Purinergic Calcium Signals in Tumor-Derived Endothelium

 ,
,  ,
, 
 , ,
, ,  , and
, and 
Abstract
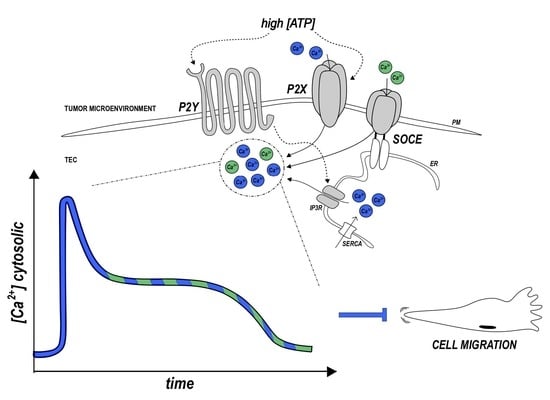
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. High ATP Stimulation Triggers Biphasic Ca2+ Signals in Human Tumor-Derived Endothelial Cells
2.2. ATP-Evoked Ca2+ Influx Includes Both Store Operated Ca2+ Entry (SOCE) and Non-SOCE Components
2.3. SOCE Component Is Not Required for the Anti-Migratory Activity of ATP
2.4. Ca2+ Signals Mediated by Other Purinergic Agonists
2.5. Contribution of ER and Other Organelles in the Calcium Release Induced by ATP and UTP
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Calcium Imaging and Experimental Protocols
4.3. Migration Assays
4.4. RNA Extraction and Real-Time PCR Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burnstock, G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol. Rev. 2006, 58, 58–86. [Google Scholar] [CrossRef] [PubMed]
- Deli, T.; Csernoch, L. Extracellular ATP and cancer: An overview with special reference to P2 purinergic receptors. Pathol. Oncol. Res. 2008, 14, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F. Purines, purinergic receptors, and cancer. Cancer Res. 2012, 72, 5441–5447. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Novak, I. Purinergic signalling in the pancreas in health and disease. J. Endocrinol. 2012, 213, 123–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta 2008, 1783, 673–694. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G. Purinergic signalling: Its unpopular beginning, its acceptance and its exciting future. Bioessays 2012, 34, 218–225. [Google Scholar] [CrossRef]
- Erlinge, D.; Burnstock, G. P2 receptors in cardiovascular regulation and disease. Purinergic. Signal. 2008, 4, 1–20. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic regulation of vascular tone and remodelling. Auton Autacoid. Pharmacol. 2009, 29, 63–72. [Google Scholar] [CrossRef]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased Level of Extracellular ATP at Tumor Sites: In Vivo Imaging with Plasma Membrane Luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Nerves. Lancet 1972. [Google Scholar] [CrossRef]
- Nishimura, A.; Sunggip, C.; Oda, S.; Numaga-Tomita, T.; Tsuda, M.; Nishida, M. Purinergic P2Y receptors: Molecular diversity and implications for treatment of cardiovascular diseases. Pharmacol. Ther. 2017, 180, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Mahaut-Smith, M.P.; Taylor, K.A.; Evans, R.J. Calcium Signalling through Ligand-Gated Ion Channels such as P2X1 Receptors in the Platelet and other Non-Excitable Cells; Springer: Cham, Switzerland, 2016; pp. 305–329. [Google Scholar]
- Burnstock, G.; Knight, G.E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic. Signal. 2018, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Paoletta, S.; Katritch, V.; Wu, B.; Gao, Z.G.; Zhao, Q.; Stevens, R.C.; Kiselev, E. Nucleotides Acting at P2Y Receptors: Connecting Structure and Function. Mol. Pharmacol. 2015, 88, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thillaiappan, N.B.; Chakraborty, P.; Hasan, G. IP3 receptors and Ca2+ entry. Biochim. Biophys. Acta Mol. Cell Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Raqeeb, A.; Sheng, J.; Ao, N.; Braun, A.P. Purinergic P2Y2 receptors mediate rapid Ca(2+) mobilization, membrane hyperpolarization and nitric oxide production in human vascular endothelial cells. Cell Calcium. 2011, 49, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Lyubchenko, T.; Woodward, H.; Veo, K.D.; Burns, N.; Nijmeh, H.; Liubchenko, G.A.; Stenmark, K.R.; Gerasimovskaya, E.V. P2Y1 and P2Y13 purinergic receptors mediate Ca2+ signaling and proliferative responses in pulmonary artery vasa vasorum endothelial cells. AJP Cell Physiol. 2011, 300, C266–C275. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Rosado, J.A. STIM and calcium channel complexes in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca2+ entry and control endoplasmic reticulum Ca2+ refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Baruffi, S.; Spaggiari, S.; Coltrini, D.; Berra-Romani, R.; Signorelli, S.; Castelli, L.; Taglietti, V.; Tanzi, F. P2y1 and P2y2 receptor-operated Ca2+ signals in primary cultures of cardiac microvascular endothelial cells. Microvasc. Res. 2001, 61, 240–252. [Google Scholar] [CrossRef]
- Xie, J.; Pan, H.; Yao, J.; Zhou, Y.; Han, W. SOCE and cancer: Recent progress and new perspectives. Int. J. Cancer 2016, 138, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal Cell-Derived Factor-1α Promotes Endothelial Colony-Forming Cell Migration Through the Ca2+-Dependent Activation of the Extracellular Signal-Regulated Kinase 1/2 and Phosphoinositide 3-Kinase/AKT Pathways. Stem. Cells Dev. 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L.; Genova, T.; Avanzato, D.; Antoniotti, S.; Fiorio Pla, A. Targeting calcium channels to block tumor vascularization. Recent Pat. Anticancer Drug Discov. 2013, 8, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Teuscher, E.; Weidlich, V. Adenosine nucleotides, adenosine and adenine as angiogenesis factors. Biomed. Biochim. Acta 1985, 44, 493–495. [Google Scholar]
- Van Daele, P.; Van Coevorden, A.; Roger, P.P.; Boeynaems, J.M. Effects of adenine nucleotides on the proliferation of aortic endothelial cells. Circ. Res. 1992, 70, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Meininger, C.J.; Schelling, M.E.; Granger, H.J. Adenosine and hypoxia stimulate proliferation and migration of endothelial cells. Am. J. Physiol. 1988, 255, H554–H562. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Di Virgilio, F. Purinergic signalling and cancer. Purinergic. Signal. 2013, 9, 491–540. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Signalling: Pathophysiology and Therapeutic Potential. Keio J. Med. 2013, 62, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Rapaport, E. Mechanisms of anticancer activities of adenine nucleotides in tumor-bearing hosts. Ann. N. Y. Acad. Sci. 1990, 603, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Jantaratnotai, N.; Serrano-Perez, M.C.; McGeer, P.L.; McLarnon, J.G. Block of purinergic P2X7R inhibits tumor growth in a C6 glioma brain tumor animal model. J. Neuropathol. Exp. Neurol. 2011, 70, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Chen, X.; Zhang, L.; Chen, J.; Liang, Y.; Li, X.; Xiang, J.; Wang, L.; Guo, G.; Zhang, B.; et al. P2X7R suppression promotes glioma growth through epidermal growth factor receptor signal pathway. Int. J. Biochem. Cell Biol. 2013, 45, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Avanzato, D.; Genova, T.; Fiorio Pla, A.; Bernardini, M.; Bianco, S.; Bussolati, B.; Mancardi, D.; Giraudo, E.; Maione, F.; Cassoni, P.; et al. Activation of P2X7 and P2Y11 purinergic receptors inhibits migration and normalizes tumor-derived endothelial cells via cAMP signaling. Sci. Rep. 2016, 6, 32602. [Google Scholar] [CrossRef] [PubMed]
- Halls, M.L. Cooper DMF. Regulation by Ca2+-signaling pathways of adenylyl cyclases. Cold Spring Harb. Perspect. Biol. 2011, 3, a004143. [Google Scholar] [CrossRef] [PubMed]
- Baumer, Y.; Spindler, V.; Werthmann, R.C.; Bünemann, M.; Waschke, J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J. Cell Physiol. 2009, 220, 716–726. [Google Scholar] [CrossRef]
- Bianco, S.; Mancardi, D.; Merlino, A.; Bussolati, B.; Munaron, L. Hypoxia and hydrogen sulfide differentially affect normal and tumor-derived vascular endothelium. Redox. Biol. 2017, 12, 499–504. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef]
- Genova, T.; Grolez, G.P.; Camillo, C.; Bernardini, M.; Bokhobza, A.; Richard, E.; Scianna, M.; Lemonnier, L.; Valdembri, D.; Munaron, L.; et al. TRPM8 inhibits endothelial cell migration via a non-channel function by trapping the small GTPase Rap1. J. Cell Biol. 2017, 216, 2107–2130. [Google Scholar] [CrossRef] [Green Version]
- Fiorio Pla, A.; Genova, T.; Pupo, E.; Tomatis, C.; Genazzani, A.; Zaninetti, R.; Munaron, L. Multiple roles of protein kinase a in arachidonic acid-mediated Ca2+ entry and tumor-derived human endothelial cell migration. Mol Cancer Res. 2010, 8, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mussano, F.; Genova, T.; Falzacappa, E.V.; Scopece, P.; Munaron, L.; Rivolo, P.; Mandracci, P.; Benedetti, A.; Carossa, S.; Patelli, A. In vitro characterization of two different atmospheric plasma jet chemical functionalizations of titanium surfaces. Appl. Surf. Sci. 2017, 409, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Fiorio Pla, A.; Brossa, A.; Bernardini, M.; Genova, T.; Grolez, G.; Villers, A.; Leroy, X.; Prevarskaya, N.; Gkika, D.; Bussolati, B. Differential sensitivity of prostate tumor derived endothelial cells to sorafenib and sunitinib. BMC Cancer 2014, 14, 939. [Google Scholar] [CrossRef] [PubMed]
- Basilico, N.; Magnetto, C.; D’Alessandro, S.; Panariti, A.; Rivolta, I.; Genova, T.; Khadjavi, A.; Gulino, G.R.; Argenziano, M.; Soster, M.; et al. Dextran-shelled oxygen-loaded nanodroplets reestablish a normoxia-like pro-angiogenic phenotype and behavior in hypoxic human dermal microvascular endothelium. Toxicol. Appl. Pharmacol. 2015, 288, 330–338. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpellino, G.; Genova, T.; Avanzato, D.; Bernardini, M.; Bianco, S.; Petrillo, S.; Tolosano, E.; de Almeida Vieira, J.R.; Bussolati, B.; Fiorio Pla, A.; et al. Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers 2019, 11, 766. https://doi.org/10.3390/cancers11060766
Scarpellino G, Genova T, Avanzato D, Bernardini M, Bianco S, Petrillo S, Tolosano E, de Almeida Vieira JR, Bussolati B, Fiorio Pla A, et al. Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers. 2019; 11(6):766. https://doi.org/10.3390/cancers11060766
Chicago/Turabian StyleScarpellino, Giorgia, Tullio Genova, Daniele Avanzato, Michela Bernardini, Serena Bianco, Sara Petrillo, Emanuela Tolosano, Joana Rita de Almeida Vieira, Benedetta Bussolati, Alessandra Fiorio Pla, and et al. 2019. "Purinergic Calcium Signals in Tumor-Derived Endothelium" Cancers 11, no. 6: 766. https://doi.org/10.3390/cancers11060766
APA StyleScarpellino, G., Genova, T., Avanzato, D., Bernardini, M., Bianco, S., Petrillo, S., Tolosano, E., de Almeida Vieira, J. R., Bussolati, B., Fiorio Pla, A., & Munaron, L. (2019). Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers, 11(6), 766. https://doi.org/10.3390/cancers11060766