The Key Roles of PTEN in T-Cell Acute Lymphoblastic Leukemia Development, Progression, and Therapeutic Response

 ,
,  , and
, and 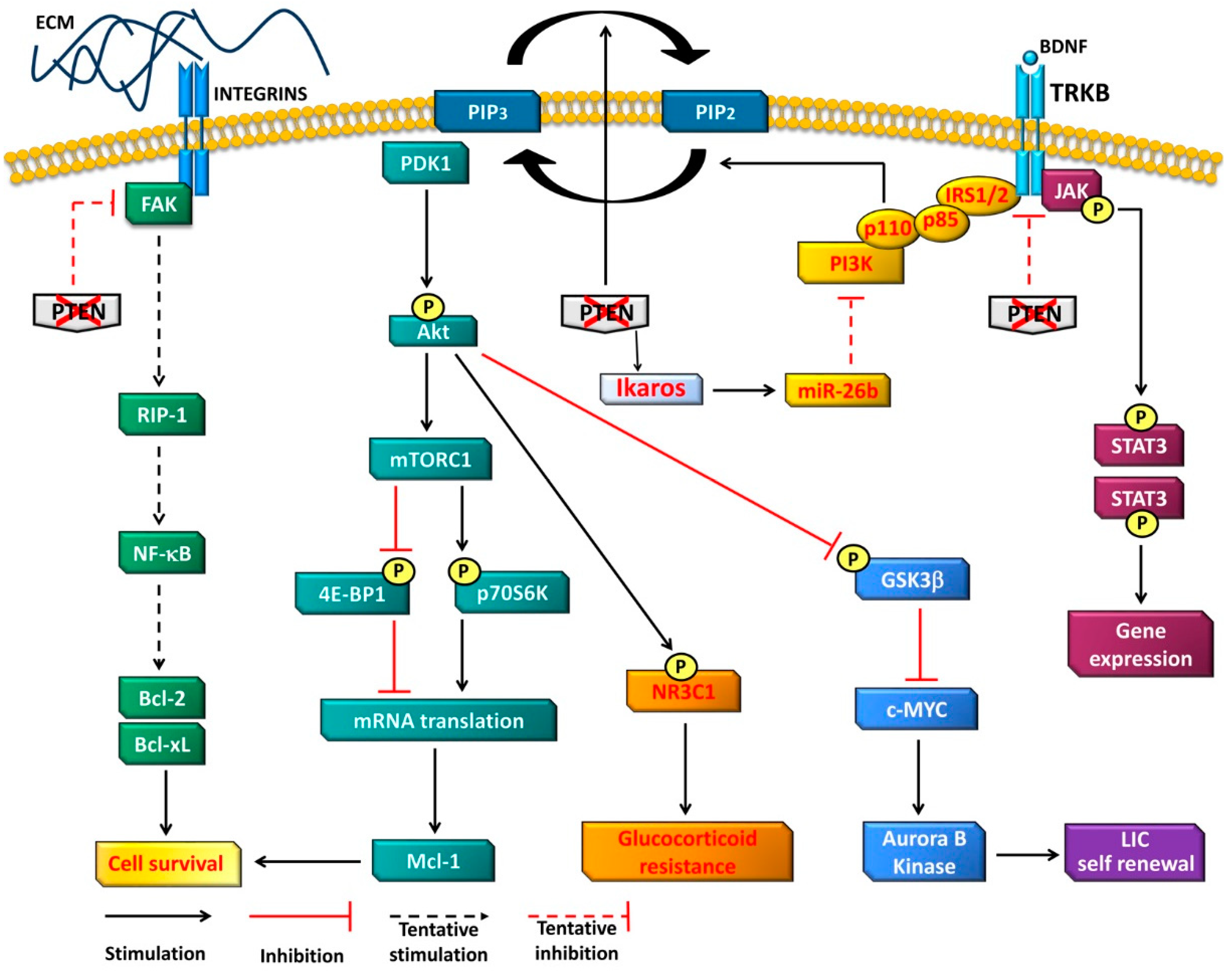{kind=link}
{kind=link}
Abstract
:1. Introduction
2. PTEN and T-ALL Development in Mice
2.1. PTEN Conditional Deletion in HSCs
2.2. PTEN Conditional Deletions in T-Cells
2.3. Studies in Human T-ALL Cells
3. PTEN Regulation in Human T-ALL
3.1. Genetic Mechanisms of PTEN Inactivation
3.2. Non-Genetic Mechanisms of PTEN Inactivation
4. Oncogenetic Functions of PTEN in T-ALL Cells
4.1. PI3K-Dependent Functions
4.2. PI3K-Independent Functions
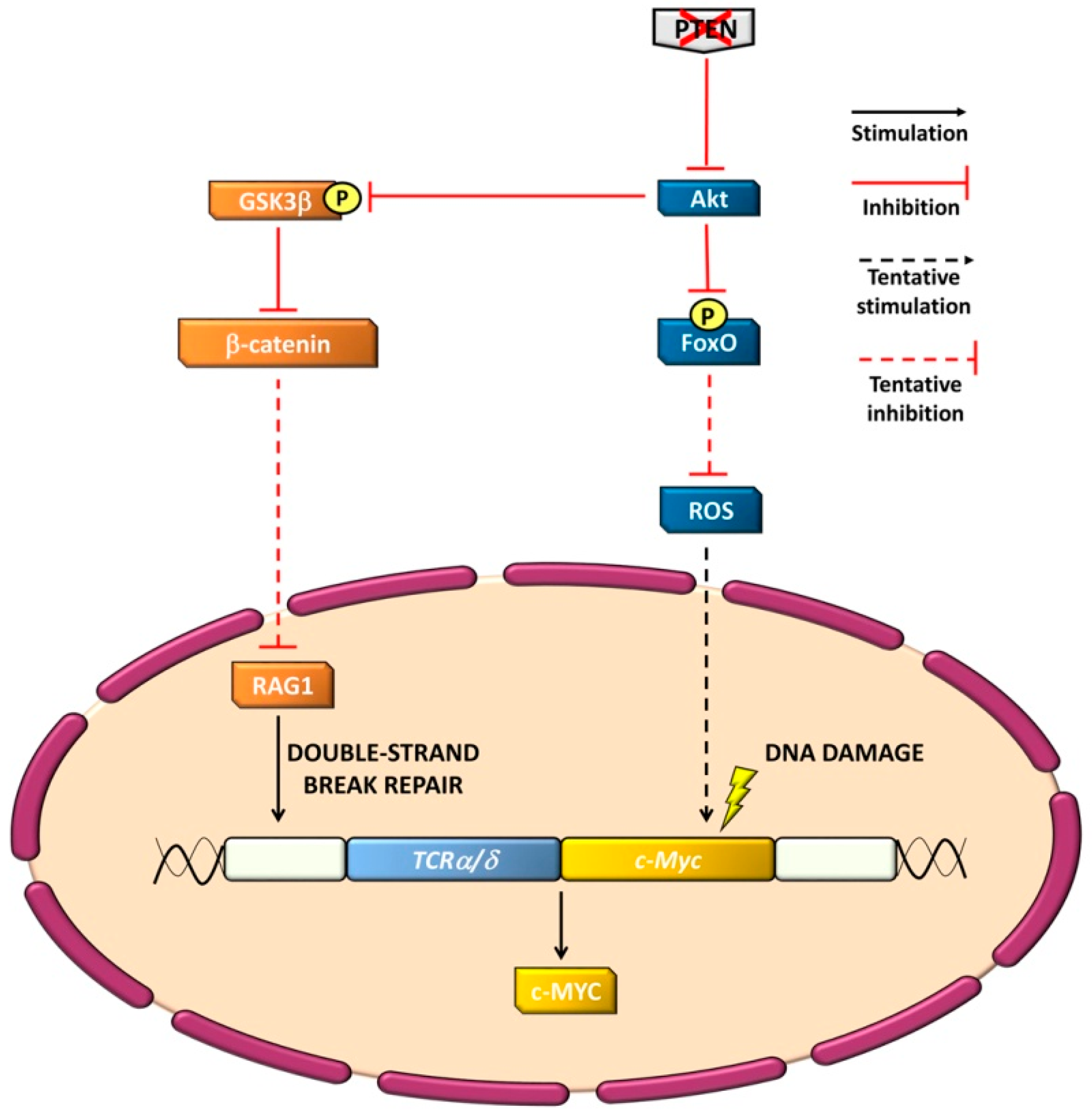
4.3. Genomic Stability
5. Clinical Impact of PTEN Loss on T-ALL Patient Outcome
6. Therapeutic Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Bcl-xL | B-cell lymphoma-extra large |
| Bcl-2 | B-cell lymphoma 2 |
| Bim | Bcl-2-like protein 11 |
| BM | Bone marrow |
| BDNF | Brain-derived neurotrophic factor |
| CCL25 | C-C chemokine ligand 25 |
| CCR9 | C-C chemokine receptor 9 |
| CK2 | Casein kinase 2 |
| c-MYC | Avian myelocytomatosis viral homolog |
| E2f | E2 transcription factor |
| ECM | Extracellular matrix |
| ERK | Extracellular signal-regulated kinase |
| ETP T-ALL | Early T-cell precursor T-ALL |
| Ets | E-twenty-six |
| 4E-BP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| FAK | Focal adhesion kinase |
| FBXW7 | F-box WD repeat-containing protein 7 |
| FLT3 | Fms-related tyrosine kinase3 |
| FoxO | Forkhead box protein O |
| GCs | Glucocorticoids |
| GSI | γ-secretase inhibitors |
| GS3Kβ | Glycogen synthase kinase 3β |
| HAVCR2 | Hepatitis A virus cellular receptor 2 |
| HES-1 | Hairy and enhancer of split-1 |
| HOX | Homeobox |
| HSCs | Hematopoietic stem cells |
| JAK | Janus kinase |
| IRS | Insulin receptor substrate |
| LMO | LIM domain only |
| Mcl-1 | Myeloid cell leukemia sequence 1 |
| MEK | Mitogen-activated protein kinase kinase |
| miRs | MicroRNAs |
| MRE11 | Meiotic recombination 11 homolog 1 |
| mTOR | Mechanistic target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| NF-κB | Nuclear factor-kB |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| NF-κB | Nuclear factor kappa B |
| NR3C1 | Nuclear receptor subfamily 3 group C member 1 |
| PDK1 | Phosphoinositide-dependent kinase 1 |
| PI3K | Phosphatidylinositol-3 kinase |
| PINK-1 | PTEN-induced putative kinase protein 1 |
| PIP2 | Phosphatidylinositol(4,5)P2 |
| PIP3 | Phosphatidylinositol(3,4,5)P3 |
| PTEN | Phosphatase and Tensin homolog, deleted on chromosome TEN |
| PU.1 | PU-box 1 protein |
| p70S6K | 70-kDa ribosomal protein S6 kinase |
| RAD50 | Rad1 homolog 50 |
| RAF | Rapidly-accelerated fibrosarcoma |
| RAG | Recombination activating gene |
| RAS | Rat sarcoma |
| RIP1 | Receptor activating protein 1 |
| Rpl24 | Ribosome protein L24 |
| ROS | Reactive oxygen species |
| SCL | Stem cell leukemia |
| STIL | SCL/TAL1 interrupting locus |
| SPI1 | Spleen focus forming virus proviral integration oncogene 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| T-ALL | T-cell acute lymphoblastic leukemia |
| TAL1 | T-cell acute lymphocytic leukemia 1 |
| TCR | T-cell receptor |
| Tcrα/δ-c-Myc | T-cell receptor α/δ-c-Myc |
| TLX | Nuclear receptor subfamily 2 group E member |
| TRK | Tropomyosin-related kinase A |
References
- Chiaretti, S.; Vitale, A.; Cazzaniga, G.; Orlando, S.M.; Silvestri, D.; Fazi, P.; Valsecchi, M.G.; Elia, L.; Testi, A.M.; Mancini, F.; et al. Clinico-biological features of 5202 patients with acute lymphoblastic leukemia enrolled in the Italian AIEOP and GIMEMA protocols and stratified in age cohorts. Haematologica 2013, 98, 1702–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paganin, M.; Ferrando, A. Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia. Blood Rev. 2011, 25, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Conter, V.; Valsecchi, M.G.; Buldini, B.; Parasole, R.; Locatelli, F.; Colombini, A.; Rizzari, C.; Putti, M.C.; Barisone, E.; Lo Nigro, L.; et al. Early T-cell precursor acute lymphoblastic leukaemia in children treated in AIEOP centres with AIEOP-BFM protocols: A retrospective analysis. Lancet. Haematol. 2016, 3, e80–e86. [Google Scholar] [CrossRef]
- Gianfelici, V.; Chiaretti, S.; Demeyer, S.; Di Giacomo, F.; Messina, M.; La Starza, R.; Peragine, N.; Paoloni, F.; Geerdens, E.; Pierini, V.; et al. RNA sequencing unravels the genetics of refractory/relapsed T-cell acute lymphoblastic leukemia. Prognostic and therapeutic implications. Haematologica 2016, 101, 941–950. [Google Scholar] [CrossRef]
- Richter-Pechanska, P.; Kunz, J.B.; Hof, J.; Zimmermann, M.; Rausch, T.; Bandapalli, O.R.; Orlova, E.; Scapinello, G.; Sagi, J.C.; Stanulla, M.; et al. Identification of a genetically defined ultra-high-risk group in relapsed pediatric T-lymphoblastic leukemia. Blood Cancer J. 2017, 7, e523. [Google Scholar] [CrossRef]
- Teepen, J.C.; van Leeuwen, F.E.; Tissing, W.J.; van Dulmen-den Broeder, E.; van den Heuvel-Eibrink, M.M.; van der Pal, H.J.; Loonen, J.J.; Bresters, D.; Versluys, B.; Neggers, S.; et al. Long-term risk of subsequent malignant neoplasms after treatment of childhood cancer in the DCOG LATER study cohort: Role of chemotherapy. J. Clin. Oncol. 2017, 35, 2288–2298. [Google Scholar] [CrossRef]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Ferrando, A. Can one target T-cell ALL? Best Pract. Res. Clin. Haematol. 2018, 31, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Vadillo, E.; Dorantes-Acosta, E.; Pelayo, R.; Schnoor, M. T cell acute lymphoblastic leukemia (T-ALL): New insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018, 32, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.K.; Zhang, C.; Sanda, T. Oncogenic transcriptional program driven by TAL1 in T-cell acute lymphoblastic leukemia. Int. J. Hematol. 2019, 109, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: it’s all about diversity. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple functions in human malignant tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Boutouja, F.; Stiehm, C.M.; Platta, H.W. mTOR: A cellular regulator interface in health and disease. Cells 2019, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the clinical development of PI3K Inhibitors: Strategies to improve their impact in solid tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Cheng, H.; Santiago, S.; Raeder, M.; Zhang, F.; Isabella, A.; Yang, J.; Semaan, D.J.; Chen, C.; Fox, E.A.; et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat. Med. 2011, 17, 1116–1120. [Google Scholar] [CrossRef]
- Kinross, K.M.; Montgomery, K.G.; Kleinschmidt, M.; Waring, P.; Ivetac, I.; Tikoo, A.; Saad, M.; Hare, L.; Roh, V.; Mantamadiotis, T.; et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J. Clin. Investig. 2012, 122, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Ngeow, J.; Sesock, K.; Eng, C. Clinical Implications for Germline PTEN spectrum disorders. Endocrinol. Metab. Clin. N. Am. 2017, 46, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.Q.; Ouyang, M.; Brandmaier, A.; Hao, H.; Shen, W.H. PTEN in the maintenance of genome integrity: From DNA replication to chromosome segregation. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Douglas, W.; Lia, M.; Edelmann, W.; Kucherlapati, R.; Podsypanina, K.; Parsons, R.; Ellenson, L.H. DNA mismatch repair deficiency accelerates endometrial tumorigenesis in Pten heterozygous mice. Am. J. Pathol. 2002, 160, 1481–1486. [Google Scholar] [CrossRef]
- Myers, M.P.; Stolarov, J.P.; Eng, C.; Li, J.; Wang, S.I.; Wigler, M.H.; Parsons, R.; Tonks, N.K. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. USA 1997, 94, 9052–9057. [Google Scholar] [CrossRef] [Green Version]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; He, S.; Yang, J.; Jia, X.; Wang, P.; Chen, X.; Zhang, Z.; Zou, X.; McNutt, M.A.; Shen, W.H.; et al. PTENα, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab 2014, 19, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Chen, X.; Yin, Q.; Ruan, D.; Zhao, X.; Zhang, C.; McNutt, M.A.; Yin, Y. PTENβ is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat. Commun. 2017, 8, 14771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cho, Y.L.; Tang, Y.; Wang, J.; Park, J.E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res 2018, 28, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jin, Y.; Liu, B.; Lu, D.; Zhu, M.; McNutt, M.A.; Yin, Y. PTENα promotes neutrophil chemotaxis through regulation of cell deformability. Blood 2019. [Google Scholar] [CrossRef] [PubMed]
- Elich, M.; Sauer, K. Regulation of hematopoietic cell development and function through phosphoinositides. Front. Immunol. 2018, 9, 931. [Google Scholar] [CrossRef]
- Richmond, C.A.; Shah, M.S.; Carlone, D.L.; Breault, D.T. Factors regulating quiescent stem cells: Insights from the intestine and other self-renewing tissues. J. Physiol. 2016, 594, 4805–4813. [Google Scholar] [CrossRef]
- Shrestha, S.; Yang, K.; Guy, C.; Vogel, P.; Neale, G.; Chi, H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat. Immunol. 2015, 16, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Seda, V.; Mraz, M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur. J. Haematol. 2015, 94, 193–205. [Google Scholar] [CrossRef]
- Wang, J.; Liu, S.; Hou, B.; Yang, M.; Dong, Z.; Qi, H.; Liu, W. PTEN-regulated AID transcription in germinal center B cells is essential for the class-switch recombination and IgG antibody responses. Front. Immunol. 2018, 9, 371. [Google Scholar] [CrossRef]
- Leong, J.W.; Schneider, S.E.; Sullivan, R.P.; Parikh, B.A.; Anthony, B.A.; Singh, A.; Jewell, B.A.; Schappe, T.; Wagner, J.A.; Link, D.C.; et al. PTEN regulates natural killer cell trafficking in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, E700–E709. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef]
- Buckler, J.L.; Liu, X.; Turka, L.A. Regulation of T-cell responses by PTEN. Immunol. Rev. 2008, 224, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Nolla, H.; Suzuki, A.; Mak, T.W.; Winoto, A. Normal development is an integral part of tumorigenesis in T cell-specific PTEN-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 2022–2027. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Karnell, J.L.; Yin, B.; Zhang, R.; Zhang, J.; Li, P.; Choi, Y.; Maltzman, J.S.; Pear, W.S.; Bassing, C.H.; et al. Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. J. Clin. Investig. 2010, 120, 2497–2507. [Google Scholar] [CrossRef] [Green Version]
- Hagenbeek, T.J.; Spits, H. T-cell lymphomas in T-cell-specific Pten-deficient mice originate in the thymus. Leukemia 2008, 22, 608–619. [Google Scholar] [CrossRef]
- Mendes, R.D.; Cante-Barrett, K.; Pieters, R.; Meijerink, J.P. The relevance of PTEN-AKT in relation to NOTCH1-directed treatment strategies in T-cell acute lymphoblastic leukemia. Haematologica 2016, 101, 1010–1017. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Tesio, M.; Trinquand, A.; Macintyre, E.; Asnafi, V. Oncogenic PTEN functions and models in T-cell malignancies. Oncogene 2016, 35, 3887–3896. [Google Scholar] [CrossRef]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef]
- Guo, W.; Lasky, J.L.; Chang, C.J.; Mosessian, S.; Lewis, X.; Xiao, Y.; Yeh, J.E.; Chen, J.Y.; Iruela-Arispe, M.L.; Varella-Garcia, M.; et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008, 453, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Tesio, M.; Oser, G.M.; Baccelli, I.; Blanco-Bose, W.; Wu, H.; Gothert, J.R.; Kogan, S.C.; Trumpp, A. Pten loss in the bone marrow leads to G-CSF-mediated HSC mobilization. J. Exp. Med. 2013, 210, 2337–2349. [Google Scholar] [CrossRef]
- Guo, W.; Schubbert, S.; Chen, J.Y.; Valamehr, B.; Mosessian, S.; Shi, H.; Dang, N.H.; Garcia, C.; Theodoro, M.F.; Varella-Garcia, M.; et al. Suppression of leukemia development caused by PTEN loss. Proc. Natl. Acad. Sci. USA 2011, 108, 1409–1414. [Google Scholar] [CrossRef] [Green Version]
- Schubbert, S.; Cardenas, A.; Chen, H.; Garcia, C.; Guo, W.; Bradner, J.; Wu, H. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014, 74, 7048–7059. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, L.; Wu, Y.; Dong, B.; Guo, W.; Wang, M.; Yang, L.; Fan, X.; Tang, Y.; Liu, N.; et al. T-ALL leukemia stem cell ‘stemness’ is epigenetically controlled by the master regulator SPI1. Elife 2018, 7. [Google Scholar] [CrossRef]
- La Starza, R.; Borga, C.; Barba, G.; Pierini, V.; Schwab, C.; Matteucci, C.; Lema Fernandez, A.G.; Leszl, A.; Cazzaniga, G.; Chiaretti, S.; et al. Genetic profile of T-cell acute lymphoblastic leukemias with MYC translocations. Blood 2014, 124, 3577–3582. [Google Scholar] [CrossRef] [Green Version]
- Magee, J.A.; Ikenoue, T.; Nakada, D.; Lee, J.Y.; Guan, K.L.; Morrison, S.J. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 2012, 11, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Miething, C.; Scuoppo, C.; Bosbach, B.; Appelmann, I.; Nakitandwe, J.; Ma, J.; Wu, G.; Lintault, L.; Auer, M.; Premsrirut, P.K.; et al. PTEN action in leukaemia dictated by the tissue microenvironment. Nature 2014, 510, 402–406. [Google Scholar] [CrossRef]
- Schwarzer, A.; Holtmann, H.; Brugman, M.; Meyer, J.; Schauerte, C.; Zuber, J.; Steinemann, D.; Schlegelberger, B.; Li, Z.; Baum, C. Hyperactivation of mTORC1 and mTORC2 by multiple oncogenic events causes addiction to eIF4E-dependent mRNA translation in T-cell leukemia. Oncogene 2015, 34, 3593–3604. [Google Scholar] [CrossRef]
- Bornschein, S.; Demeyer, S.; Stirparo, R.; Gielen, O.; Vicente, C.; Geerdens, E.; Ghesquiere, B.; Aerts, S.; Cools, J.; de Bock, C.E. Defining the molecular basis of oncogenic cooperation between TAL1 expression and Pten deletion in T-ALL using a novel pro-T-cell model system. Leukemia 2018, 32, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Gehre, N.; Nusser, A.; von Muenchow, L.; Tussiwand, R.; Engdahl, C.; Capoferri, G.; Bosco, N.; Ceredig, R.; Rolink, A.G. A stromal cell free culture system generates mouse pro-T cells that can reconstitute T-cell compartments in vivo. Eur. J. Immunol. 2015, 45, 932–942. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef]
- Vicente, C.; Schwab, C.; Broux, M.; Geerdens, E.; Degryse, S.; Demeyer, S.; Lahortiga, I.; Elliott, A.; Chilton, L.; La Starza, R.; et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Hagenbeek, T.J.; Naspetti, M.; Malergue, F.; Garcon, F.; Nunes, J.A.; Cleutjens, K.B.; Trapman, J.; Krimpenfort, P.; Spits, H. The loss of PTEN allows TCR αβ lineage thymocytes to bypass IL-7 and Pre-TCR-mediated signaling. J. Exp. Med. 2004, 200, 883–894. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamaguchi, M.T.; Ohteki, T.; Sasaki, T.; Kaisho, T.; Kimura, Y.; Yoshida, R.; Wakeham, A.; Higuchi, T.; Fukumoto, M.; et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity 2001, 14, 523–534. [Google Scholar] [CrossRef]
- Gon, S.; Loosveld, M.; Crouzet, T.; Potier, D.; Bonnet, M.; Morin, S.O.; Michel, G.; Vey, N.; Nunes, J.A.; Malissen, B.; et al. Fit αβ T-cell receptor suppresses leukemogenesis of Pten-deficient thymocytes. Haematologica 2018, 103, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Gascoigne, N.R.; Rybakin, V.; Acuto, O.; Brzostek, J. TCR signal strength and T cell development. Annu Rev. Cell Dev. Biol. 2016, 32, 327–348. [Google Scholar] [CrossRef]
- Burger, M.L.; Xue, L.; Sun, Y.; Kang, C.; Winoto, A. Premalignant PTEN-deficient thymocytes activate microRNAs miR-146a and miR-146b as a cellular defense against malignant transformation. Blood 2014, 123, 4089–4100. [Google Scholar] [CrossRef] [Green Version]
- Lo, W.L.; Allen, P.M. Self-peptides in TCR repertoire selection and peripheral T cell function. Curr. Top. Microbiol. Immunol. 2014, 373, 49–67. [Google Scholar] [CrossRef]
- Hagenbeek, T.J.; Wu, X.; Choy, L.; Sanchez-Irizarry, C.; Seshagiri, S.; Stinson, J.; Siebel, C.W.; Spits, H. Murine Pten(−/−) T-ALL requires non-redundant PI3K/mTOR and DLL4/Notch1 signals for maintenance and γc/TCR signals for thymic exit. Cancer Lett. 2014, 346, 237–248. [Google Scholar] [CrossRef]
- Soond, D.R.; Garcon, F.; Patton, D.T.; Rolf, J.; Turner, M.; Scudamore, C.; Garden, O.A.; Okkenhaug, K. Pten loss in CD4 T cells enhances their helper function but does not lead to autoimmunity or lymphoma. J. Immunol. 2012, 188, 5935–5943. [Google Scholar] [CrossRef]
- Locke, F.L.; Zha, Y.Y.; Zheng, Y.; Driessens, G.; Gajewski, T.F. Conditional deletion of PTEN in peripheral T cells augments TCR-mediated activation but does not abrogate CD28 dependency or prevent anergy induction. J. Immunol. 2013, 191, 1677–1685. [Google Scholar] [CrossRef]
- Clappier, E.; Gerby, B.; Sigaux, F.; Delord, M.; Touzri, F.; Hernandez, L.; Ballerini, P.; Baruchel, A.; Pflumio, F.; Soulier, J. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J. Exp. Med. 2011, 208, 653–661. [Google Scholar] [CrossRef] [Green Version]
- D’Angio, M.; Valsecchi, M.G.; Testi, A.M.; Conter, V.; Nunes, V.; Parasole, R.; Colombini, A.; Santoro, N.; Varotto, S.; Caniglia, M.; et al. Clinical features and outcome of SIL/TAL1-positive T-cell acute lymphoblastic leukemia in children and adolescents: A 10-year experience of the AIEOP group. Haematologica 2015, 100, e10–e13. [Google Scholar] [CrossRef]
- Furness, C.L.; Mansur, M.B.; Weston, V.J.; Ermini, L.; van Delft, F.W.; Jenkinson, S.; Gale, R.; Harrison, C.J.; Pombo-de-Oliveira, M.S.; Sanchez-Martin, M.; et al. The subclonal complexity of STIL-TAL1+ T-cell acute lymphoblastic leukaemia. Leukemia 2018, 32, 1984–1993. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, A.; Sanda, T.; Grebliunaite, R.; Carracedo, A.; Salmena, L.; Ahn, Y.; Dahlberg, S.; Neuberg, D.; Moreau, L.A.; Winter, S.S.; et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009, 114, 647–650. [Google Scholar] [CrossRef] [Green Version]
- Jotta, P.Y.; Ganazza, M.A.; Silva, A.; Viana, M.B.; da Silva, M.J.; Zambaldi, L.J.; Barata, J.T.; Brandalise, S.R.; Yunes, J.A. Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia 2010, 24, 239–242. [Google Scholar] [CrossRef]
- Zuurbier, L.; Petricoin, E.F., 3rd; Vuerhard, M.J.; Calvert, V.; Kooi, C.; Buijs-Gladdines, J.G.; Smits, W.K.; Sonneveld, E.; Veerman, A.J.; Kamps, W.A.; et al. The significance of PTEN and AKT aberrations in pediatric T-cell acute lymphoblastic leukemia. Haematologica 2012, 97, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- Trinquand, A.; Tanguy-Schmidt, A.; Ben Abdelali, R.; Lambert, J.; Beldjord, K.; Lengline, E.; De Gunzburg, N.; Payet-Bornet, D.; Lhermitte, L.; Mossafa, H.; et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: A Group for Research in Adult Acute Lymphoblastic Leukemia study. J. Clin. Oncol. 2013, 31, 4333–4342. [Google Scholar] [CrossRef]
- Bandapalli, O.R.; Zimmermann, M.; Kox, C.; Stanulla, M.; Schrappe, M.; Ludwig, W.D.; Koehler, R.; Muckenthaler, M.U.; Kulozik, A.E. NOTCH1 activation clinically antagonizes the unfavorable effect of PTEN inactivation in BFM-treated children with precursor T-cell acute lymphoblastic leukemia. Haematologica 2013, 98, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Mendes, R.D.; Sarmento, L.M.; Cante-Barrett, K.; Zuurbier, L.; Buijs-Gladdines, J.G.; Povoa, V.; Smits, W.K.; Abecasis, M.; Yunes, J.A.; Sonneveld, E.; et al. PTEN microdeletions in T-cell acute lymphoblastic leukemia are caused by illegitimate RAG-mediated recombination events. Blood 2014, 124, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Jenkinson, S.; Kirkwood, A.A.; Goulden, N.; Vora, A.; Linch, D.C.; Gale, R.E. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia 2016, 30, 39–47. [Google Scholar] [CrossRef]
- Doerrenberg, M.; Kloetgen, A.; Hezaveh, K.; Wossmann, W.; Bleckmann, K.; Stanulla, M.; Schrappe, M.; McHardy, A.C.; Borkhardt, A.; Hoell, J.I. T-cell acute lymphoblastic leukemia in infants has distinct genetic and epigenetic features compared to childhood cases. Genes Chromosomes Cancer 2017, 56, 159–167. [Google Scholar] [CrossRef]
- Tesio, M.; Trinquand, A.; Ballerini, P.; Hypolite, G.; Lhermitte, L.; Petit, A.; Ifrah, N.; Baruchel, A.; Dombret, H.; Macintyre, E.; et al. Age-related clinical and biological features of PTEN abnormalities in T-cell acute lymphoblastic leukaemia. Leukemia 2017, 31, 2594–2600. [Google Scholar] [CrossRef]
- Paganin, M.; Grillo, M.F.; Silvestri, D.; Scapinello, G.; Buldini, B.; Cazzaniga, G.; Biondi, A.; Valsecchi, M.G.; Conter, V.; Te Kronnie, G.; et al. The presence of mutated and deleted PTEN is associated with an increased risk of relapse in childhood T cell acute lymphoblastic leukaemia treated with AIEOP-BFM ALL protocols. Br. J. Haematol. 2018, 182, 705–711. [Google Scholar] [CrossRef]
- Dawidowska, M.; Jaksik, R.; Drobna, M.; Szarzynska-Zawadzka, B.; Kosmalska, M.; Sedek, L.; Machowska, L.; Lalik, A.; Lejman, M.; Ussowicz, M.; et al. Comprehensive investigation of miRNome identifies novel candidate miRNA-mRNA interactions implicated in T-cell acute lymphoblastic leukemia. Neoplasia 2019, 21, 294–310. [Google Scholar] [CrossRef]
- Aguissa-Toure, A.H.; Li, G. Genetic alterations of PTEN in human melanoma. Cell Mol. Life Sci. 2012, 69, 1475–1491. [Google Scholar] [CrossRef]
- Kechagioglou, P.; Papi, R.M.; Provatopoulou, X.; Kalogera, E.; Papadimitriou, E.; Grigoropoulos, P.; Nonni, A.; Zografos, G.; Kyriakidis, D.A.; Gounaris, A. Tumor suppressor PTEN in breast cancer: Heterozygosity, mutations and protein expression. Anticancer Res. 2014, 34, 1387–1400. [Google Scholar] [PubMed]
- Wang, X.; Shi, Y.; Wang, J.; Huang, G.; Jiang, X. Crucial role of the C-terminus of PTEN in antagonizing NEDD4-1-mediated PTEN ubiquitination and degradation. Biochem. J. 2008, 414, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef]
- Li, W.; Zhang, T.; Guo, L.; Huang, L. Regulation of PTEN expression by noncoding RNAs. J. Exp. Clin. Cancer Res. 2018, 37, 223. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; Wolfe, A.L.; Oricchio, E.; Palomero, T.; de Keersmaecker, K.; McJunkin, K.; Zuber, J.; James, T.; Khan, A.A.; Leslie, C.S.; et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat. Cell Biol. 2010, 12, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Liu, X.; Lv, M.; Wu, Y.; Kuang, S.; Gong, J.; Yuan, P.; Zhong, Z.; Li, Q.; Jia, H.; et al. MicroRNA and transcription factor co-regulatory network analysis reveals miR-19 inhibits CYLD in T-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2012, 40, 5201–5214. [Google Scholar] [CrossRef] [Green Version]
- Wallaert, A.; Van Loocke, W.; Hernandez, L.; Taghon, T.; Speleman, F.; Van Vlierberghe, P. Comprehensive miRNA expression profiling in human T-cell acute lymphoblastic leukemia by small RNA-sequencing. Sci. Rep. 2017, 7, 7901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallaert, A.; Durinck, K.; Taghon, T.; Van Vlierberghe, P.; Speleman, F. T-ALL and thymocytes: A message of noncoding RNAs. J. Hematol. Oncol. 2017, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; Van Der Meulen, J.; Wolfe, A.L.; Liu, X.; Mets, E.; Taghon, T.; Khan, A.A.; Setty, M.; Rondou, P.; Vandenberghe, P.; et al. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat. Genet. 2011, 43, 673–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulci, V.; Colombo, T.; Chiaretti, S.; Messina, M.; Citarella, F.; Tavolaro, S.; Guarini, A.; Foa, R.; Macino, G. Characterization of B- and T-lineage acute lymphoblastic leukemia by integrated analysis of MicroRNA and mRNA expression profiles. Genes Chromosomes Cancer 2009, 48, 1069–1082. [Google Scholar] [CrossRef]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Investig. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [Green Version]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Evangelisti, C.; Melchionda, F.; et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: Targeting the unfolded protein response signaling. Leukemia 2014, 28, 543–553. [Google Scholar] [CrossRef]
- Fragoso, R.; Barata, J.T. Kinases, tails and more: Regulation of PTEN function by phosphorylation. Methods 2015, 77–78, 75–81. [Google Scholar] [CrossRef]
- Buontempo, F.; McCubrey, J.A.; Orsini, E.; Ruzzene, M.; Cappellini, A.; Lonetti, A.; Evangelisti, C.; Chiarini, F.; Evangelisti, C.; Barata, J.T.; et al. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia 2018, 32, 1–10. [Google Scholar] [CrossRef]
- Miller, S.J.; Lou, D.Y.; Seldin, D.C.; Lane, W.S.; Neel, B.G. Direct identification of PTEN phosphorylation sites. FEBS Lett. 2002, 528, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Piovan, E.; Yu, J.; Tosello, V.; Herranz, D.; Ambesi-Impiombato, A.; Da Silva, A.C.; Sanchez-Martin, M.; Perez-Garcia, A.; Rigo, I.; Castillo, M.; et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell 2013, 24, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Bayascas, J.R.; Leslie, N.R.; Parsons, R.; Fleming, S.; Alessi, D.R. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/−) mice. Curr. Biol. 2005, 15, 1839–1846. [Google Scholar] [CrossRef]
- Finlay, D.K.; Sinclair, L.V.; Feijoo, C.; Waugh, C.M.; Hagenbeek, T.J.; Spits, H.; Cantrell, D.A. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J. Exp. Med. 2009, 206, 2441–2454. [Google Scholar] [CrossRef] [Green Version]
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M.; Khandan, T.; Paktinat, M.; Gilliland, D.G.; Gritsman, K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415. [Google Scholar] [CrossRef]
- Subramaniam, P.S.; Whye, D.W.; Efimenko, E.; Chen, J.; Tosello, V.; De Keersmaecker, K.; Kashishian, A.; Thompson, M.A.; Castillo, M.; Cordon-Cardo, C.; et al. Targeting nonclassical oncogenes for therapy in T-ALL. Cancer Cell 2012, 21, 459–472. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, Y.; Chen, J.; Li, W.; Li, W.; Zhang, Q.; Mi, Y.; Goswami, R.S.; You, J.Q.; Lin, D.; et al. Regulation of PI3K signaling in T-cell acute lymphoblastic leukemia: A novel PTEN/Ikaros/miR-26b mechanism reveals a critical targetable role for PIK3CD. Leukemia 2017, 31, 2355–2364. [Google Scholar] [CrossRef]
- Malstrom, S.; Tili, E.; Kappes, D.; Ceci, J.D.; Tsichlis, P.N. Tumor induction by an Lck-MyrAkt transgene is delayed by mechanisms controlling the size of the thymus. Proc. Natl. Acad. Sci. USA 2001, 98, 14967–14972. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, P.P. GSK-3 as a novel prognostic indicator in leukemia. Adv. Biol. Regul. 2017, 65, 26–35. [Google Scholar] [CrossRef]
- Bonnet, M.; Loosveld, M.; Montpellier, B.; Navarro, J.M.; Quilichini, B.; Picard, C.; Di Cristofaro, J.; Bagnis, C.; Fossat, C.; Hernandez, L.; et al. Posttranscriptional deregulation of MYC via PTEN constitutes a major alternative pathway of MYC activation in T-cell acute lymphoblastic leukemia. Blood 2011, 117, 6650–6659. [Google Scholar] [CrossRef] [Green Version]
- Rothenberg, E.V.; Hosokawa, H.; Ungerback, J. Mechanisms of action of hematopoietic transcription factor PU.1 in initiation of T-Cell development. Front. Immunol. 2019, 10, 228. [Google Scholar] [CrossRef]
- Anderson, M.K.; Weiss, A.H.; Hernandez-Hoyos, G.; Dionne, C.J.; Rothenberg, E.V. Constitutive expression of PU.1 in fetal hematopoietic progenitors blocks T cell development at the pro-T cell stage. Immunity 2002, 16, 285–296. [Google Scholar] [CrossRef]
- Seki, M.; Kimura, S.; Isobe, T.; Yoshida, K.; Ueno, H.; Nakajima-Takagi, Y.; Wang, C.; Lin, L.; Kon, A.; Suzuki, H.; et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1274–1281. [Google Scholar] [CrossRef]
- Evangelisti, C.; Chiarini, F.; McCubrey, J.A.; Martelli, A.M. Therapeutic Targeting of mTOR in T-Cell Acute Lymphoblastic Leukemia: An Update. Int. J. Mol. Sci. 2018, 19, 1878. [Google Scholar] [CrossRef]
- Martelli, A.M.; Buontempo, F.; McCubrey, J.A. Drug discovery targeting the mTOR pathway. Clin. Sci. (Lond.) 2018, 132, 543–568. [Google Scholar] [CrossRef]
- Blackburn, J.S.; Liu, S.; Wilder, J.L.; Dobrinski, K.P.; Lobbardi, R.; Moore, F.E.; Martinez, S.A.; Chen, E.Y.; Lee, C.; Langenau, D.M. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell 2014, 25, 366–378. [Google Scholar] [CrossRef]
- Kalaitzidis, D.; Sykes, S.M.; Wang, Z.; Punt, N.; Tang, Y.; Ragu, C.; Sinha, A.U.; Lane, S.W.; Souza, A.L.; Clish, C.B.; et al. mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell 2012, 11, 429–439. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Mao, Z.; Zhang, W. Role of mTOR in glucose and lipid metabolism. Int. J. Mol. Sci. 2018, 19, 2043. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in cancer and autophagy. Cancers (Basel) 2018, 10, 18. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. Who does TORC2 talk to? Biochem. J. 2018, 475, 1721–1738. [Google Scholar] [CrossRef]
- Signer, R.A.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Thoreen, C.C. The molecular basis of mTORC1-regulated translation. Biochem. Soc. Trans. 2017, 45, 213–221. [Google Scholar] [CrossRef]
- Tandon, P.; Gallo, C.A.; Khatri, S.; Barger, J.F.; Yepiskoposyan, H.; Plas, D.R. Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 2361–2365. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Tamura, M.; Pankov, R.; Danen, E.H.; Takino, T.; Matsumoto, K.; Yamada, K.M. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell Biol. 1999, 146, 389–403. [Google Scholar] [CrossRef]
- You, D.; Xin, J.; Volk, A.; Wei, W.; Schmidt, R.; Scurti, G.; Nand, S.; Breuer, E.K.; Kuo, P.C.; Breslin, P.; et al. FAK mediates a compensatory survival signal parallel to PI3K-AKT in PTEN-null T-ALL cells. Cell Rep. 2015, 10, 2055–2068. [Google Scholar] [CrossRef]
- Kamarajan, P.; Bunek, J.; Lin, Y.; Nunez, G.; Kapila, Y.L. Receptor-interacting protein shuttles between cell death and survival signaling pathways. Mol. Biol Cell 2010, 21, 481–488. [Google Scholar] [CrossRef]
- Yin, Y.; Shen, W.H. PTEN: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef]
- Newton, R.H.; Lu, Y.; Papa, A.; Whitcher, G.H.; Kang, Y.J.; Yan, C.; Pandolfi, P.P.; Turka, L.A. Suppression of T-cell lymphomagenesis in mice requires PTEN phosphatase activity. Blood 2015, 125, 852–855. [Google Scholar] [CrossRef]
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Bartell, S.M.; Kim, H.N.; Ambrogini, E.; Han, L.; Iyer, S.; Serra Ucer, S.; Rabinovitch, P.; Jilka, R.L.; Weinstein, R.S.; Zhao, H.; et al. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat. Commun. 2014, 5, 3773. [Google Scholar] [CrossRef]
- Monsalve, M.; Prieto, I.; de Bem, A.F.; Olmos, Y. Methodological approach for the evaluation of FOXO as a positive regulator of antioxidant genes. Methods Mol. Biol. 2019, 1890, 61–76. [Google Scholar] [CrossRef]
- Brown, A.K.; Webb, A.E. Regulation of FOXO Factors in mammalian cells. Curr. Top. Dev. Biol. 2018, 127, 165–192. [Google Scholar] [CrossRef]
- Dose, M.; Emmanuel, A.O.; Chaumeil, J.; Zhang, J.; Sun, T.; Germar, K.; Aghajani, K.; Davis, E.M.; Keerthivasan, S.; Bredemeyer, A.L.; et al. β-Catenin induces T-cell transformation by promoting genomic instability. Proc. Natl. Acad. Sci. USA 2014, 111, 391–396. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Dedhar, S.; Wu, H.; Nelson, C.C. PTEN and GSK3β: Key regulators of progression to androgen-independent prostate cancer. Oncogene 2006, 25, 329–337. [Google Scholar] [CrossRef]
- Larson Gedman, A.; Chen, Q.; Kugel Desmoulin, S.; Ge, Y.; LaFiura, K.; Haska, C.L.; Cherian, C.; Devidas, M.; Linda, S.B.; Taub, J.W.; et al. The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Leukemia 2009, 23, 1417–1425. [Google Scholar] [CrossRef]
- Petit, A.; Trinquand, A.; Chevret, S.; Ballerini, P.; Cayuela, J.M.; Grardel, N.; Touzart, A.; Brethon, B.; Lapillonne, H.; Schmitt, C.; et al. Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 2018, 131, 289–300. [Google Scholar] [CrossRef]
- Szarzynska-Zawadzka, B.; Kunz, J.B.; Sedek, L.; Kosmalska, M.; Zdon, K.; Biecek, P.; Bandapalli, O.R.; Kraszewska-Hamilton, M.; Jaksik, R.; Drobna, M.; et al. PTEN abnormalities predict poor outcome in children with T-cell acute lymphoblastic leukemia treated according to ALL IC-BFM protocols. Am. J. Hematol. 2019. [Google Scholar] [CrossRef]
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef]
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Wei, G.; Twomey, D.; Lamb, J.; Schlis, K.; Agarwal, J.; Stam, R.W.; Opferman, J.T.; Sallan, S.E.; den Boer, M.L.; Pieters, R.; et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 2006, 10, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Beesley, A.H.; Firth, M.J.; Ford, J.; Weller, R.E.; Freitas, J.R.; Perera, K.U.; Kees, U.R. Glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia is associated with a proliferative metabolism. Br. J. Cancer 2009, 100, 1926–1936. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Yang, A.; Ma, J.; Wu, M.; Xu, H.; Wu, K.; Jin, Y.; Xie, Y. Akt2 mediates glucocorticoid resistance in lymphoid malignancies through FoxO3a/Bim axis and serves as a direct target for resistance reversal. Cell Death Dis. 2019, 9, 1013. [Google Scholar] [CrossRef]
- Hall, C.P.; Reynolds, C.P.; Kang, M.H. Modulation of glucocorticoid resistance in pediatric T-cell acute lymphoblastic leukemia by increasing BIM expression with the PI3K/mTOR Inhibitor BEZ235. Clin. Cancer Res. 2016, 22, 621–632. [Google Scholar] [CrossRef]
- Lonetti, A.; Antunes, I.L.; Chiarini, F.; Orsini, E.; Buontempo, F.; Ricci, F.; Tazzari, P.L.; Pagliaro, P.; Melchionda, F.; Pession, A.; et al. Activity of the pan-class I phosphoinositide 3-kinase inhibitor NVP-BKM120 in T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1196–1206. [Google Scholar] [CrossRef]
- Stengel, C.; Jenner, E.; Meja, K.; Mayekar, S.; Khwaja, A. Proliferation of PTEN-deficient haematopoietic tumour cells is not affected by isoform-selective inhibition of p110 PI3-kinase and requires blockade of all class 1 PI3K activity. Br. J. Haematol. 2013, 162, 285–289. [Google Scholar] [CrossRef]
- Lonetti, A.; Cappellini, A.; Sparta, A.M.; Chiarini, F.; Buontempo, F.; Evangelisti, C.; Evangelisti, C.; Orsini, E.; McCubrey, J.A.; Martelli, A.M. PI3K pan-inhibition impairs more efficiently proliferation and survival of T-cell acute lymphoblastic leukemia cell lines when compared to isoform-selective PI3K inhibitors. Oncotarget 2015, 6, 10399–10414. [Google Scholar] [CrossRef] [Green Version]
- Yuzugullu, H.; Von, T.; Thorpe, L.M.; Walker, S.R.; Roberts, T.M.; Frank, D.A.; Zhao, J.J. NTRK2 activation cooperates with PTEN deficiency in T-ALL through activation of both the PI3K-AKT and JAK-STAT3 pathways. Cell Discov. 2016, 2, 16030. [Google Scholar] [CrossRef]
- Sinkevicius, K.W.; Kriegel, C.; Bellaria, K.J.; Lee, J.; Lau, A.N.; Leeman, K.T.; Zhou, P.; Beede, A.M.; Fillmore, C.M.; Caswell, D.; et al. Neurotrophin receptor TrkB promotes lung adenocarcinoma metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, 10299–10304. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Lee, W.S.; Jeong, J.; Kim, S.J.; Jin, W. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 2015, 6, 40158–40171. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Chen, H.J.; Li, T.M.; Fong, Y.C.; Liu, S.C.; Chen, P.C.; Tang, C.H. β5 integrin up-regulation in brain-derived neurotrophic factor promotes cell motility in human chondrosarcoma. PLoS ONE 2013, 8, e67990. [Google Scholar] [CrossRef]
- Yamada, M.; Ohnishi, H.; Sano, S.; Nakatani, A.; Ikeuchi, T.; Hatanaka, H. Insulin receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured cerebral cortical neurons. J. Biol. Chem. 1997, 272, 30334–30339. [Google Scholar] [CrossRef]
- Holgado-Madruga, M.; Moscatello, D.K.; Emlet, D.R.; Dieterich, R.; Wong, A.J. Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc. Natl. Acad. Sci. USA 1997, 94, 12419–12424. [Google Scholar] [CrossRef] [Green Version]
- Vaishnavi, A.; Le, A.T.; Doebele, R.C. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015, 5, 25–34. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.R.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef]
- Lange, A.M.; Lo, H.W. Inhibiting TRK Proteins in clinical cancer therapy. Cancers (Basel) 2018, 10, 105. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.t.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Hales, E.C.; Taub, J.W.; Matherly, L.H. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: Targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal. 2014, 26, 149–161. [Google Scholar] [CrossRef]
- Herranz, D.; Ambesi-Impiombato, A.; Sudderth, J.; Sanchez-Martin, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef] [Green Version]
- Papayannidis, C.; DeAngelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel γ-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350. [Google Scholar] [CrossRef]
- Efimenko, E.; Dave, U.P.; Lebedeva, I.V.; Shen, Y.; Sanchez-Quintero, M.J.; Diolaiti, D.; Kung, A.; Lannutti, B.J.; Chen, J.; Realubit, R.; et al. PI3Kg/d and NOTCH1 cross-regulate pathways that define the T-cell acute lymphoblastic leukemia disease signature. Mol. Cancer Ther. 2017, 16, 2069–2082. [Google Scholar] [CrossRef]
- Evangelisti, C.; Ricci, F.; Tazzari, P.; Tabellini, G.; Battistelli, M.; Falcieri, E.; Chiarini, F.; Bortul, R.; Melchionda, F.; Pagliaro, P.; et al. Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia 2011, 25, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Dastur, A.; Choi, A.; Costa, C.; Yin, X.; Williams, A.; McClanaghan, J.; Greenberg, M.; Roderick, J.; Patel, N.U.; Boisvert, J.; et al. NOTCH1 represses MCL-1 levels in GSI-resistant T-ALL, making them susceptible to ABT-263. Clin. Cancer Res. 2019, 25, 312–324. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martelli, A.M.; Paganelli, F.; Fazio, A.; Bazzichetto, C.; Conciatori, F.; McCubrey, J.A. The Key Roles of PTEN in T-Cell Acute Lymphoblastic Leukemia Development, Progression, and Therapeutic Response. Cancers 2019, 11, 629. https://doi.org/10.3390/cancers11050629
Martelli AM, Paganelli F, Fazio A, Bazzichetto C, Conciatori F, McCubrey JA. The Key Roles of PTEN in T-Cell Acute Lymphoblastic Leukemia Development, Progression, and Therapeutic Response. Cancers. 2019; 11(5):629. https://doi.org/10.3390/cancers11050629
Chicago/Turabian StyleMartelli, Alberto M., Francesca Paganelli, Antonietta Fazio, Chiara Bazzichetto, Fabiana Conciatori, and James A. McCubrey. 2019. "The Key Roles of PTEN in T-Cell Acute Lymphoblastic Leukemia Development, Progression, and Therapeutic Response" Cancers 11, no. 5: 629. https://doi.org/10.3390/cancers11050629
APA StyleMartelli, A. M., Paganelli, F., Fazio, A., Bazzichetto, C., Conciatori, F., & McCubrey, J. A. (2019). The Key Roles of PTEN in T-Cell Acute Lymphoblastic Leukemia Development, Progression, and Therapeutic Response. Cancers, 11(5), 629. https://doi.org/10.3390/cancers11050629