A Comparison of DNA Mutation and Copy Number Profiles of Primary Breast Cancers and Paired Brain Metastases for Identifying Clinically Relevant Genetic Alterations in Brain Metastases


 ,
,
Abstract
:1. Introduction
2. Results
2.1. Population
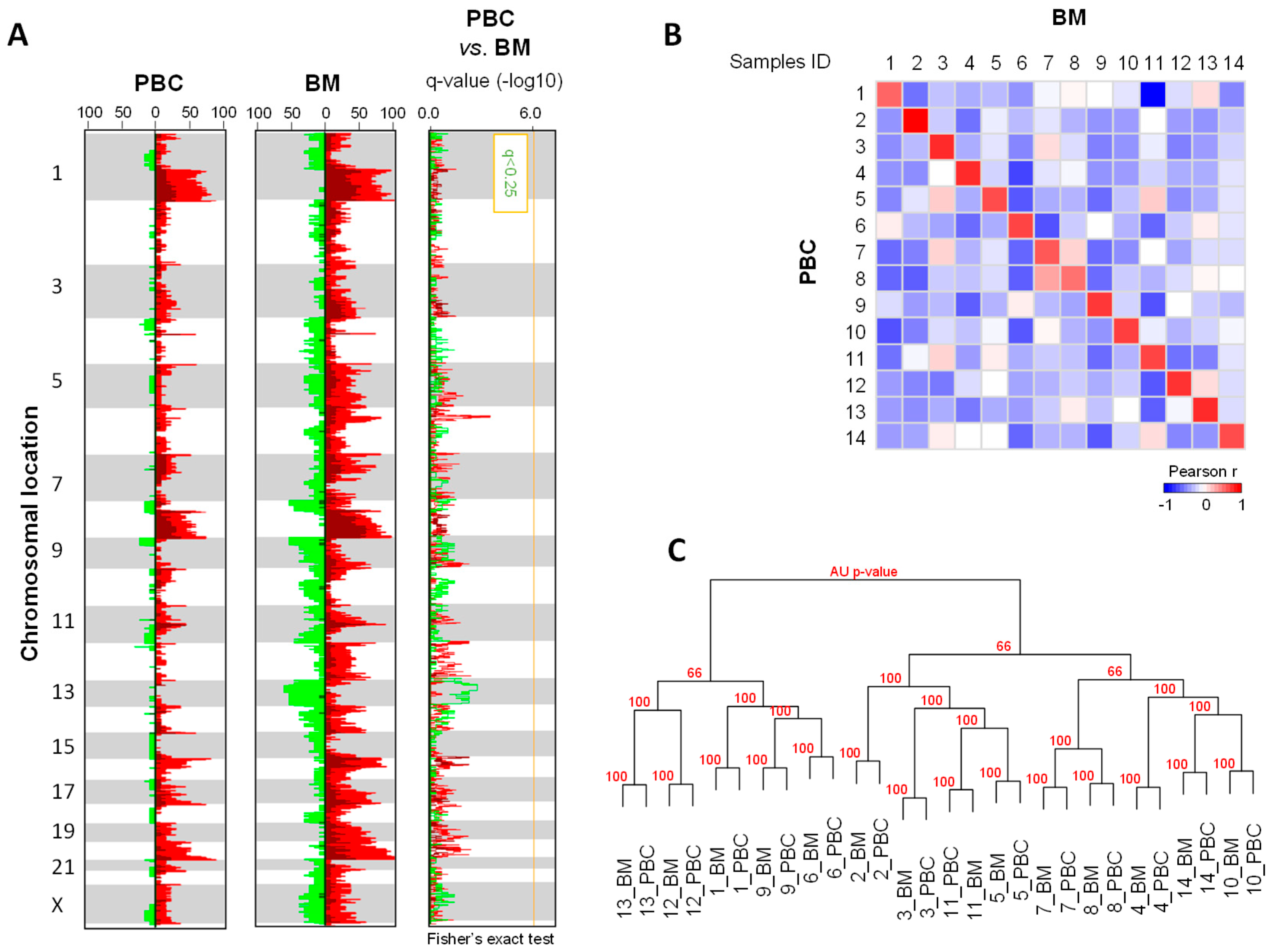
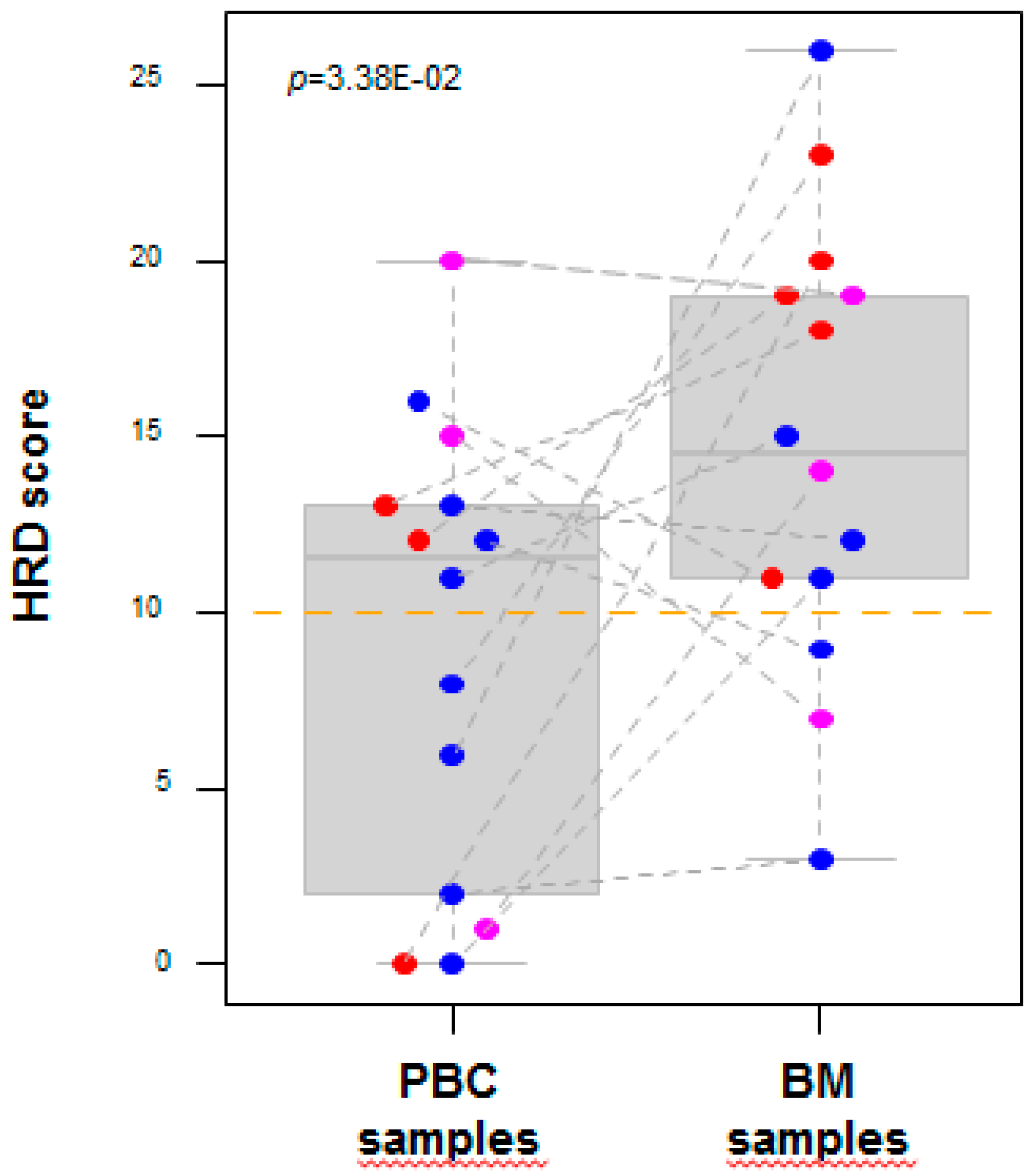
2.2. DNA Copy Number Profiles
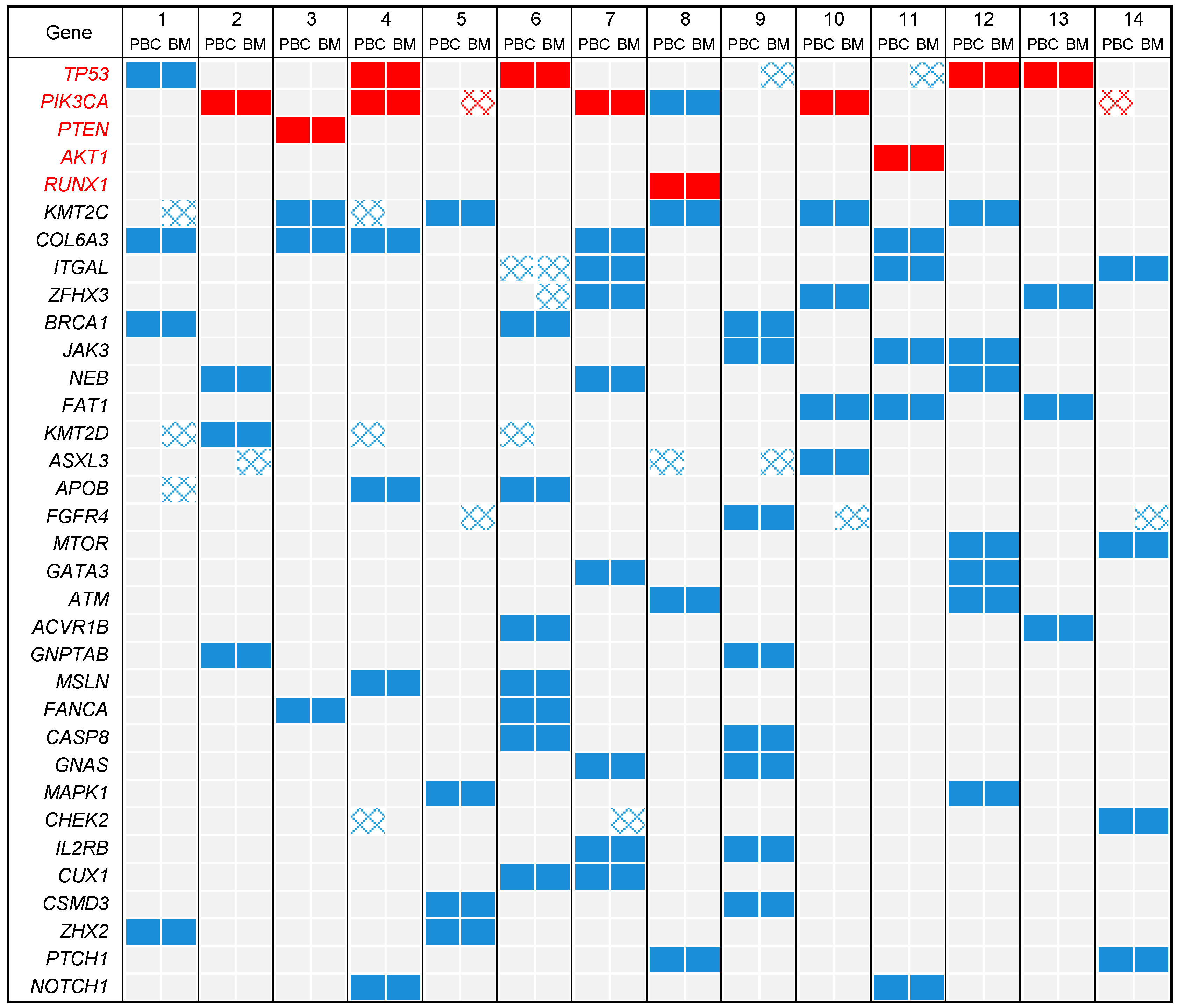
2.3. DNA Mutational Profiles
2.4. Profile of Actionable Genetic Alterations
3. Discussion
4. Materials and Methods
4.1. Patients and Samples Selection
4.2. DNA Copy Number Profiling
4.3. Mutational Profiling
4.4. Definition of Actionable Genetic Alterations
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lin, N.U.; Gaspar, L.E.; Soffietti, R. Breast Cancer in the Central Nervous System: Multidisciplinary Considerations and Management. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.-R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massagué, J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.-F.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massagué, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Weigelt, B.; Peterse, J.L.; van ’t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- André, F.; Bachelot, T.; Commo, F.; Campone, M.; Arnedos, M.; Dieras, V.; Lacroix-Triki, M.; Lacroix, L.; Cohen, P.; Gentien, D.; et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: A multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol. 2014, 15, 267–274. [Google Scholar] [CrossRef]
- Adélaïde, J.; Finetti, P.; Bekhouche, I.; Repellini, L.; Geneix, J.; Sircoulomb, F.; Charafe-Jauffret, E.; Cervera, N.; Desplans, J.; Parzy, D.; et al. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007, 67, 11565–11575. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Orsetti, B.; Cervera, N.; Finetti, P.; Sircoulomb, F.; Rougé, C.; Lasorsa, L.; Letessier, A.; Ginestier, C.; Monville, F.; et al. Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol. Cancer Res. 2005, 3, 655–667. [Google Scholar] [CrossRef]
- Sircoulomb, F.; Nicolas, N.; Ferrari, A.; Finetti, P.; Bekhouche, I.; Rousselet, E.; Lonigro, A.; Adélaïde, J.; Baudelet, E.; Esteyriès, S.; et al. ZNF703 gene amplification at 8p12 specifies luminal B breast cancer. EMBO Mol. Med. 2011, 3, 153–166. [Google Scholar] [CrossRef]
- Perera-Bel, J.; Hutter, B.; Heining, C.; Bleckmann, A.; Fröhlich, M.; Fröhling, S.; Glimm, H.; Brors, B.; Beißbarth, T. From somatic variants towards precision oncology: Evidence-driven reporting of treatment options in molecular tumor boards. Genome Med. 2018, 10. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Guille, A.; Adélaïde, J.; Garnier, S.; Carbuccia, N.; Monneur, A.; Charafe-Jauffret, E.; Goncalves, A.; Viens, P.; et al. Comparative genomic analysis of primary tumors and metastases in breast cancer. Oncotarget 2016, 7, 27208–27219. [Google Scholar] [CrossRef]
- Goswami, R.S.; Patel, K.P.; Singh, R.R.; Meric-Bernstam, F.; Kopetz, E.S.; Subbiah, V.; Alvarez, R.H.; Davies, M.A.; Jabbar, K.J.; Roy-Chowdhuri, S.; et al. Hotspot mutation panel testing reveals clonal evolution in a study of 265 paired primary and metastatic tumors. Clin. Cancer Res. 2015, 21, 2644–2651. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Beck, J.T.; Royce, M. Everolimus-based combination therapies for HR+, HER2− metastatic breast cancer. Cancer Treat. Rev. 2018, 69, 204–214. [Google Scholar] [CrossRef]
- Arthur, L.M.; Turnbull, A.K.; Renshaw, L.; Keys, J.; Thomas, J.S.; Wilson, T.R.; Lackner, M.R.; Sims, A.H.; Dixon, J.M. Changes in PIK3CA mutation status are not associated with recurrence, metastatic disease or progression in endocrine-treated breast cancer. Breast Cancer Res. Treat. 2014, 147, 211–219. [Google Scholar] [CrossRef]
- Martin, L.-A.; Ribas, R.; Simigdala, N.; Schuster, E.; Pancholi, S.; Tenev, T.; Gellert, P.; Buluwela, L.; Harrod, A.; Thornhill, A.; et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Duchnowska, R.; Jassem, J.; Goswami, C.P.; Dundar, M.; Gökmen-Polar, Y.; Li, L.; Woditschka, S.; Biernat, W.; Sosińska-Mielcarek, K.; Czartoryska-Arłukowicz, B.; et al. Predicting early brain metastases based on clinicopathological factors and gene expression analysis in advanced HER2-positive breast cancer patients. J. Neuro-Oncol. 2015, 122, 205–216. [Google Scholar] [CrossRef]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, K.; Lim, S.H.; Kim, H.S.; Yoo, K.H.; Jung, K.S.; Song, H.-N.; Hong, M.; Do, I.-G.; Ahn, T.; et al. Mutational profiling of brain metastasis from breast cancer: Matched pair analysis of targeted sequencing between brain metastasis and primary breast cancer. Oncotarget 2015, 6, 43731–43742. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef]
- Da Silva, L.; Simpson, P.T.; Smart, C.E.; Cocciardi, S.; Waddell, N.; Lane, A.; Morrison, B.J.; Vargas, A.C.; Healey, S.; Beesley, J.; et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res. BCR 2010, 12. [Google Scholar] [CrossRef]
- Saunus, J.M.; Quinn, M.C.J.; Patch, A.M.; Pearson, J.V.; Bailey, P.J.; Nones, K.; McCart Reed, A.E.; Miller, D.; Wilson, P.J.; Al-Ejeh, F.; et al. Integrated genomic and transcriptomic analysis of human brain metastases identifies alterations of potential clinical significance. J. Pathol. 2015, 237, 363–378. [Google Scholar] [CrossRef]
- Woditschka, S.; Evans, L.; Duchnowska, R.; Reed, L.T.; Palmieri, D.; Qian, Y.; Badve, S.; Sledge, G.; Gril, B.; Aladjem, M.I.; et al. DNA double-strand break repair genes and oxidative damage in brain metastasis of breast cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Salhia, B.; Kiefer, J.; Ross, J.T.D.; Metapally, R.; Martinez, R.A.; Johnson, K.N.; DiPerna, D.M.; Paquette, K.M.; Jung, S.; Nasser, S.; et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Pangeni, R.P.; Channathodiyil, P.; Huen, D.S.; Eagles, L.W.; Johal, B.K.; Pasha, D.; Hadjistephanou, N.; Nevell, O.; Davies, C.L.; Adewumi, A.I.; et al. The GALNT9, BNC1 and CCDC8 genes are frequently epigenetically dysregulated in breast tumours that metastasise to the brain. Clin. Epigenet. 2015, 7, 57. [Google Scholar] [CrossRef]
- Park, E.S.; Kim, S.J.; Kim, S.W.; Yoon, S.-L.; Leem, S.-H.; Kim, S.-B.; Kim, S.M.; Park, Y.-Y.; Cheong, J.-H.; Woo, H.G.; et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc. Natl. Acad. Sci. USA 2011, 108, 17456–17461. [Google Scholar] [CrossRef]
- Burnett, R.M.; Craven, K.E.; Krishnamurthy, P.; Goswami, C.P.; Badve, S.; Crooks, P.; Mathews, W.P.; Bhat-Nakshatri, P.; Nakshatri, H. Organ-specific adaptive signaling pathway activation in metastatic breast cancer cells. Oncotarget 2015, 6, 12682–12696. [Google Scholar] [CrossRef]
- Dun, M.D.; Chalkley, R.J.; Faulkner, S.; Keene, S.; Avery-Kiejda, K.A.; Scott, R.J.; Falkenby, L.G.; Cairns, M.J.; Larsen, M.R.; Bradshaw, R.A.; et al. Proteotranscriptomic Profiling of 231-BR Breast Cancer Cells: Identification of Potential Biomarkers and Therapeutic Targets for Brain Metastasis. Mol. Cell. Proteom. MCP 2015, 14, 2316–2330. [Google Scholar] [CrossRef]
- Duchnowska, R.; Jarząb, M.; Żebracka-Gala, J.; Matkowski, R.; Kowalczyk, A.; Radecka, B.; Kowalska, M.; Pfeifer, A.; Foszczyńska-Kłoda, M.; Musolino, A.; et al. Brain Metastasis Prediction by Transcriptomic Profiling in Triple-Negative Breast Cancer. Clin. Breast Cancer 2017, 17. [Google Scholar] [CrossRef]
- Sjøbakk, T.E.; Johansen, R.; Bathen, T.F.; Sonnewald, U.; Kvistad, K.A.; Lundgren, S.; Gribbestad, I.S. Metabolic profiling of human brain metastases using in vivo proton MR spectroscopy at 3T. BMC Cancer 2007, 7. [Google Scholar] [CrossRef]
- Bollig-Fischer, A.; Michelhaugh, S.K.; Wijesinghe, P.; Dyson, G.; Kruger, A.; Palanisamy, N.; Choi, L.; Alosh, B.; Ali-Fehmi, R.; Mittal, S. Cytogenomic profiling of breast cancer brain metastases reveals potential for repurposing targeted therapeutics. Oncotarget 2015, 6, 14614–14624. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Ng, C.K.Y.; Piscuoglio, S.; Gonzalez-Cao, M.; Lim, R.S.; De Filippo, M.R.; Fusco, N.; Schultheis, A.M.; Ortiz, C.; Viteri, S.; et al. Genetic heterogeneity and actionable mutations in HER2-positive primary breast cancers and their brain metastases. Oncotarget 2018, 9, 20617–20630. [Google Scholar]
- Diossy, M.; Reiniger, L.; Sztupinszki, Z.; Krzystanek, M.; Timms, K.M.; Neff, C.; Solimeno, C.; Pruss, D.; Eklund, A.C.; Tóth, E.; et al. Breast cancer brain metastases show increased levels of genomic aberration-based homologous recombination deficiency scores relative to their corresponding primary tumors. Ann. Oncol. 2018, 29, 1948–1954. [Google Scholar] [CrossRef]
- McMullin, R.P.; Wittner, B.S.; Yang, C.; Denton-Schneider, B.R.; Hicks, D.; Singavarapu, R.; Moulis, S.; Lee, J.; Akbari, M.R.; Narod, S.A.; et al. A BRCA1 deficient-like signature is enriched in breast cancer brain metastases and predicts DNA damage-induced poly (ADP-ribose) polymerase inhibitor sensitivity. Breast Cancer Res. BCR 2014, 16. [Google Scholar] [CrossRef]
- Zhou, D.; Ouyang, Q.; Liu, L.; Liu, J.; Tang, Y.; Xiao, M.; Wang, Y.; He, Q.; Hu, Z.Y. Chemotherapy Modulates Endocrine Therapy-Related Resistance Mutations in Metastatic Breast Cancer. Transl. Oncol. 2019, 12, 764–774. [Google Scholar] [CrossRef]
- Rugo, H.S.; Vidula, N.; Ma, C. Improving Response to Hormone Therapy in Breast Cancer: New Targets, New Therapeutic Options. Am. Soc. Clin. Oncol. Educ. Book 2016, 35. [Google Scholar] [CrossRef]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef]
- Tomlinson, D.C.; Knowles, M.A.; Speirs, V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int. J. Cancer 2012, 130, 2857–2866. [Google Scholar] [CrossRef]
- Musolino, A.; Campone, M.; Neven, P.; Denduluri, N.; Barrios, C.H.; Cortes, J.; Blackwell, K.; Soliman, H.; Kahan, Z.; Bonnefoi, H.; et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR(+), HER2(−) breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res. 2017, 19. [Google Scholar] [CrossRef]
- Hanker, A.B.; Garrett, J.T.; Estrada, M.V.; Moore, P.D.; Ericsson, P.G.; Koch, J.P.; Langley, E.; Singh, S.; Kim, P.S.; Frampton, G.M.; et al. HER2–Overexpressing Breast Cancers Amplify FGFR Signaling upon Acquisition of Resistance to Dual Therapeutic Blockade of HER2. Clin. Cancer Res. 2017, 23, 4323–4334. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Booher, R.N.; Hatch, H.; Dolinski, B.M.; Nguyen, T.; Harmonay, L.; Al-Assaad, A.-S.; Ayers, M.; Nebozhyn, M.; Loboda, A.; Hirsch, H.A.; et al. MCL1 and BCL-xL levels in solid tumors are predictive of dinaciclib-induced apoptosis. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Guo, L.; Li, W.; Zhu, X.; Ling, Y.; Qiu, T.; Dong, L.; Fang, Y.; Yang, H.; Ying, J. PD-L1 expression and CD274 gene alteration in triple-negative breast cancer: Implication for prognostic biomarker. SpringerPlus 2016, 5. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Learn, C.A.; Archer, G.E.; McLendon, R.E.; Chewning, T.A.; Tuck, F.L.; Pracyk, J.B.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; et al. Brain tumors in mice are susceptible to blockade of epidermal growth factor receptor (EGFR) with the oral, specific, EGFR-tyrosine kinase inhibitor ZD1839 (iressa). Clin. Cancer Res. 2002, 8, 3496–3502. [Google Scholar]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef]
- Narayana, A.; Mathew, M.; Tam, M.; Kannan, R.; Madden, K.M.; Golfinos, J.G.; Parker, E.C.; Ott, P.A.; Pavlick, A.C. Vemurafenib and radiation therapy in melanoma brain metastases. J. Neurooncol. 2013, 113, 411–416. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Costa, R.; Carneiro, B.A.; Wainwright, D.A.; Santa-Maria, C.A.; Kumthekar, P.; Chae, Y.K.; Gradishar, W.J.; Cristofanilli, M.; Giles, F.J. Developmental therapeutics for patients with breast cancer and central nervous system metastasis: Current landscape and future perspectives. Ann. Oncol. 2017, 28, 44–56. [Google Scholar] [CrossRef]
- Shah, N.; Mohammad, A.S.; Saralkar, P.; Sprowls, S.A.; Vickers, S.D.; John, D.; Tallman, R.M.; Lucke-Wold, B.P.; Jarrell, K.E.; Pinti, M.; et al. Investigational chemotherapy and novel pharmacokinetic mechanisms for the treatment of breast cancer brain metastases. Pharmacol. Res. 2018, 132, 47–68. [Google Scholar] [CrossRef]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S.; et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Collette, Y.; Prébet, T.; Goubard, A.; Adélaïde, J.; Castellano, R.; Carbuccia, N.; Garnier, S.; Guille, A.; Arnoulet, C.; Charbonier, A.; et al. Drug response profiling can predict response to ponatinib in a patient with t(1;9)(q24;q34)-associated B-cell acute lymphoblastic leukemia. Blood Cancer J. 2015, 5. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef]
- Song, F.; Li, X.; Song, F.; Zhao, Y.; Li, H.; Zheng, H.; Gao, Z.; Wang, J.; Zhang, W.; Chen, K. Comparative genomic analysis reveals bilateral breast cancers are genetically independent. Oncotarget 2015, 6, 31820–31829. [Google Scholar] [CrossRef]
- Van Poznak, C.; Somerfield, M.R.; Bast, R.C.; Cristofanilli, M.; Goetz, M.P.; Gonzalez-Angulo, A.M.; Hicks, D.G.; Hill, E.G.; Liu, M.C.; Lucas, W.; et al. Use of Biomarkers to Guide Decisions on Systemic Therapy for Women With Metastatic Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2015, 33, 2695–2704. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Samples | Age (Years) | Time Before BM Occurrence (Months) | No. and Type of Systemic Therapies Before BM Occurrence | Tumor Cellularity (%) | ER Status | PR Status | ERBB2 Status | Molecular Subtype | Survival After BM Occurrence (Months) |
|---|---|---|---|---|---|---|---|---|---|
| PBC1 | 51 | 4 (CT, beva) | 100 | NEG | NEG | NEG | TN | ||
| BM1 | 54 | 100 | NEG | NEG | NEG | TN | 11 | ||
| PBC2 | 66 | 4 (CT, trastu) | 100 | NEG | NEG | POS | HER2+ | ||
| BM2 | 53 | 100 | NEG | POS | POS | HER2+ | 18 | ||
| PBC3 | 62 | 5 (CT, HT) | 100 | POS | POS | NEG | HR+/HER2– | ||
| BM3 | 49 | 70 | POS | NEG | NEG | HR+/HER2– | 30 | ||
| PBC4 | 61 | 2 (CT, HT) | 100 | POS | POS | NEG | HR+/HER2– | ||
| BM4 | 28 | 80 | POS | POS | NEG | HR+/HER2– | 2 | ||
| PBC5 | 51 | 5 (CT, HT, beva) | 100 | POS | NEG | NEG | HR+/HER2– | ||
| BM5 | 173 | 80 | POS | NEG | NEG | HR+/HER2– | 24 | ||
| PBC6 | 43 | 2 (CT) | 80 | NEG | NEG | NEG | TN | ||
| BM6 | 22 | 70 | NEG | NEG | NEG | TN | 7 | ||
| PBC7 | 39 | 6 (CT, HT) | 100 | POS | POS | NEG | HR+/HER2– | ||
| BM7 | 185 | 60 | POS | POS | NEG | HR+/HER2– | 22 | ||
| PBC8 | 43 | 5 (CT, HT, trastu) | 100 | POS | POS | POS | HER2+ | ||
| BM8 | 250 | 60 | POS | POS | POS | HER2+ | 56 | ||
| PBC9 | 29 | 1 (CT) | 100 | NEG | NEG | NEG | TN | ||
| BM9 | 43 | 60 | NEG | NEG | NEG | TN | 67 | ||
| PBC10 | 58 | 2 (CT, HT) | 100 | POS | NEG | NEG | HR+/HER2– | ||
| BM10 | 0 | 100 | NEG | NEG | NEG | TN | 12 | ||
| PBC11 | 40 | 13 (CT, HT) | 100 | POS | POS | NEG | HR+/HER2– | ||
| BM11 | 106 | 100 | NR | NR | NEG | HR+/HER2– | 6 | ||
| PBC12 | 41 | 6 (CT, HT, beva) | 100 | POS | NEG | NEG | HR+/HER2– | ||
| BM12 | 75 | 100 | POS | POS | NEG | HR+/HER2– | 11 | ||
| PBC13 | 61 | 2 (CT, HT, trastu) | 100 | POS | POS | POS | HER2+ | ||
| BM13 | 19 | 100 | POS | POS | POS | HER2+ | 26 | ||
| PBC14 | 65 | 3 (CT, HT, éverolimus) | 75 | POS | POS | NEG | HR+/HER2– | ||
| BM14 | 32 | 50 | NEG | NEG | NEG | TN | 4 |
| Total Number of Mutations | Number of Shared Mutations | Number of Unshared Mutations | Concordance Rate between Paired PBC and BM | |
|---|---|---|---|---|
| All mutations | 478 | 343 | 135 | 72% |
| Recurrent mutations | 25 | 22 | 3 | 88% |
| Non-recurrent mutations | 453 | 320 | 133 | 71% |
| Therapeutic Class | Gene Symbol | Alteration | Highest Evidence Level | Pt 1 | Pt 2 | Pt 3 | Pt 4 | Pt 5 | Pt 6 | Pt 7 | Pt 8 | Pt 9 | Pt 10 | Pt 11 | Pt 12 | Pt 13 | Pt 14 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | PBC | BM | ||||
| PIK3/AKT/MTOR inhibitors | AKT1 | E17K | B2 | ||||||||||||||||||||||||||||
| D44N | B2 | ||||||||||||||||||||||||||||||
| MTOR | A607V | B2 | |||||||||||||||||||||||||||||
| PIK3CA | E542K H1047R | A2 | |||||||||||||||||||||||||||||
| E545K | A2 | ||||||||||||||||||||||||||||||
| H1047R | A2 | ||||||||||||||||||||||||||||||
| Q546E | A2 | ||||||||||||||||||||||||||||||
| NF1 | D176E | B2 | |||||||||||||||||||||||||||||
| PTEN | Q171X | A2 | |||||||||||||||||||||||||||||
| PARP inhibitors | ATM | G2023R | B2 | ||||||||||||||||||||||||||||
| BRCA1 | E1282fs 1280-1281del | B1 | |||||||||||||||||||||||||||||
| E881X | B1 | ||||||||||||||||||||||||||||||
| Q1811R | B1 | ||||||||||||||||||||||||||||||
| BRCA2 | Y2222C | B1 | |||||||||||||||||||||||||||||
| CHEK2 | T367fs | B2 | |||||||||||||||||||||||||||||
| TKR inhibitors | EGFR | amplification | B2 | ||||||||||||||||||||||||||||
| M137I | B1 | ||||||||||||||||||||||||||||||
| ERBB2 | amplification | A1 | |||||||||||||||||||||||||||||
| RET | L389F | B1 | |||||||||||||||||||||||||||||
| FGF3 | amplification | A2 | |||||||||||||||||||||||||||||
| FGF4 | amplification | A2 | |||||||||||||||||||||||||||||
| FGFR1 | amplification | A2 | |||||||||||||||||||||||||||||
| Hormone therapy | ESR1 | E380Q | A2 | ||||||||||||||||||||||||||||
| Y537C | A2 | ||||||||||||||||||||||||||||||
| Epigenetic therapy | SMARCA4 | D1607N | B2 | ||||||||||||||||||||||||||||
| Others | TP53 | R196Q | B2 | ||||||||||||||||||||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyran, M.; Carbuccia, N.; Garnier, S.; Guille, A.; Adelaïde, J.; Finetti, P.; Touzlian, J.; Viens, P.; Tallet, A.; Goncalves, A.; et al. A Comparison of DNA Mutation and Copy Number Profiles of Primary Breast Cancers and Paired Brain Metastases for Identifying Clinically Relevant Genetic Alterations in Brain Metastases. Cancers 2019, 11, 665. https://doi.org/10.3390/cancers11050665
Tyran M, Carbuccia N, Garnier S, Guille A, Adelaïde J, Finetti P, Touzlian J, Viens P, Tallet A, Goncalves A, et al. A Comparison of DNA Mutation and Copy Number Profiles of Primary Breast Cancers and Paired Brain Metastases for Identifying Clinically Relevant Genetic Alterations in Brain Metastases. Cancers. 2019; 11(5):665. https://doi.org/10.3390/cancers11050665
Chicago/Turabian StyleTyran, Marguerite, Nadine Carbuccia, Séverine Garnier, Arnaud Guille, José Adelaïde, Pascal Finetti, Julien Touzlian, Patrice Viens, Agnès Tallet, Anthony Goncalves, and et al. 2019. "A Comparison of DNA Mutation and Copy Number Profiles of Primary Breast Cancers and Paired Brain Metastases for Identifying Clinically Relevant Genetic Alterations in Brain Metastases" Cancers 11, no. 5: 665. https://doi.org/10.3390/cancers11050665
APA StyleTyran, M., Carbuccia, N., Garnier, S., Guille, A., Adelaïde, J., Finetti, P., Touzlian, J., Viens, P., Tallet, A., Goncalves, A., Metellus, P., Birnbaum, D., Chaffanet, M., & Bertucci, F. (2019). A Comparison of DNA Mutation and Copy Number Profiles of Primary Breast Cancers and Paired Brain Metastases for Identifying Clinically Relevant Genetic Alterations in Brain Metastases. Cancers, 11(5), 665. https://doi.org/10.3390/cancers11050665