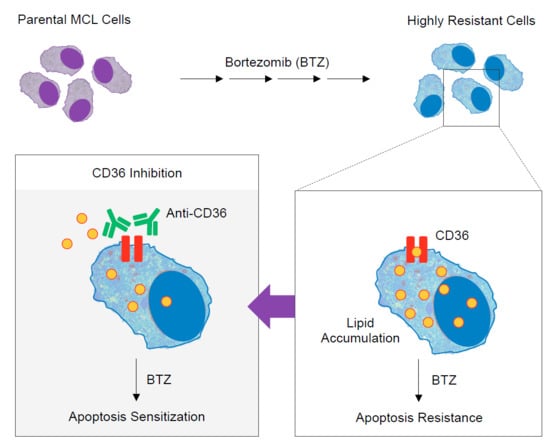
Deciphering the Elevated Lipid via CD36 in Mantle Cell Lymphoma with Bortezomib Resistance Using Synchrotron-Based Fourier Transform Infrared Spectroscopy of Single Cells

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
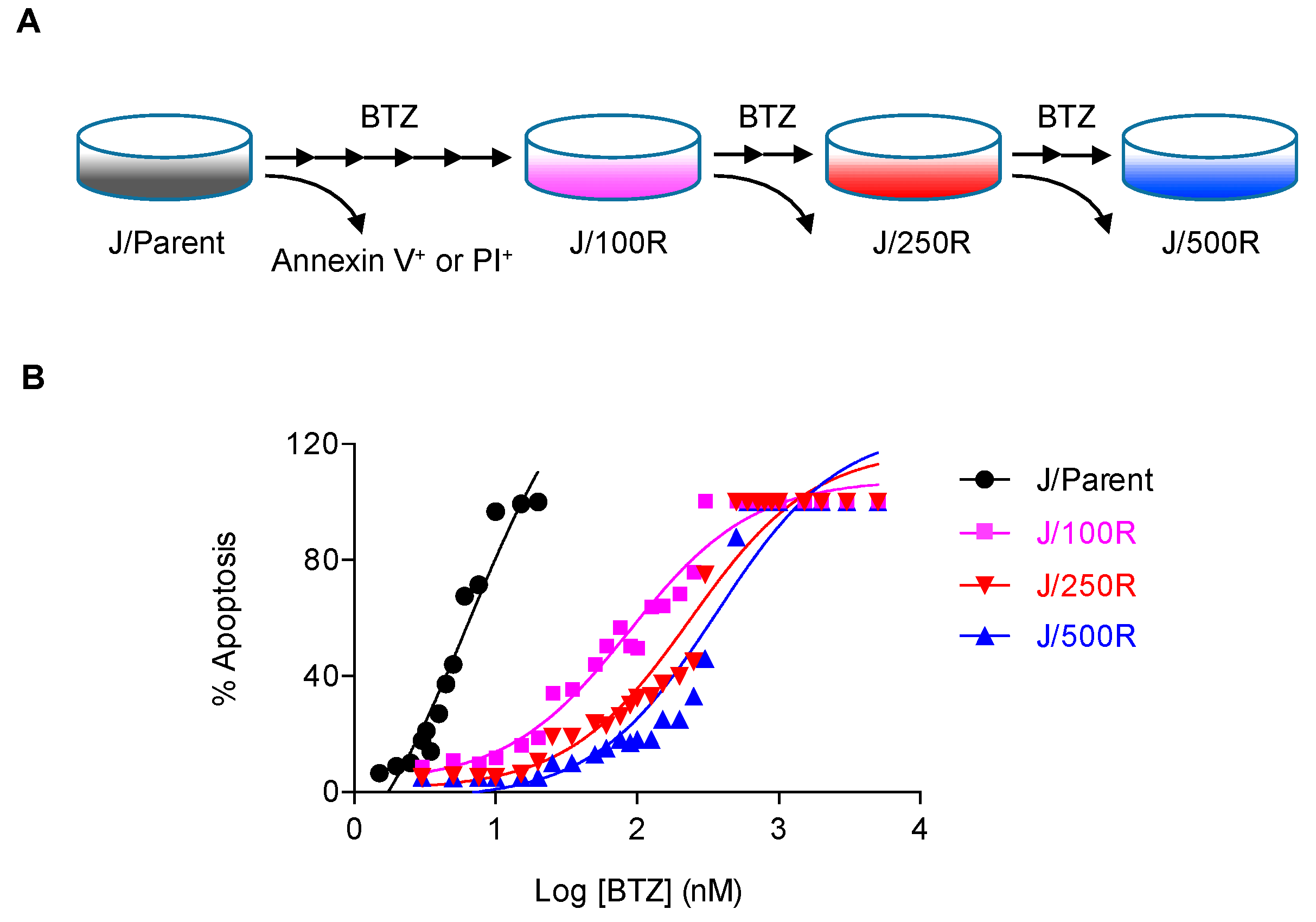
2.1. Establishment of BTZ-Resistant MCL Cells and Their Sensitivity to BTZ
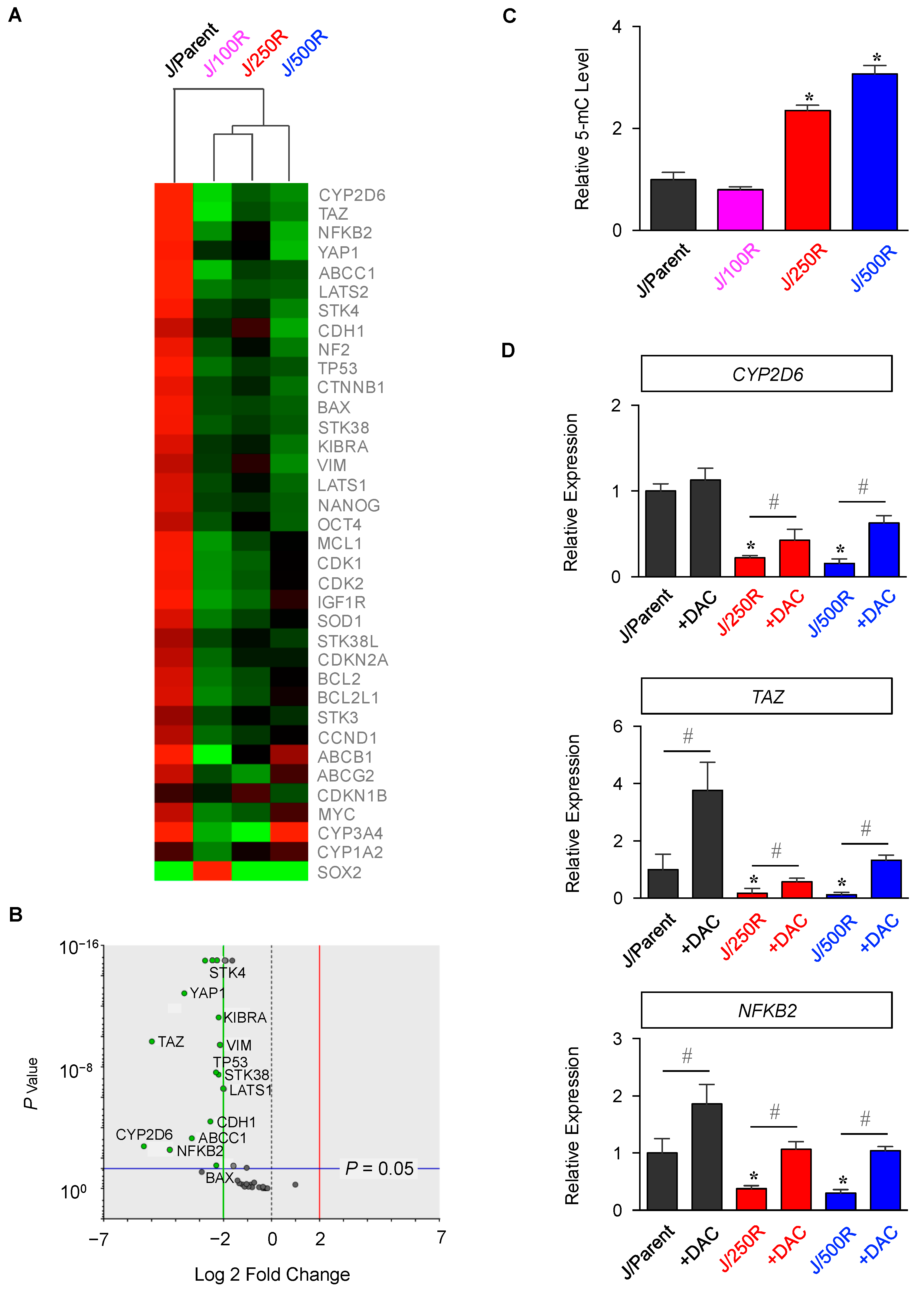
2.2. Gene Expression Profiling of BTZ-Resistant MCL
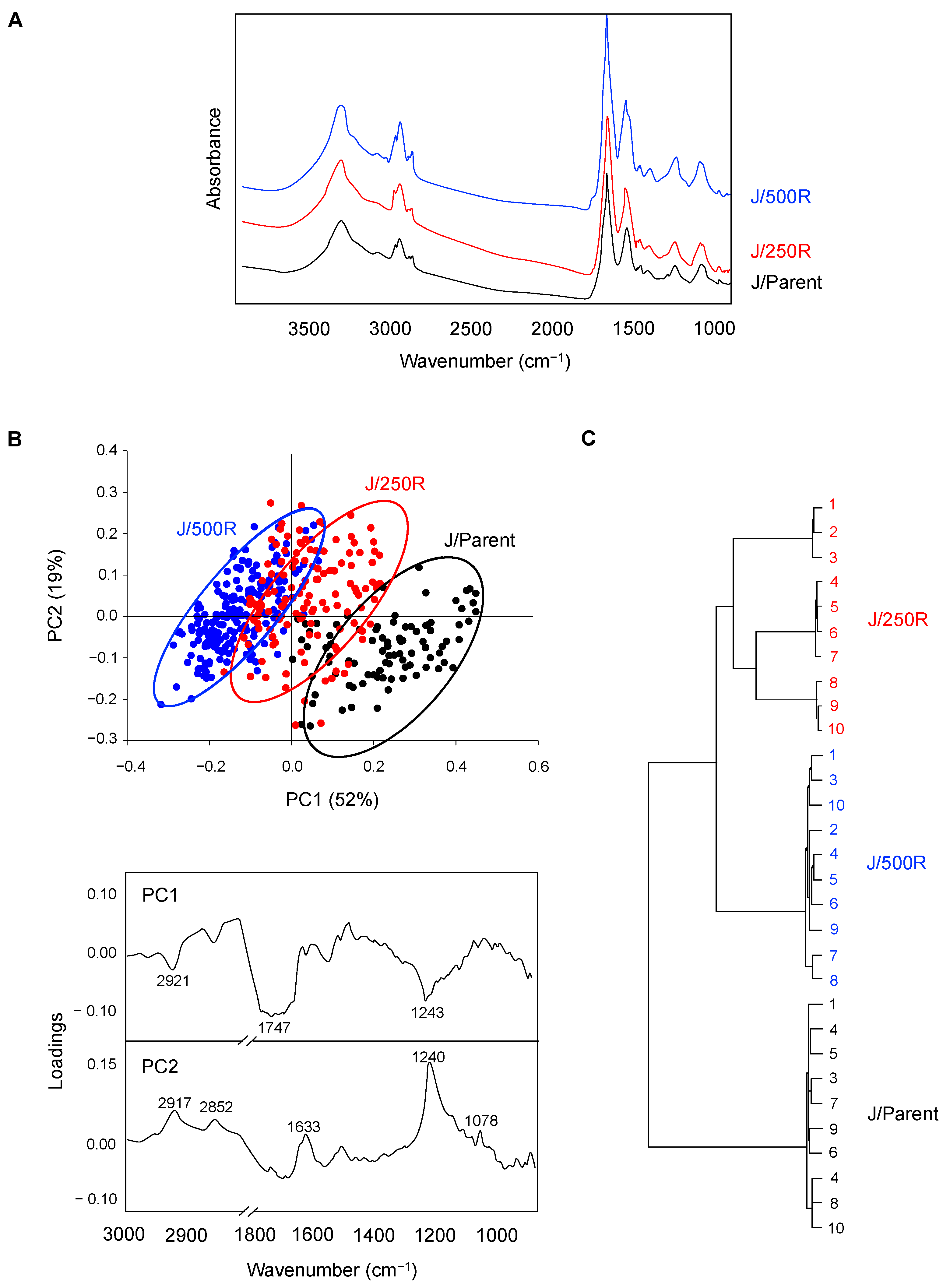
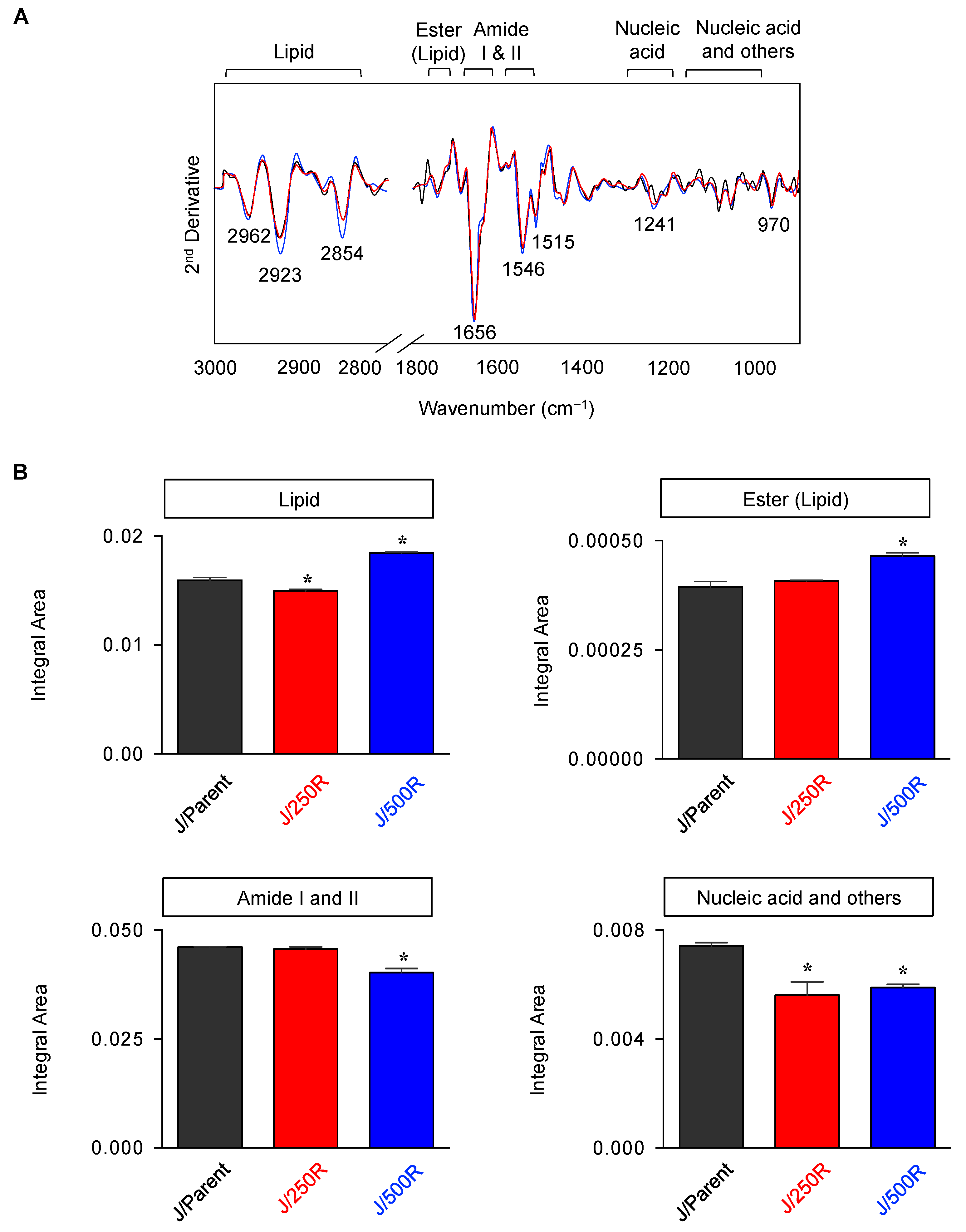
2.3. FTIR Signatures of BTZ-Resistant MCL
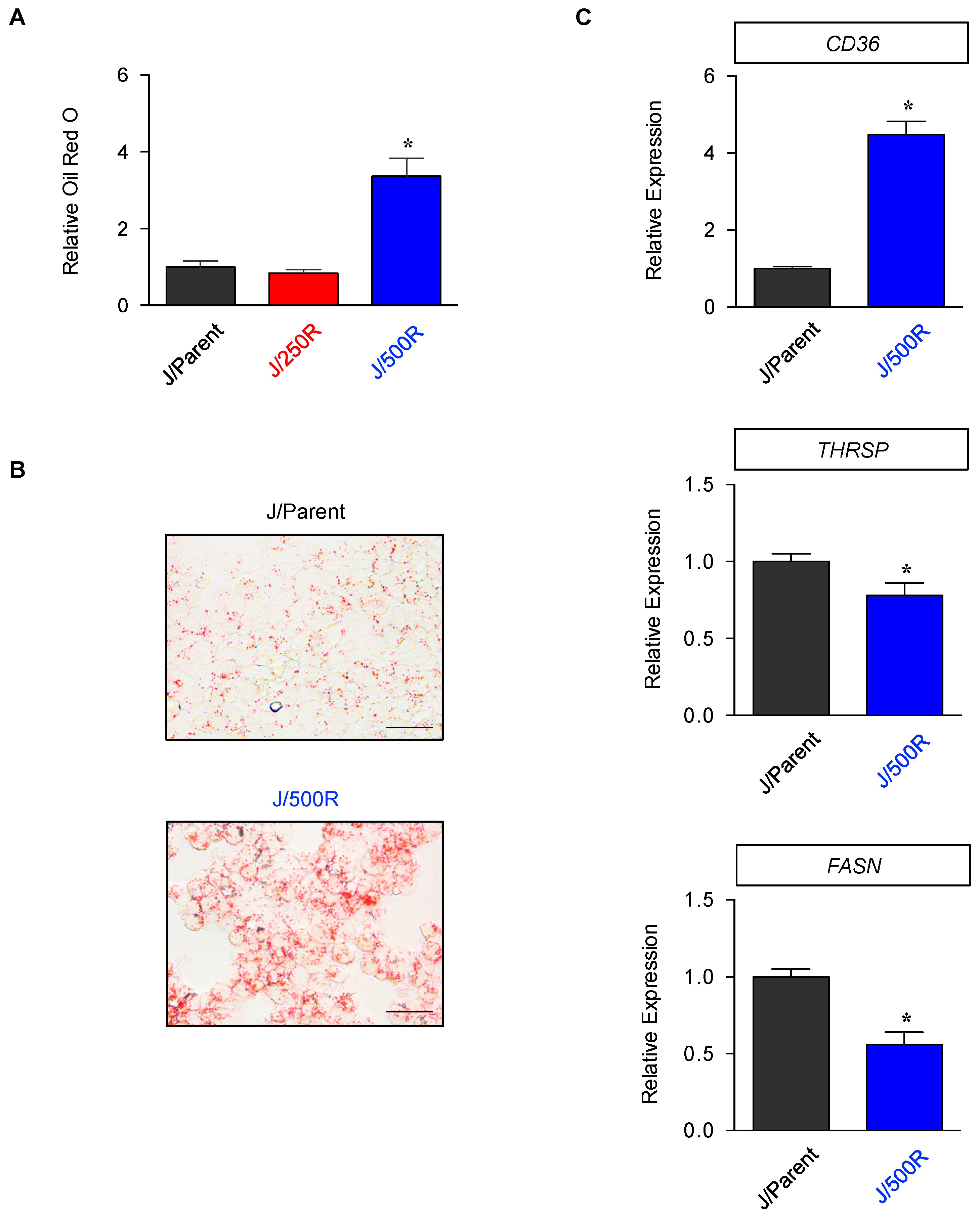
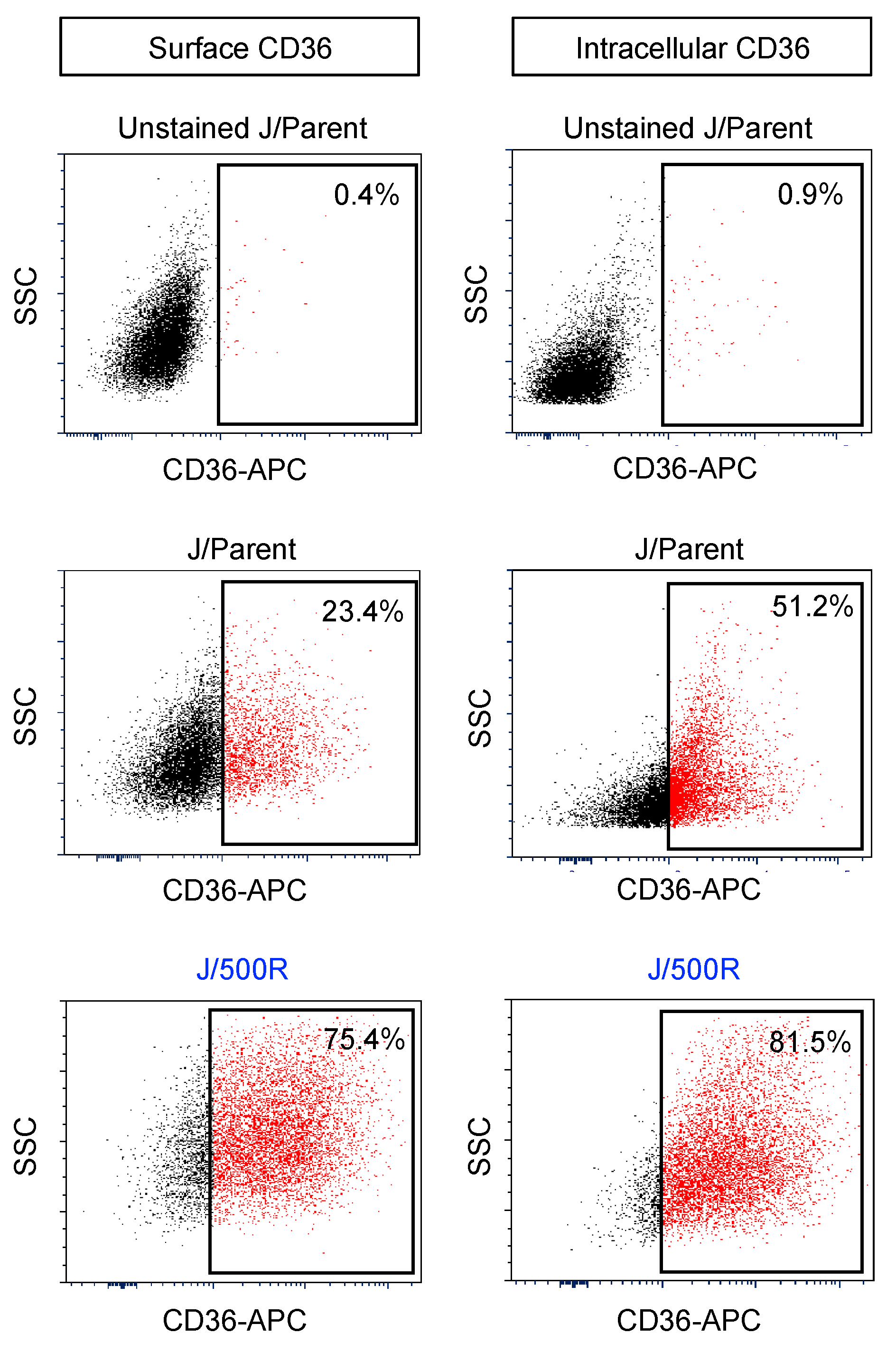
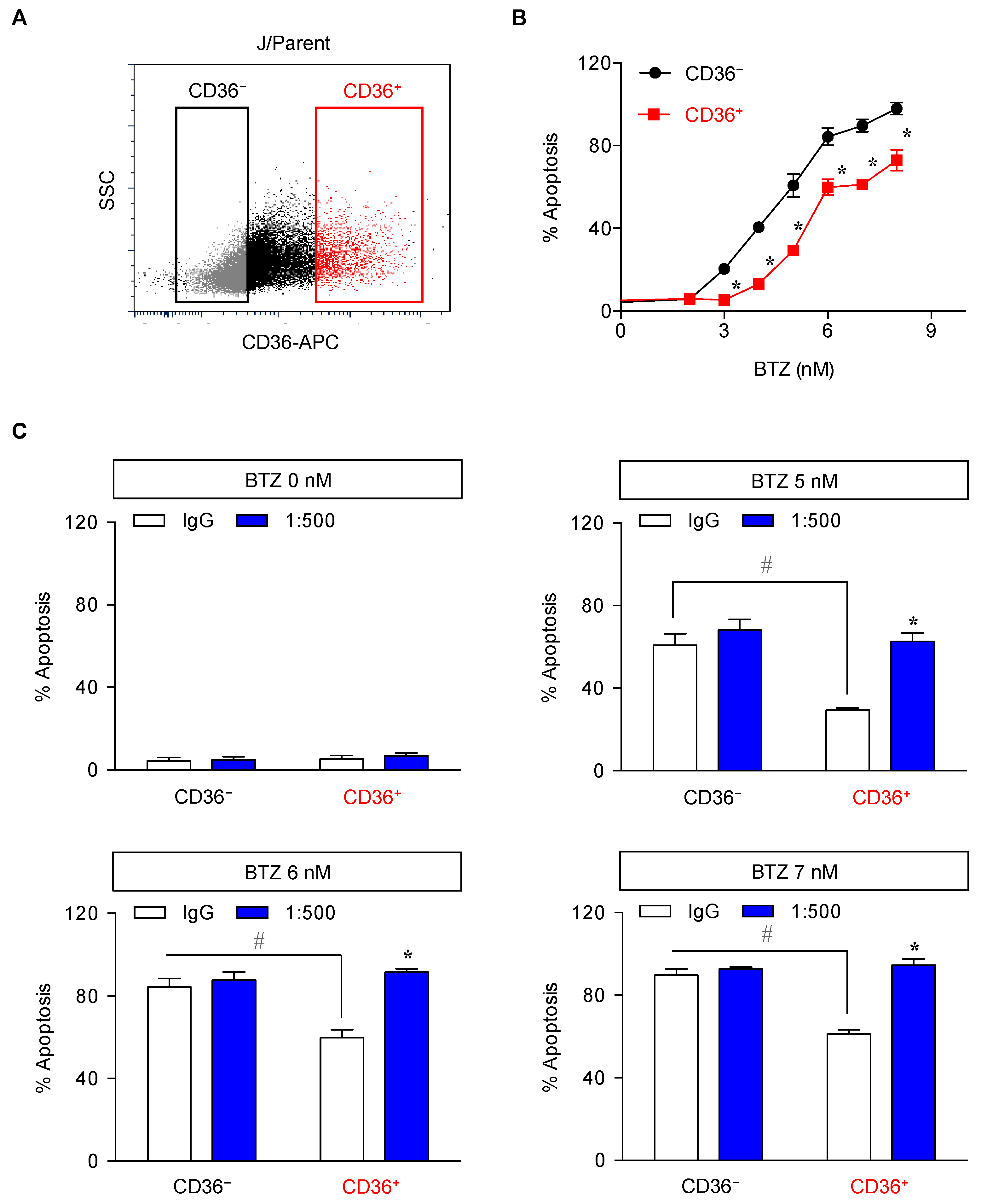
2.4. Elevated Lipid Content and CD36 Upregulation in BTZ-Resistant MCL
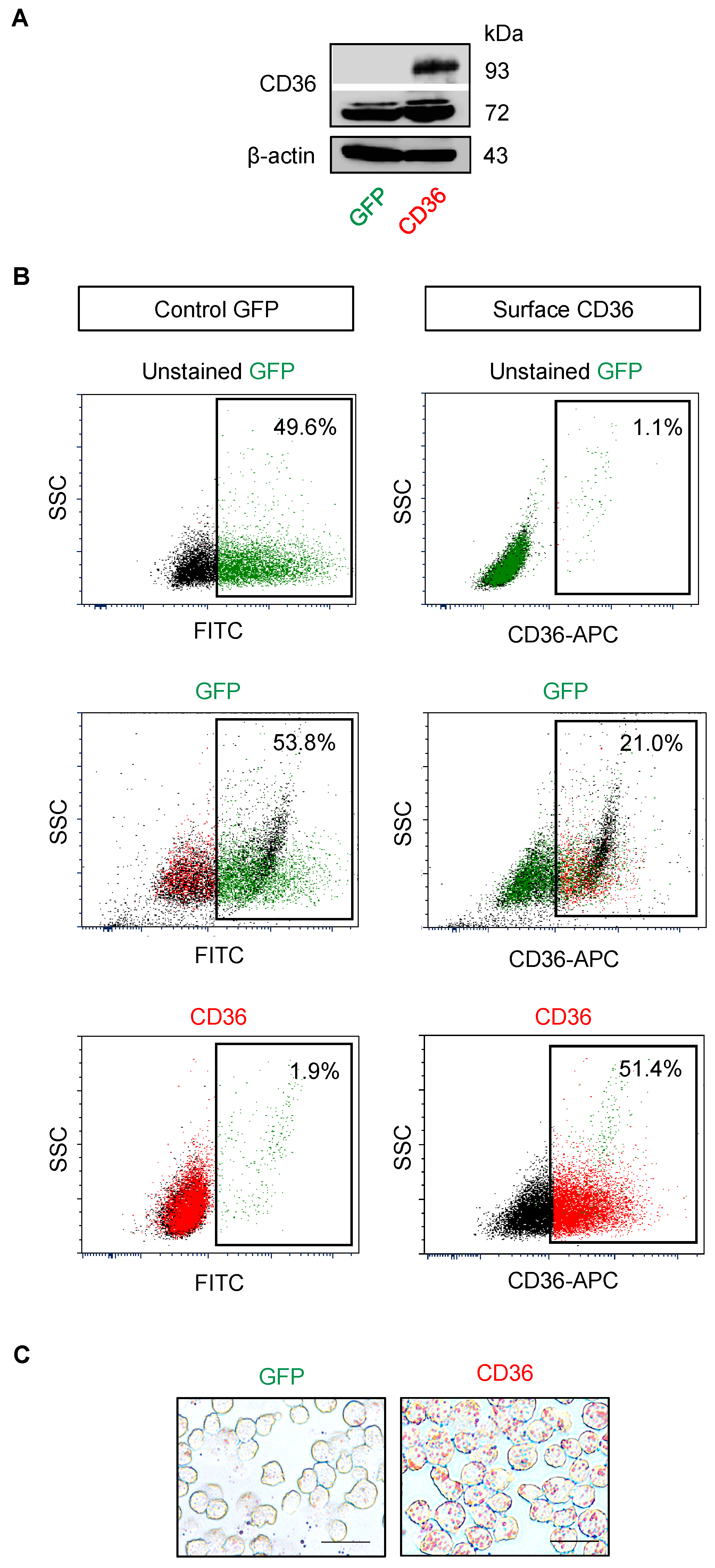
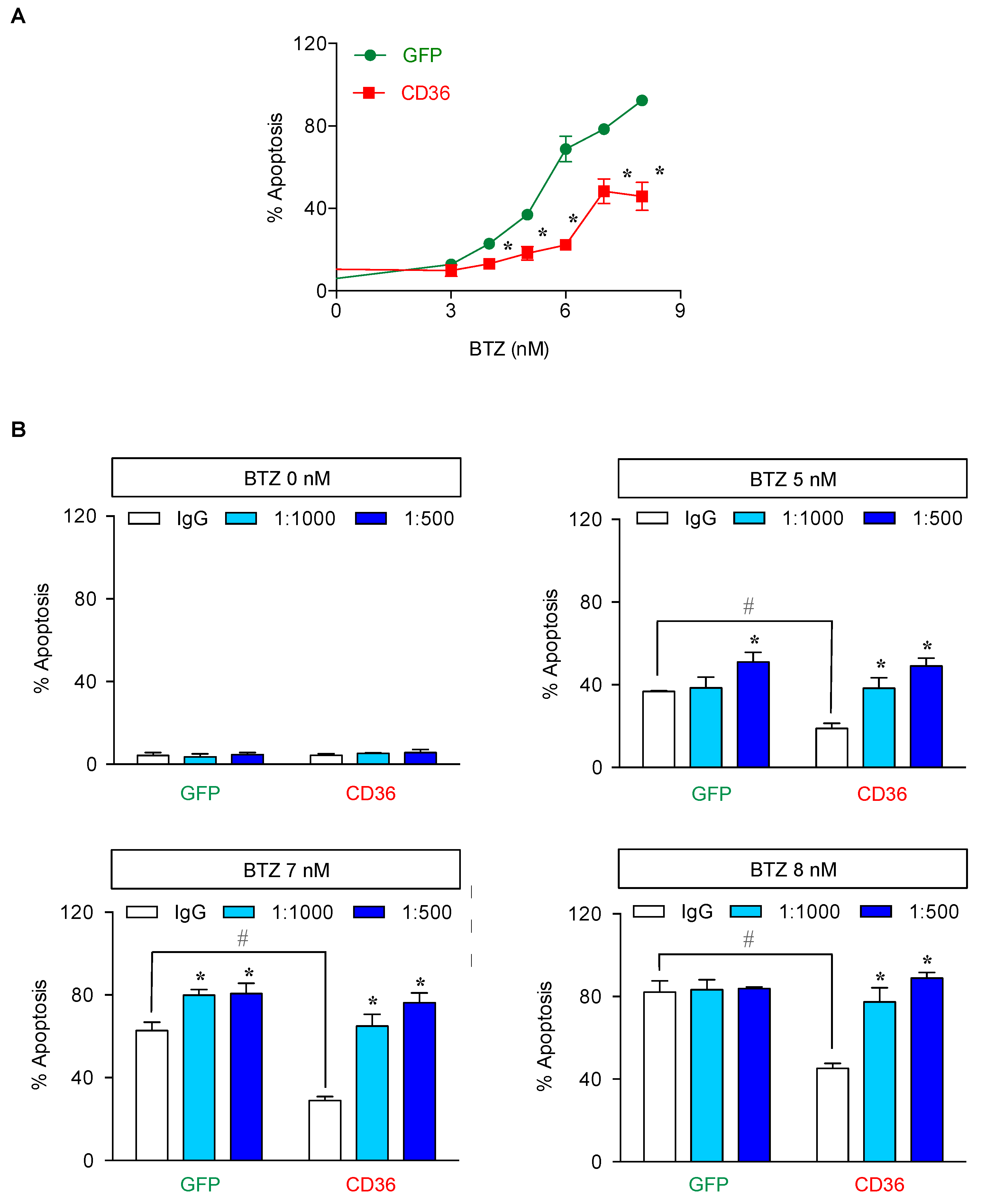
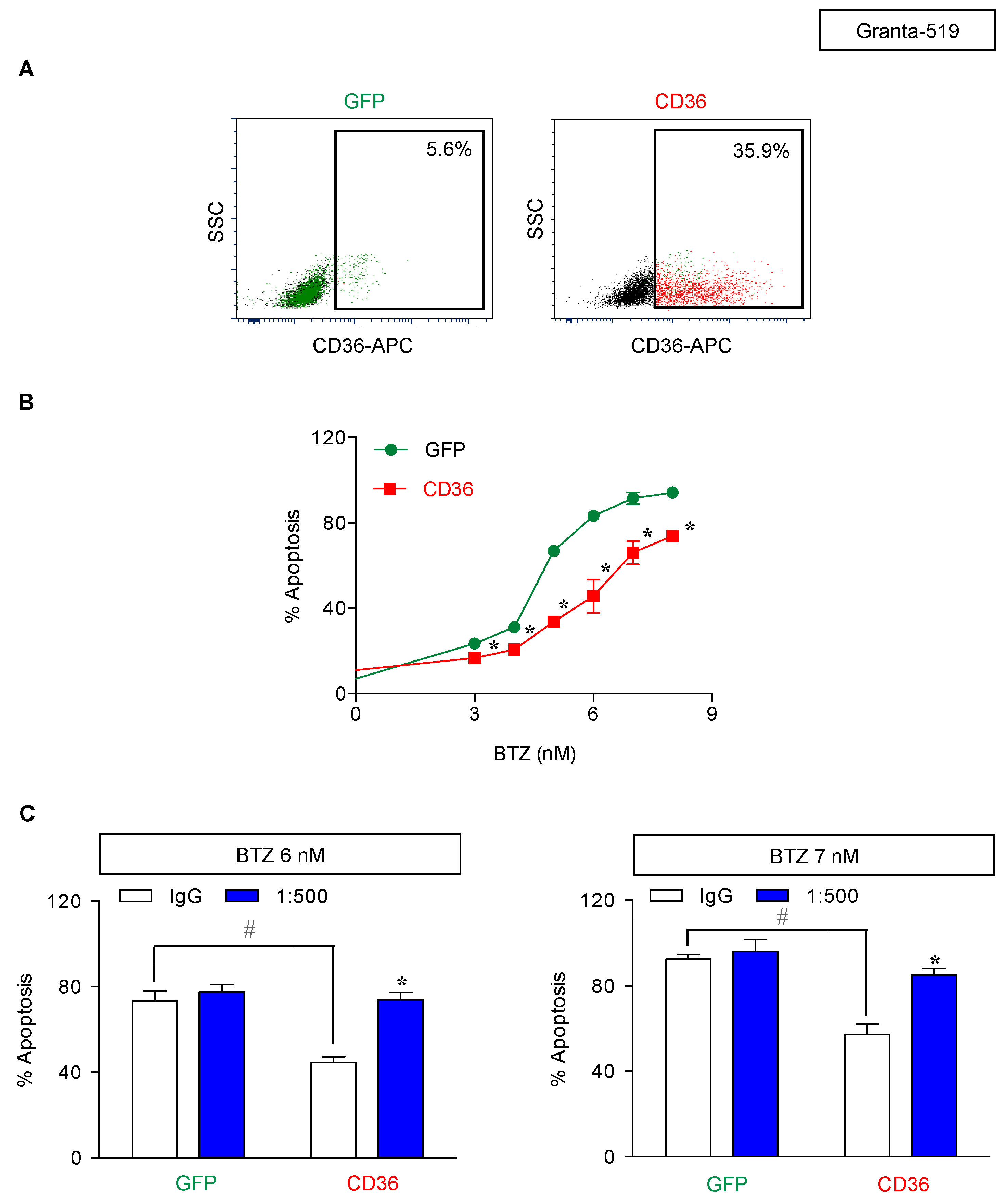
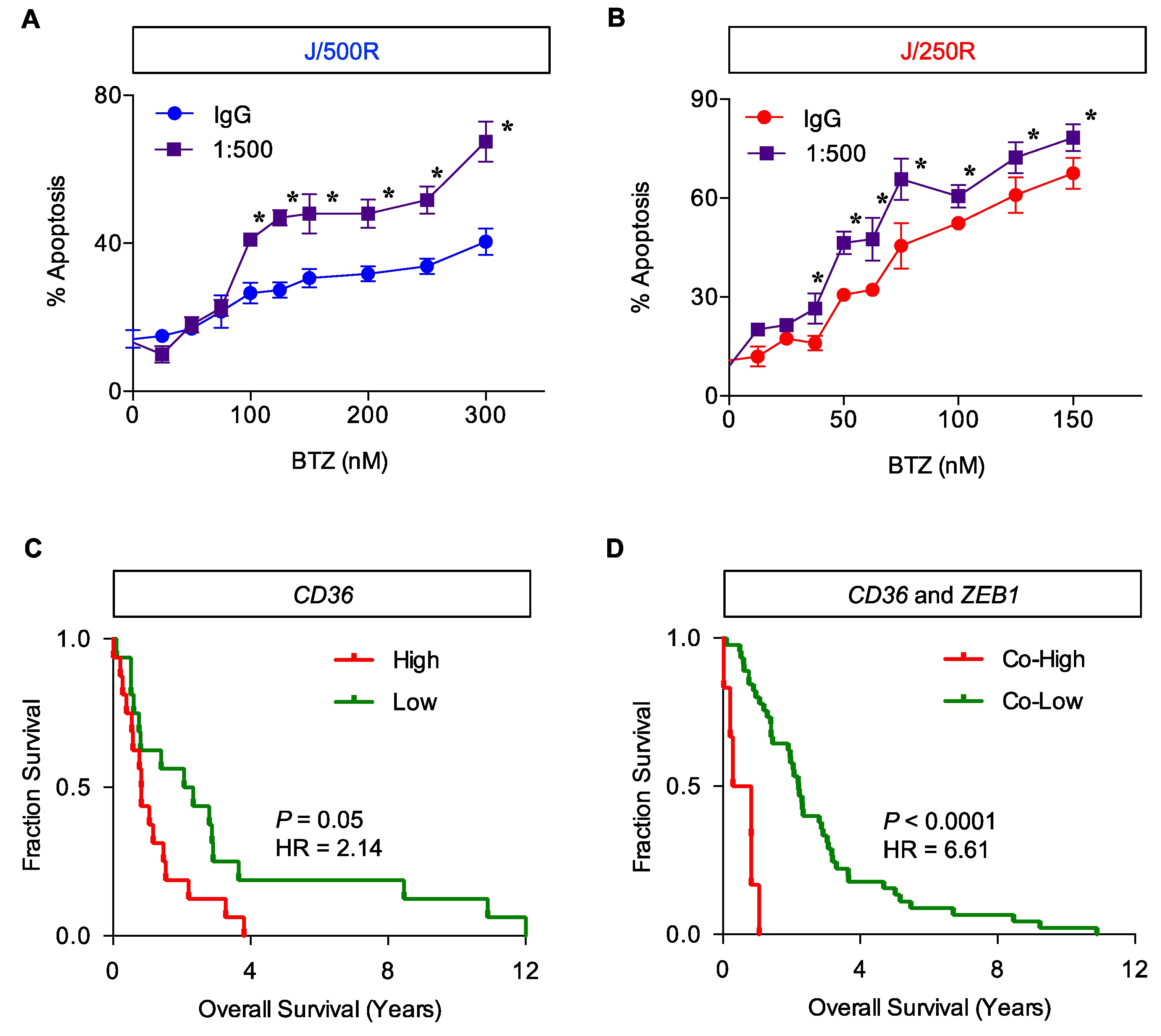
2.5. CD36 Renders MCL Cells to BTZ Resistance and Is an Attractive Co-Target
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Generation of De Novo BTZ-Resistant Cells
4.3. Apoptosis Assay
4.4. RNA Isolation and Quantitative PCR
4.5. FTIR Sample Preparation and Microspectroscopy Analysis
4.6. DNA Methylation Status
4.7. Oil Red O Staining and Quantification
4.8. CD36 Staining and Flow Cytometric Analysis
4.9. Plasmids and Transfection
4.10. Western Blotting
4.11. Gene Microarray Dataset and Survival Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perez-Galan, P.; Dreyling, M.; Wiestner, A. Mantle cell lymphoma: Biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood 2011, 117, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.D.; Martin, P.; Sotomayor, E.M. Mantle cell lymphoma: A clinically heterogeneous disease in need of tailored approaches. Cancer Control 2012, 19, 227–235. [Google Scholar] [CrossRef]
- Prichard, M.; Harris, T.; Williams, M.E.; Densmore, J.J. Treatment strategies for relapsed and refractory aggressive non-Hodgkin’s lymphoma. Expert. Opin. Pharmacother. 2009, 10, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Zaja, F.; Federico, M.; Vitolo, U.; Zinzani, P.L. Management of relapsed/refractory mantle cell lymphoma: A review of current therapeutic strategies. Leuk. Lymphoma 2013, 55, 988–998. [Google Scholar] [CrossRef]
- Diefenbach, C.S.; O’Connor, O.A. Mantle cell lymphoma in relapse: The role of emerging new drugs. Curr. Opin. Oncol. 2010, 22, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Hambley, B.; Caimi, P.F.; William, B.M. Bortezomib for the treatment of mantle cell lymphoma: An update. Ther. Adv. Hematol. 2016, 7, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Arkwright, R.; Pham, T.M.; Zonder, J.A.; Dou, Q.P. The preclinical discovery and development of bortezomib for the treatment of mantle cell lymphoma. Expert Opin. Drug Discov. 2017, 12, 225–235. [Google Scholar] [CrossRef]
- Yazbeck, V.; Shafer, D.; Perkins, E.B.; Coppola, D.; Sokol, L.; Richards, K.L.; Shea, T.; Ruan, J.; Parekh, S.; Strair, R.; et al. A phase II trial of bortezomib and vorinostat in mantle cell lymphoma and diffuse large b-cell lymphoma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 569–575. [Google Scholar] [CrossRef]
- Kruh, G.D. Introduction to resistance to anticancer agents. Oncogene 2003, 22, 7262–7264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, M.C.E.; Melvin, W.T.; Murray, G.I. Cytochrome P450 enzymes: Novel options for cancer therapeutics. Mol. Cancer Ther. 2004, 3, 363–371. [Google Scholar]
- Pommier, Y.; Sorder, O.; Antony, S.; Hayward, R.L.; Kohn, K.W. Apoptosis defects and chemotherapy resistance: Molecular interaction maps and networks. Oncogene 2004, 23, 2934–2949. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Galán, P.; Mora-Jensen, H.; Weniger, M.A.; Shaffer III, A.L.; Rizzatti, E.G.; Chapman, C.M.; Mo, C.C.; Stennett, L.S.; Rader, C.; Liu, P.; et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood 2011, 117, 542–552. [Google Scholar] [CrossRef]
- Kim, A.; Seong, K.M.; Kang, H.J.; Park, S.; Lee, S.S. Inhibition of Lyn is a promising treatment for mantle cell lymphoma with bortezomib resistance. Oncotarget 2015, 6, 38225–38238. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.A.; Schwartz, G.K. Cell cycle-mediated drug resistance: An emerging concept in cancer therapy. Clin. Cancer Res. 2001, 7, 2168–2181. [Google Scholar] [PubMed]
- Dai, B.; Grau, M.; Juilland, M.; Klener, P.; Höring, E.; Molinsky, J.; Schimmack, G.; Aukema, S.M.; Hoster, E.; Vogt, N.; et al. B-cell receptor-driven MALT1 activity regulates MYC signaling in mantle cell lymphoma. Blood 2017, 129, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Bisikirska, B.C.; Adam, S.J.; Alvarez, M.J.; Rajbhandari, P.; Cox, R.; Lefebvre, C.; Wang, K.; Rieckhof, G.E.; Felsher, D.W.; Califano, A. STK38 is a critical upstream regulator of MYC’s oncogenic activity in human B-cell lymphoma. Oncogene 2013, 32, 5283–5291. [Google Scholar] [CrossRef]
- Vishwamitra, D.; Shi, P.; Wilson, D.; Manshouri, R.; Vega, F.; Schlette, E.J.; Amin, H.M. Expression and effects of inhibition of type I insulin-like growth factor receptor tyrosine kinasein mantle cell lymphoma. Haematologica 2011, 96, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Auer, R. Discovery of Hippo in MCL. Blood 2010, 116, 861–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Tilló, E.; Fanlo, L.; Siles, L.; Montes-Moreno, S.; Moros, A.; Chiva-Blanch, G.; Estruch, R.; Martinez, A.; Colomer, D.; Győrffy, B.; et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. 2014, 21, 247–257. [Google Scholar] [CrossRef]
- Samaniego, F.; Sehgal, L.; Braun, F.K.; Berkova, Z.; Romaguera, J.E.; Wang, M.; Rodriguez, M.A.; Neelapu, S.S.; Mathur, R. Molecular signatures of tumor-initiating cells unveil Wnt pathway as a therapeutic target in mantle cell lymphoma. Blood 2014, 124, 2148. [Google Scholar]
- Curradi, M.; Izzo, A.; Badaracco, G.; Landsberger, N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol. Cell. Biol. 2002, 22, 3157–3173. [Google Scholar] [CrossRef]
- Breiling, A.; Lyko, F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics Chromatin. 2015, 8, 24. [Google Scholar] [CrossRef]
- Güler, G.; Acikgoz, E.; Karabay Yavasoglu, N.Ü.; Bakan, B.; Goormaghtigh, E.; Aktug, H. Deciphering the biochemical similarities and differences among mouse embryonic stem cells, somatic and cancer cells using ATR-FTIR spectroscopy. Analyst 2018, 143, 1624–1634. [Google Scholar] [CrossRef]
- Mourant, J.R.; Yamada, Y.R.; Carpenter, S.; Dominique, L.R.; Freyer, J.P. FTIR spectroscopy demonstrates biochemical differences in mammalian cell cultures at different growth stages. Biophys. J. 2003, 85, 1938–1947. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Sunshine, H.; Iruela-Arispe, M.L. Membrane lipids and cell signaling. Curr. Opin. Lipidol. 2017, 28, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Bozza, P.T.; Viola, J.P.B. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fatty Acids 2010, 4–6, 243–250. [Google Scholar] [CrossRef]
- Danilova, O.V.; Dumont, L.J.; Levy, N.B.; Lansigan, F.; Kinlaw, W.B.; Danilov, A.V.; Kaur, P. FASN and CD36 predict survival in rituximab-treated diffuse large B-cell lymphoma. J. Hematop. 2013, 6, 11–18. [Google Scholar] [CrossRef]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Luanpitpong, S.; Poohadsuan, J.; Samart, P.; Kiratipaiboon, C.; Rojanasakul, Y.; Issaragrisil, S. Reactive oxygen species mediate cancer stem-like cells and determine bortezomib sensitivity via Mcl-1 and Zeb-1 in mantle cell lymphoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3739–3753. [Google Scholar] [CrossRef]
- Blenk, S.; Engelmann, J.C.; Pinkert, S.; Weniger, M.; Schultz, J.; Rosenwald, A.; Müller-Hermelink, H.K.; Müller, T.; Dandekar, T. Explorative data analysis of MCL reveals gene expression networks implicated in survival and prognosis supported by explorative CGH analysis. BMC Cancer 2008, 8, 106. [Google Scholar] [CrossRef]
- Nyce, J. Drug-induced DNA hypermethylation and drug resistance in human tumors. Cancer Res. 1989, 49, 5826–5836. [Google Scholar]
- Nyce, J.W. Drug-induced DNA hypermethylation: A potential mediator of acquired drug resistance during cancer chemotherapy. Mutat. Res. 1997, 386, 153–161. [Google Scholar] [CrossRef]
- Segura-Pacheco, B.; Perez-Cardenas, E.; Taja-Chayeb, L.; Chavez-Blanco, A.; Revilla-Vazquez, A.; Benitez-Bribiesca, L.; Duenas-González, A. Global DNA hypermethylation-associated cancer chemotherapy resistance and its reversion with the demethylating agent hydralazine. J. Transl. Med. 2006, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.Y.; Mirkin, B.L.; Dwivedi, R.S. Differential expression of DNA-methyltransferases in drug resistant murine neuroblastoma cells. Cancer Detect. Prev. 2002, 26, 444–453. [Google Scholar] [CrossRef]
- Qiu, Y.Y.; Mirkin, B.L.; Dwivedi, R.S. Inhibition of DNA methyltransferase reverses cisplatin induced drug resistance in murine neuroblastoma cells. Cancer Detect. Prev. 2005, 29, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 645–674. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Lu, C.; Lindsten, T.; Thompson, C.B. Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol. 2012, 30, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Luanpitpong, S.; Chanthra, N.; Janan, M.; Poohadsuan, J.; Samart, P.; U-Pratya, Y.; Rojanasakul, Y.; Issaragrisil, S. Inhibition of O-GlcNAcase sensitizes apoptosis and reverses bortezomib resistance in mantle cell lymphoma through modification of truncated Bid. Mol. Cancer Ther. 2018, 17, 484–496. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Miao, H.; Cheng, X.L.; Wei, F. Lipidomics: Novel insight into the biochemical mechanism of lipid metabolism and dysregulation-associated disease. Chem. Biol. Interact. 2015, 240, 220–238. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Yeon, A.; Shahid, M.; Cho, E.; Sairam, V.; Figlin, R.; Kim, K.H.; Kim, J. Reprogrammed lipid metabolism in bladder cancer with cisplatin resistance. Oncotarget 2018, 9, 13231–13243. [Google Scholar] [CrossRef]
- Rak, S.; De Zan, T.; Stefulj, J.; Kosović, M.; Gamulin, O.; Osmak, M. FTIR spectroscopy reveals lipid droplets in drug resistant laryngeal carcinoma cells through detection of increased ester vibrational bands intensity. Analyst 2014, 139, 3407–3415. [Google Scholar] [CrossRef]
- Enciu, A.M.; Radu, E.; Popescu, I.D.; Hinescu, M.E.; Ceafalan, L.C. Targeting CD36 as biomarker for metastasis prognostic: How far from translation into clinical practice? Biomed Res. Int. 2018, 2018, 7801202. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Alaterre, E.; Raimbault, S.; Goldschmidt, H.; Bouhya, S.; Requirand, G.; Robert, N.; Boireau, S.; Seckinger, A.; Hose, D.; Klein, B.; et al. CD24, CD27, CD36 and CD302 gene expression for outcome prediction in patients with multiple myeloma. Oncotarget 2017, 8, 98931–98944. [Google Scholar] [CrossRef] [Green Version]
- Gubelmann, C.; Schwalie, P.C.; Raghav, S.K.; Röder, E.; Delessa, T.; Kiehlmann, E.; Waszak, S.M.; Corsinotti, A.; Udin, G.; Holcombe, W.; et al. Identification of the transcription factor ZEB1 as a central component of the adipogenic generegulatory network. Elife 2014, 3, e03346. [Google Scholar] [CrossRef]
- Amin, H.M.; McDonnell, T.J.; Medeiros, L.J.; Rassidakis, G.Z.; Leventaki, V.; O’Connor, S.L.; Keating, M.J.; Lai, R. Characterization of 4 mantle cell lymphoma cell lines. Arch. Pathol. Lab. Med. 2003, 127, 424–431. [Google Scholar] [PubMed]
- Kridel, R.; Meissner, B.; Rogic, S.; Boyle, M.; Telenius, A.; Woolcock, B.; Gunawardana, J.; Jenkins, C.; Cochrane, C.; Ben-Neriah, S.; et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 2012, 119, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.R.; Mo, H.; Bieberich, A.A.; Alavanja, T.; Colby, D.A. Fluorinated amino-derivatives of the sesquiterpene lactone, parthenolide, as (19)f NMR probes in deuterium-free environments. J. Med. Chem. 2011, 54, 7934–7941. [Google Scholar] [CrossRef] [PubMed]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luanpitpong, S.; Janan, M.; Thumanu, K.; Poohadsuan, J.; Rodboon, N.; Klaihmon, P.; Issaragrisil, S. Deciphering the Elevated Lipid via CD36 in Mantle Cell Lymphoma with Bortezomib Resistance Using Synchrotron-Based Fourier Transform Infrared Spectroscopy of Single Cells. Cancers 2019, 11, 576. https://doi.org/10.3390/cancers11040576
Luanpitpong S, Janan M, Thumanu K, Poohadsuan J, Rodboon N, Klaihmon P, Issaragrisil S. Deciphering the Elevated Lipid via CD36 in Mantle Cell Lymphoma with Bortezomib Resistance Using Synchrotron-Based Fourier Transform Infrared Spectroscopy of Single Cells. Cancers. 2019; 11(4):576. https://doi.org/10.3390/cancers11040576
Chicago/Turabian StyleLuanpitpong, Sudjit, Montira Janan, Kanjana Thumanu, Jirarat Poohadsuan, Napachai Rodboon, Phatchanat Klaihmon, and Surapol Issaragrisil. 2019. "Deciphering the Elevated Lipid via CD36 in Mantle Cell Lymphoma with Bortezomib Resistance Using Synchrotron-Based Fourier Transform Infrared Spectroscopy of Single Cells" Cancers 11, no. 4: 576. https://doi.org/10.3390/cancers11040576
APA StyleLuanpitpong, S., Janan, M., Thumanu, K., Poohadsuan, J., Rodboon, N., Klaihmon, P., & Issaragrisil, S. (2019). Deciphering the Elevated Lipid via CD36 in Mantle Cell Lymphoma with Bortezomib Resistance Using Synchrotron-Based Fourier Transform Infrared Spectroscopy of Single Cells. Cancers, 11(4), 576. https://doi.org/10.3390/cancers11040576