Profiling of Epigenetic Features in Clinical Samples Reveals Novel Widespread Changes in Cancer


 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
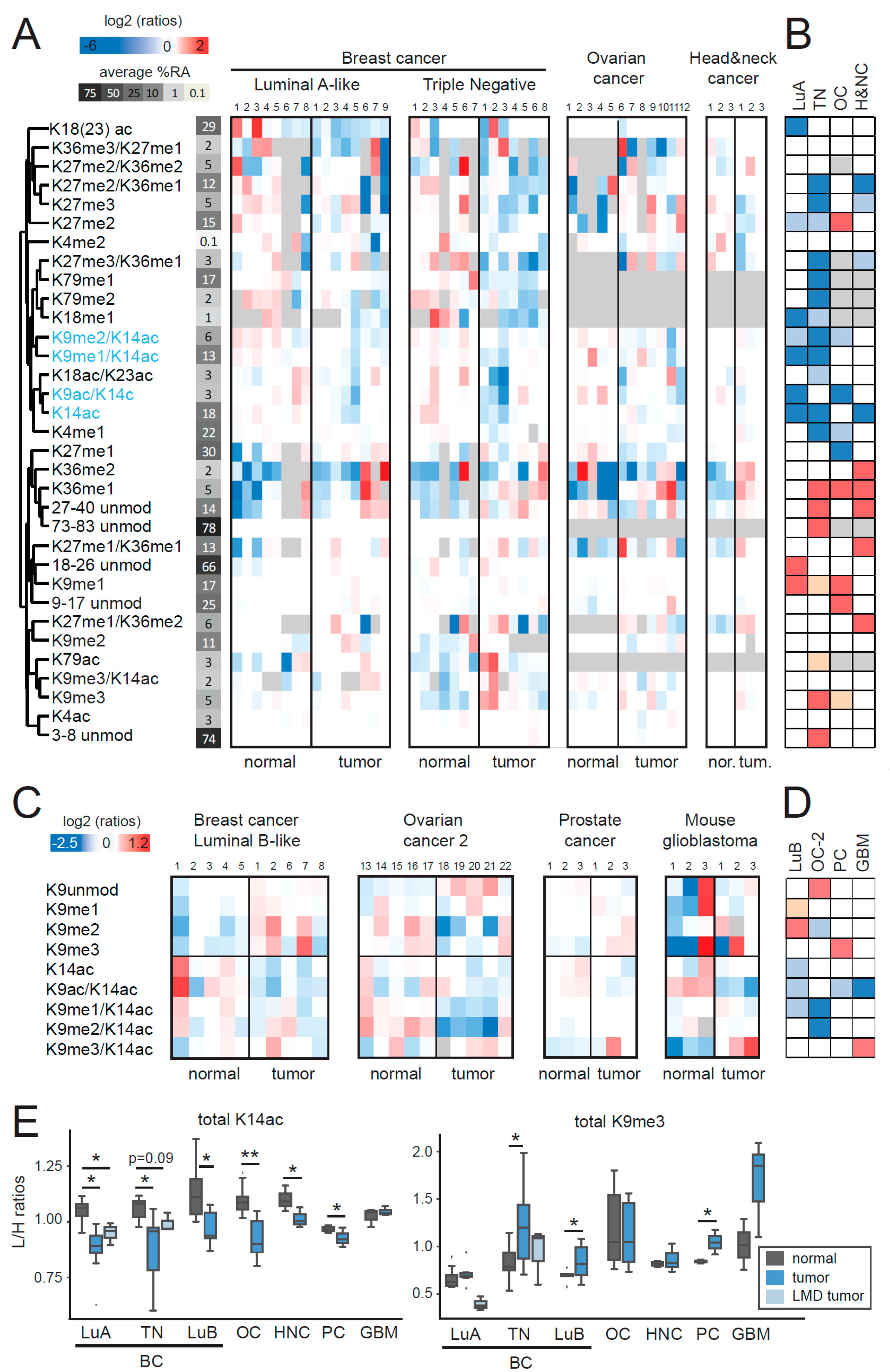
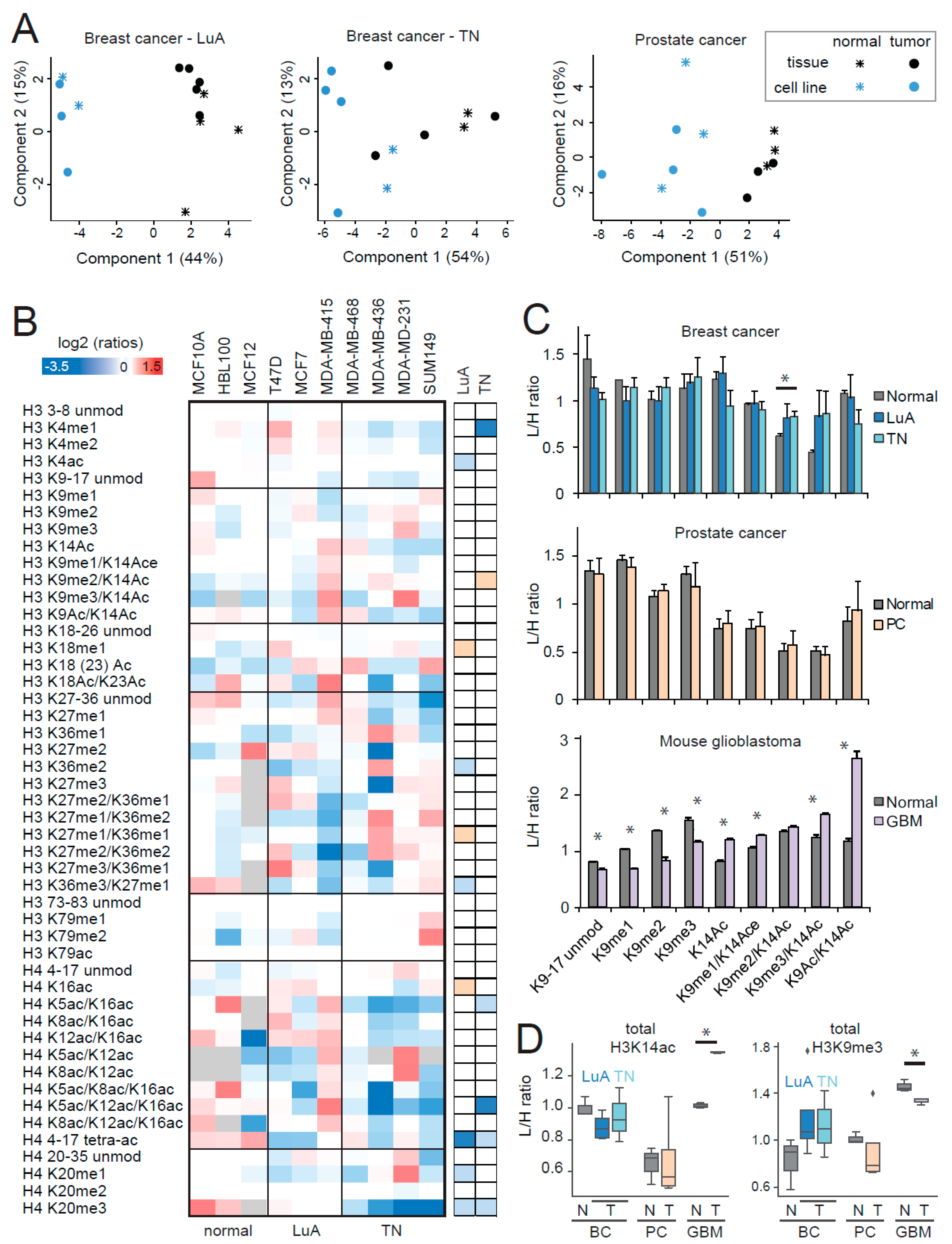
2.1. MS-Based Profiling of Normal and Tumor Tissues
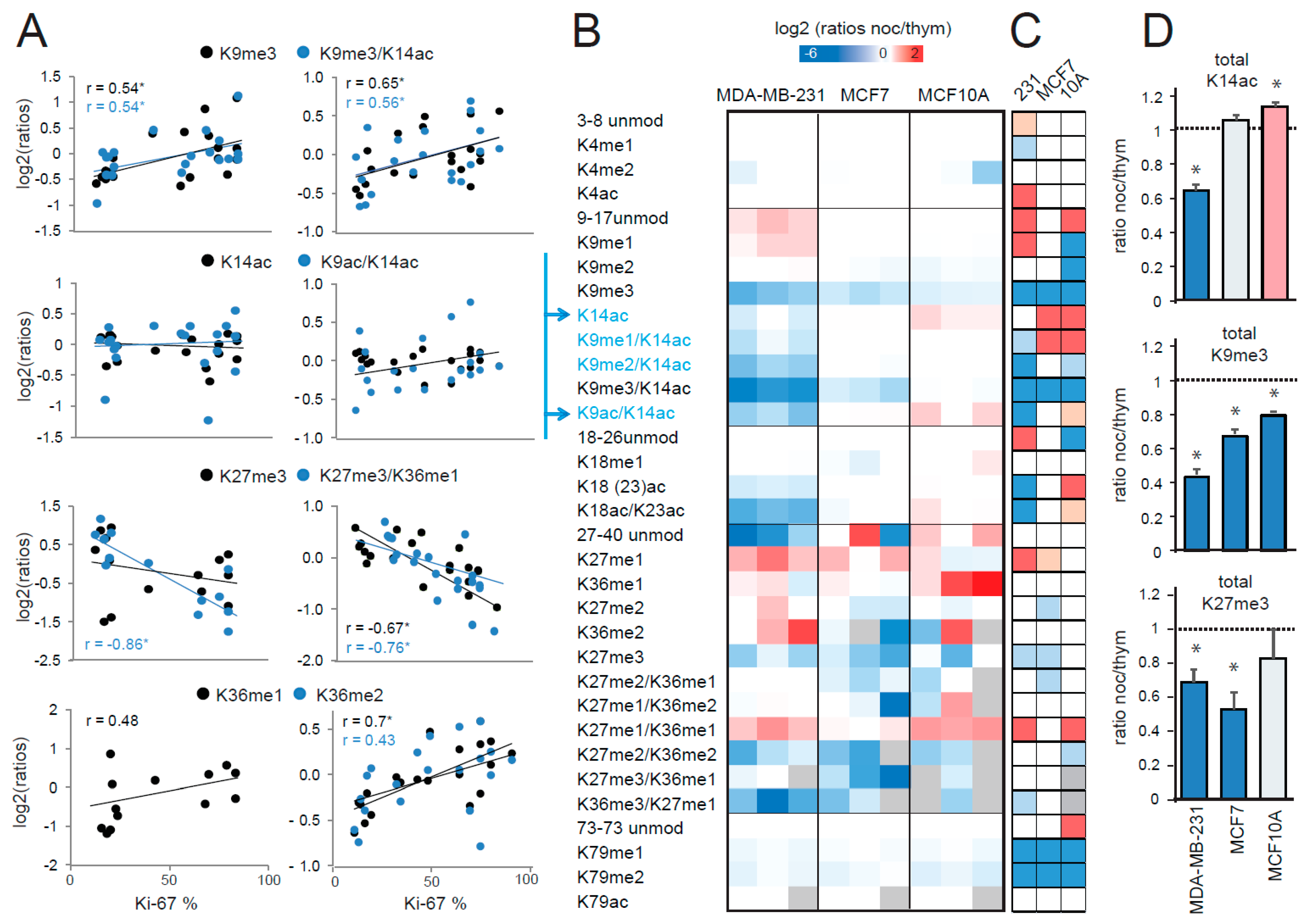
2.2. MS-Based Profiling of Cycling Cells
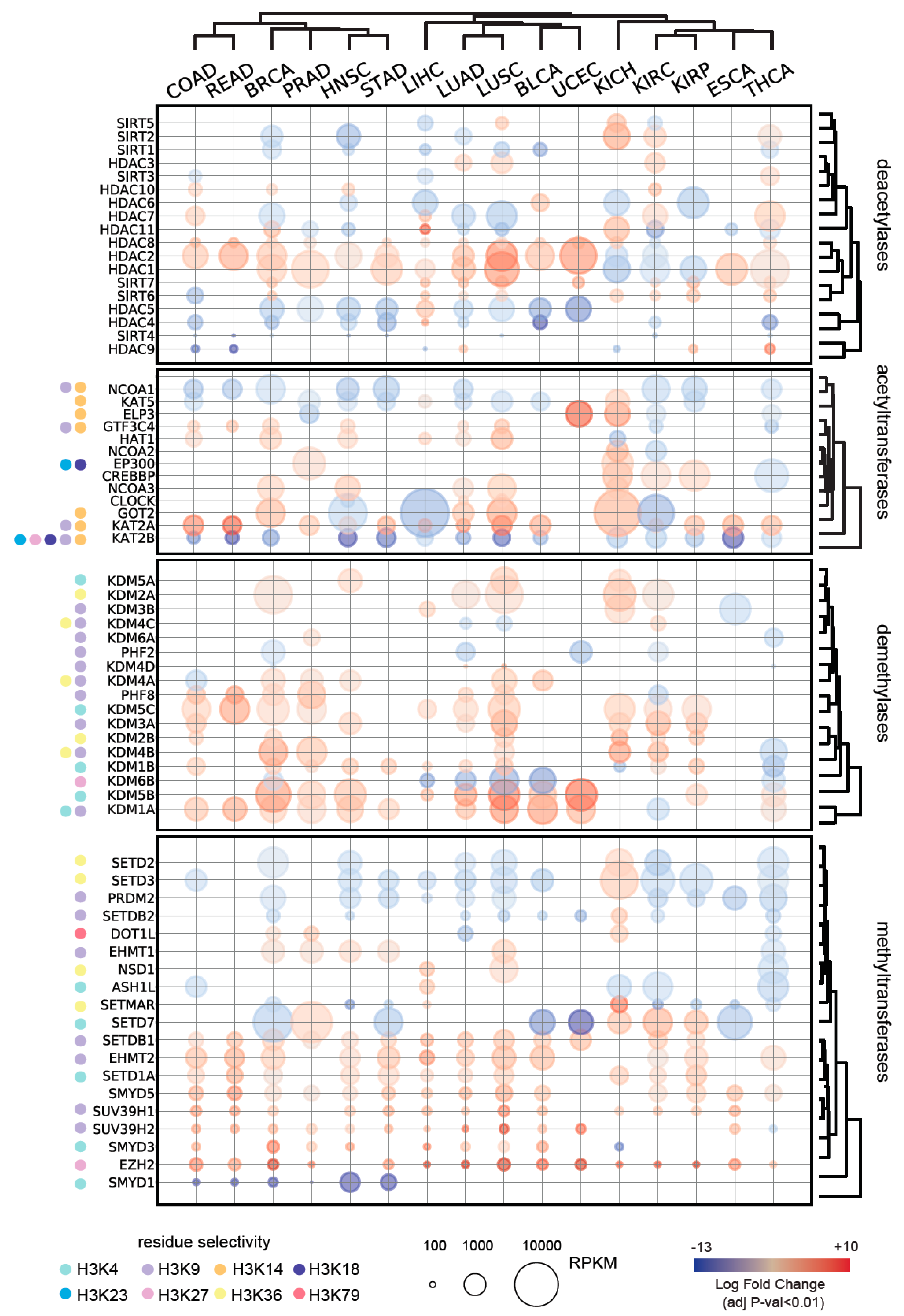
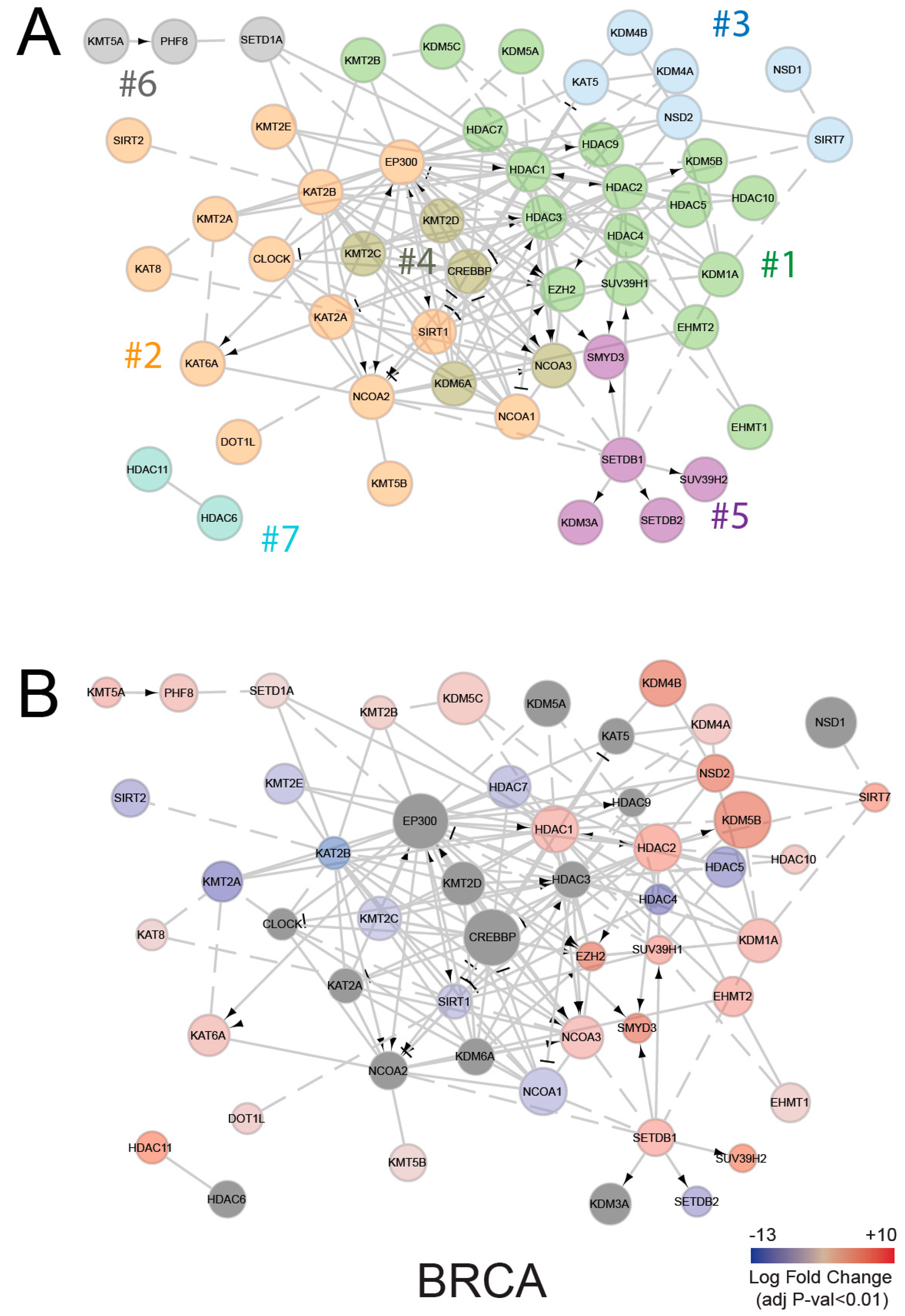
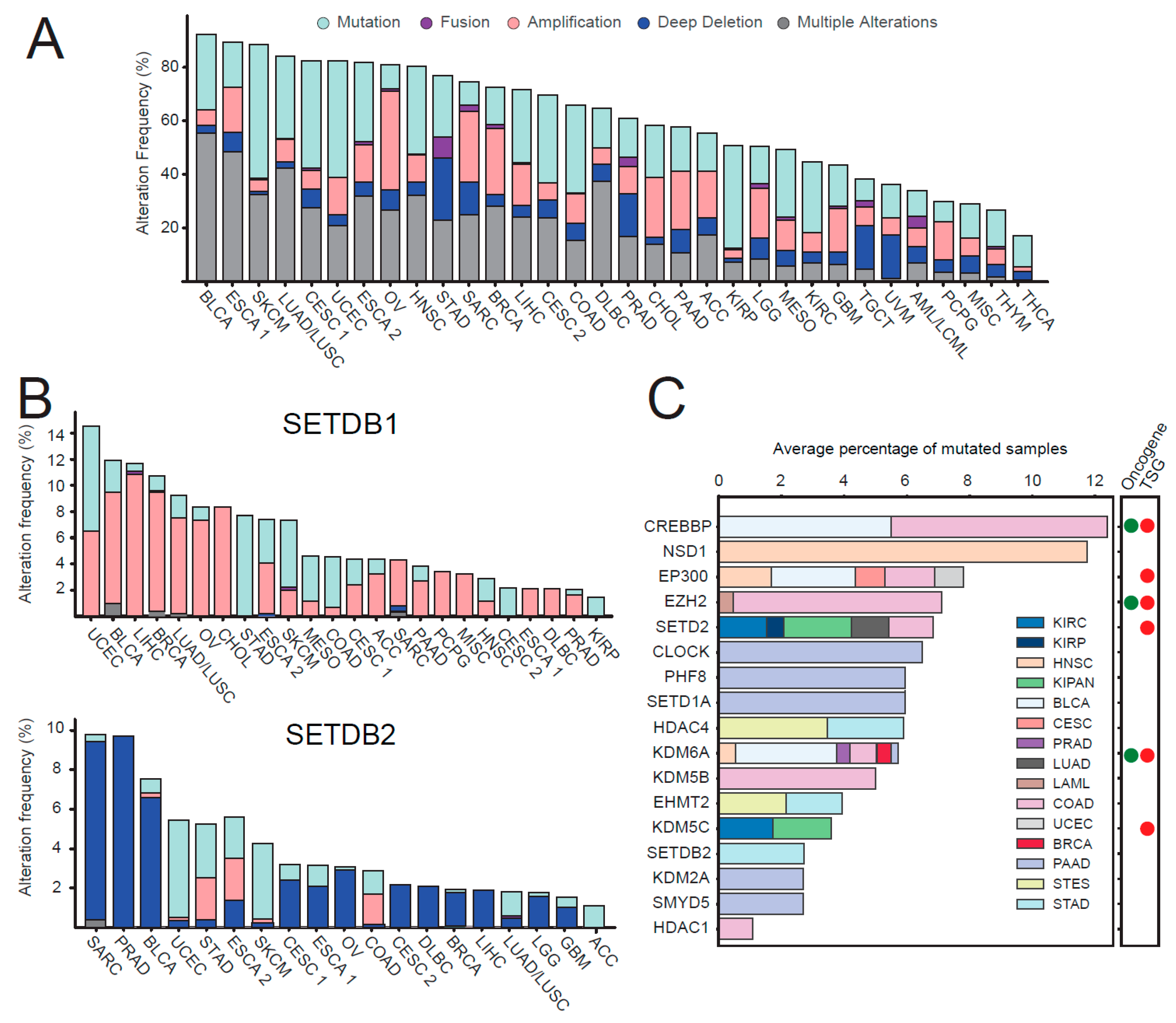
2.3. Expression of Histone Modifying Enzymes in Normal and Tumor Tissues
2.4. Comparison of Histone PTMs in Normal and Tumor Cell Lines
3. Materials and Methods
3.1. Tissue Specimens
3.2. Cell Lines and Primary Cultures
3.3. Mouse Tissues
3.4. Cell Synchronization
3.5. Histone Enrichment
3.6. Super-SILAC
3.7. Histone Digestion
3.8. LC-MS/MS Analysis of Histone PTMs
3.9. Histone PTM Data Analysis
3.10. Analysis of TCGA RNAseq Expression Data
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Garcia, B.A. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotorynski, E. Histone serotonylation boosts neuronal transcription. Nat. Rev. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Shen, L.; Kondo, Y.; Guo, Y.; Zhang, J.; Zhang, L.; Ahmed, S.; Shu, J.; Chen, X.; Waterland, R.A.; Issa, J.P. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007, 3, 2023–2036. [Google Scholar] [CrossRef]
- Sincic, N.; Herceg, Z. DNA methylation and cancer: Ghosts and angels above the genes. Curr. Opin. Oncol. 2011, 23, 69–76. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, A.; Moretti, S.; Minucci, S.; Bonaldi, T. SILAC-based proteomic analysis to dissect the “histone modification signature” of human breast cancer cells. Amino Acids 2011, 41, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Leroy, G.; Dimaggio, P.A.; Chan, E.Y.; Zee, B.M.; Blanco, M.A.; Bryant, B.; Flaniken, I.Z.; Liu, S.; Kang, Y.; Trojer, P.; et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 2013, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, J.; Bachmann, A.; Goke, F.; Behbahani, T.E.; Baumann, C.; Heukamp, L.C.; Rogenhofer, S.; Muller, S.C. Alterations of global histone H3K9 and H3K27 methylation levels in bladder cancer. Urol. Int. 2014, 93, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Nestor, C.E.; Ottaviano, R.; Reinhardt, D.; Cruickshanks, H.A.; Mjoseng, H.K.; McPherson, R.C.; Lentini, A.; Thomson, J.P.; Dunican, D.S.; Pennings, S.; et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 2015, 16, 11. [Google Scholar] [CrossRef]
- Noberini, R.; Osti, D.; Miccolo, C.; Richichi, C.; Lupia, M.; Corleone, G.; Hong, S.P.; Colombo, P.; Pollo, B.; Fornasari, L.; et al. Extensive and systematic rewiring of histone post-translational modifications in cancer model systems. Nucleic Acids Res. 2018, 46, 3817–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noberini, R.; Longuespee, R.; Richichi, C.; Pruneri, G.; Kriegsmann, M.; Pelicci, G.; Bonaldi, T. PAT-H-MS coupled with laser microdissection to study histone post-translational modifications in selected cell populations from pathology samples. Clin. Epigenetics 2017, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Noberini, R.; Uggetti, A.; Pruneri, G.; Minucci, S.; Bonaldi, T. Pathology Tissue-quantitative Mass Spectrometry Analysis to Profile Histone Post-translational Modification Patterns in Patient Samples. Mol. Cell. Proteom. 2016, 15, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Soldi, M.; Cuomo, A.; Bonaldi, T. Improved bottom-up strategy to efficiently separate hypermodified histone peptides through ultra-HPLC separation on a bench top Orbitrap instrument. Proteomics 2014, 14, 2212–2225. [Google Scholar] [CrossRef]
- Restellini, C.; Cuomo, A.; Lupia, M.; Giordano, M.; Bonaldi, T.; Noberini, R. Alternative digestion approaches improve histone modification mapping by mass spectrometry in clinical samples. Proteom. Clin. Appl. 2019, 13, e1700166. [Google Scholar] [CrossRef]
- Prat, A.; Perou, C.M. Mammary development meets cancer genomics. Nat. Med. 2009, 15, 842–844. [Google Scholar] [CrossRef]
- Bonenfant, D.; Towbin, H.; Coulot, M.; Schindler, P.; Mueller, D.R.; van Oostrum, J. Analysis of dynamic changes in post-translational modifications of human histones during cell cycle by mass spectrometry. Mol. Cell. Proteom. 2007, 6, 1917–1932. [Google Scholar] [CrossRef]
- Zane, L.; Chapus, F.; Pegoraro, G.; Misteli, T. HiHiMap: Single-cell quantitation of histones and histone posttranslational modifications across the cell cycle by high-throughput imaging. Mol. Biol. Cell 2017, 28, 2290–2302. [Google Scholar] [CrossRef]
- Blais, A.; van Oevelen, C.J.; Margueron, R.; Acosta-Alvear, D.; Dynlacht, B.D. Retinoblastoma tumor suppressor protein-dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. J. Cell Biol. 2007, 179, 1399–1412. [Google Scholar] [CrossRef]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011, 286, 7983–7989. [Google Scholar] [CrossRef]
- Schmitges, F.W.; Prusty, A.B.; Faty, M.; Stutzer, A.; Lingaraju, G.M.; Aiwazian, J.; Sack, R.; Hess, D.; Li, L.; Zhou, S.; et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 2011, 42, 330–341. [Google Scholar] [CrossRef]
- Zheng, Y.; Sweet, S.M.; Popovic, R.; Martinez-Garcia, E.; Tipton, J.D.; Thomas, P.M.; Licht, J.D.; Kelleher, N.L. Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc. Natl. Acad. Sci. USA 2012, 109, 13549–13554. [Google Scholar] [CrossRef] [Green Version]
- Suva, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef]
- Albert, M.; Helin, K. Histone methyltransferases in cancer. Semin. Cell Dev. Biol. 2010, 21, 209–220. [Google Scholar] [CrossRef]
- D’Oto, A.; Tian, Q.W.; Davidoff, A.M.; Yang, J. Histone demethylases and their roles in cancer epigenetics. J. Med. Oncol. Ther. 2016, 1, 34–40. [Google Scholar]
- Nottke, A.; Colaiacovo, M.P.; Shi, Y. Developmental roles of the histone lysine demethylases. Development 2009, 136, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Dell’Aversana, C.; Lepore, I.; Altucci, L. HDAC modulation and cell death in the clinic. Exp. Cell Res. 2012, 318, 1229–1244. [Google Scholar] [CrossRef]
- Di Cerbo, V.; Schneider, R. Cancers with wrong HATs: The impact of acetylation. Brief. Funct. Genom. 2013, 12, 231–243. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Anamika, K.; Krebs, A.R.; Thompson, J.; Poch, O.; Devys, D.; Tora, L. Lessons from genome-wide studies: An integrated definition of the coactivator function of histone acetyl transferases. Epigenetics Chromatin 2010, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Koutsogiannouli, E.A.; Wagner, N.; Hader, C.; Pinkerneil, M.; Hoffmann, M.J.; Schulz, W.A. Differential Effects of Histone Acetyltransferase GCN5 or PCAF Knockdown on Urothelial Carcinoma Cells. Int. J. Mol. Sci. 2017, 18, 1449. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.W.; Jin, H.J.; Zhao, W.; Gao, B.; Fang, J.; Wei, J.; Zhang, D.D.; Zhang, J.; Fang, D. The Histone Acetyltransferase GCN5 Expression Is Elevated and Regulated by c-Myc and E2F1 Transcription Factors in Human Colon Cancer. Gene Expr. 2015, 16, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Gai, X.; Tu, K.; Li, C.; Lu, Z.; Roberts, L.R.; Zheng, X. Histone acetyltransferase PCAF accelerates apoptosis by repressing a GLI1/BCL2/BAX axis in hepatocellular carcinoma. Cell Death Dis. 2015, 6, e1712. [Google Scholar] [CrossRef]
- Zheng, X.; Gai, X.; Ding, F.; Lu, Z.; Tu, K.; Yao, Y.; Liu, Q. Histone acetyltransferase PCAF up-regulated cell apoptosis in hepatocellular carcinoma via acetylating histone H4 and inactivating AKT signaling. Mol. Cancer 2013, 12, 96. [Google Scholar] [CrossRef]
- Fei, H.J.; Zu, L.D.; Wu, J.; Jiang, X.S.; Wang, J.L.; Chin, Y.E.; Fu, G.H. PCAF acts as a gastric cancer suppressor through a novel PCAF-p16-CDK4 axis. Am. J. Cancer Res. 2016, 6, 2772–2786. [Google Scholar] [PubMed]
- Wan, J.; Xu, W.; Zhan, J.; Ma, J.; Li, X.; Xie, Y.; Wang, J.; Zhu, W.G.; Luo, J.; Zhang, H. PCAF-mediated acetylation of transcriptional factor HOXB9 suppresses lung adenocarcinoma progression by targeting oncogenic protein JMJD6. Nucleic Acids Res. 2016, 44, 10662–10675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, M.Z.; Wang, J.J.; Li, D.W.; Yu, G.; Wang, X.; Pan, J.; Chen, Y.; He, M.X. The p300/CBP associated factor is frequently downregulated in intestinal-type gastric carcinoma and constitutes a biomarker for clinical outcome. Cancer Biol. Ther. 2010, 9, 312–320. [Google Scholar] [CrossRef]
- Spencer, T.E.; Jenster, G.; Burcin, M.M.; Allis, C.D.; Zhou, J.; Mizzen, C.A.; McKenna, N.J.; Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 1997, 389, 194–198. [Google Scholar] [CrossRef]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DesJarlais, R.; Tummino, P.J. Role of Histone-Modifying Enzymes and Their Complexes in Regulation of Chromatin Biology. Biochemistry 2016, 55, 1584–1599. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Dawson, E.; Duong, A.; Haw, R.; Stein, L. ReactomeFIViz: A Cytoscape app for pathway and network-based data analysis. F1000Research 2014, 3, 146. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Shilatifard, A. Chromatin signatures of cancer. Genes Dev. 2015, 29, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, F.; Ding, J.; Liang, Y.; Zhan, Z.; Zhan, Y.; Chen, L.H.; Ding, Y. Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53. J. Cancer 2017, 8, 3318–3330. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, J.; Li, Q.; Chen, K.; Liang, Y.; Zhan, Z.; Ye, F.; Ni, W.; Chen, L.; Ding, Y. Histone methyltransferase SETDB1 promotes cells proliferation and migration by interacting withTiam1 in hepatocellular carcinoma. BMC Cancer 2018, 18, 539. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Nishikawaji, T.; Akiyama, Y.; Shimada, S.; Kojima, K.; Kawano, T.; Eishi, Y.; Yuasa, Y.; Tanaka, S. Oncogenic roles of the SETDB2 histone methyltransferase in gastric cancer. Oncotarget 2016, 7, 67251–67265. [Google Scholar] [CrossRef] [Green Version]
- Bui, N.; Huang, J.K.; Bojorquez-Gomez, A.; Licon, K.; Sanchez, K.S.; Tang, S.N.; Beckett, A.N.; Wang, T.; Zhang, W.; Shen, J.P.; et al. Disruption of NSD1 in Head and Neck Cancer Promotes Favorable Chemotherapeutic Responses Linked to Hypomethylation. Mol. Cancer Ther. 2018, 17, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Sanchini, V.; Bonizzi, G.; Disalvatore, D.; Monturano, M.; Pece, S.; Viale, G.; Di Fiore, P.P.; Boniolo, G. A Trust-Based Pact in Research Biobanks. From Theory to Practice. Bioethics 2016, 30, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Althouse, A.D. Adjust for Multiple Comparisons? It’s Not That Simple. Ann. Thorac. Surg. 2016, 101, 1644–1645. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noberini, R.; Restellini, C.; Savoia, E.O.; Raimondi, F.; Ghiani, L.; Jodice, M.G.; Bertalot, G.; Bonizzi, G.; Capra, M.; Maffini, F.A.; et al. Profiling of Epigenetic Features in Clinical Samples Reveals Novel Widespread Changes in Cancer. Cancers 2019, 11, 723. https://doi.org/10.3390/cancers11050723
Noberini R, Restellini C, Savoia EO, Raimondi F, Ghiani L, Jodice MG, Bertalot G, Bonizzi G, Capra M, Maffini FA, et al. Profiling of Epigenetic Features in Clinical Samples Reveals Novel Widespread Changes in Cancer. Cancers. 2019; 11(5):723. https://doi.org/10.3390/cancers11050723
Chicago/Turabian StyleNoberini, Roberta, Camilla Restellini, Evelyn Oliva Savoia, Francesco Raimondi, Lavinia Ghiani, Maria Giovanna Jodice, Giovanni Bertalot, Giuseppina Bonizzi, Maria Capra, Fausto Antonio Maffini, and et al. 2019. "Profiling of Epigenetic Features in Clinical Samples Reveals Novel Widespread Changes in Cancer" Cancers 11, no. 5: 723. https://doi.org/10.3390/cancers11050723
APA StyleNoberini, R., Restellini, C., Savoia, E. O., Raimondi, F., Ghiani, L., Jodice, M. G., Bertalot, G., Bonizzi, G., Capra, M., Maffini, F. A., Tagliabue, M., Ansarin, M., Lupia, M., Giordano, M., Osti, D., Pelicci, G., Chiocca, S., & Bonaldi, T. (2019). Profiling of Epigenetic Features in Clinical Samples Reveals Novel Widespread Changes in Cancer. Cancers, 11(5), 723. https://doi.org/10.3390/cancers11050723