durek, P.; Merchant, M.; Schäfer, R.; et al. Network quantification of EGFR signaling unveils potential for targeted combination therapy. Mol. Syst. Biol. 2013, 9, 673. [Google Scholar] [CrossRef]
- Kirouac, D.C.; Schaefer, G.; Chan, J.; Merchant, M.; Orr, C.; Huang, S.-M.A.; Moffat, J.; Liu, L.; Gadkar, K.; Ramanujan, S. Clinical responses to ERK inhibition in BRAFV600E-mutant colorectal cancer predicted using a computational model. NPJ Syst. Biol. Appl. 2017, 3, 14. [Google Scholar] [CrossRef]
- Kim, E.; Kim, J.-Y.; Smith, M.A.; Haura, E.B.; Anderson, A.R.A. Cell signaling heterogeneity is modulated by both cell-intrinsic and -extrinsic mechanisms: An integrated approach to understanding targeted therapy. PLoS Biol. 2018, 16, e2002930. [Google Scholar] [CrossRef]
- Alla, V.; Engelmann, D.; Niemetz, A.; Pahnke, J.; Schmidt, A.; Kunz, M.; Emmrich, S.; Steder, M.; Koczan, D.; Pützer, B.M. E2F1 in melanoma progression and metastasis. J. Natl. Cancer Inst. 2010, 102, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Gerber, T.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Schadendorf, D.; Schartl, M.; Anderegg, U.; Camp, G.; Treutlein, B.; Binder, H.; et al. Mapping heterogeneity in patient-derived melanoma cultures by single-cell RNA-seq. Oncotarget 2017, 8, 846–862. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunz, M.; Vera, J. Modelling of Protein Kinase Signaling Pathways in Melanoma and Other Cancers. Cancers 2019, 11, 465. https://doi.org/10.3390/cancers11040465
Kunz M, Vera J. Modelling of Protein Kinase Signaling Pathways in Melanoma and Other Cancers. Cancers. 2019; 11(4):465. https://doi.org/10.3390/cancers11040465
Chicago/Turabian StyleKunz, Manfred, and Julio Vera. 2019. "Modelling of Protein Kinase Signaling Pathways in Melanoma and Other Cancers" Cancers 11, no. 4: 465. https://doi.org/10.3390/cancers11040465
APA StyleKunz, M., & Vera, J. (2019). Modelling of Protein Kinase Signaling Pathways in Melanoma and Other Cancers. Cancers, 11(4), 465. https://doi.org/10.3390/cancers11040465