Frugoside Induces Mitochondria-Mediated Apoptotic Cell Death through Inhibition of Sulfiredoxin Expression in Melanoma Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
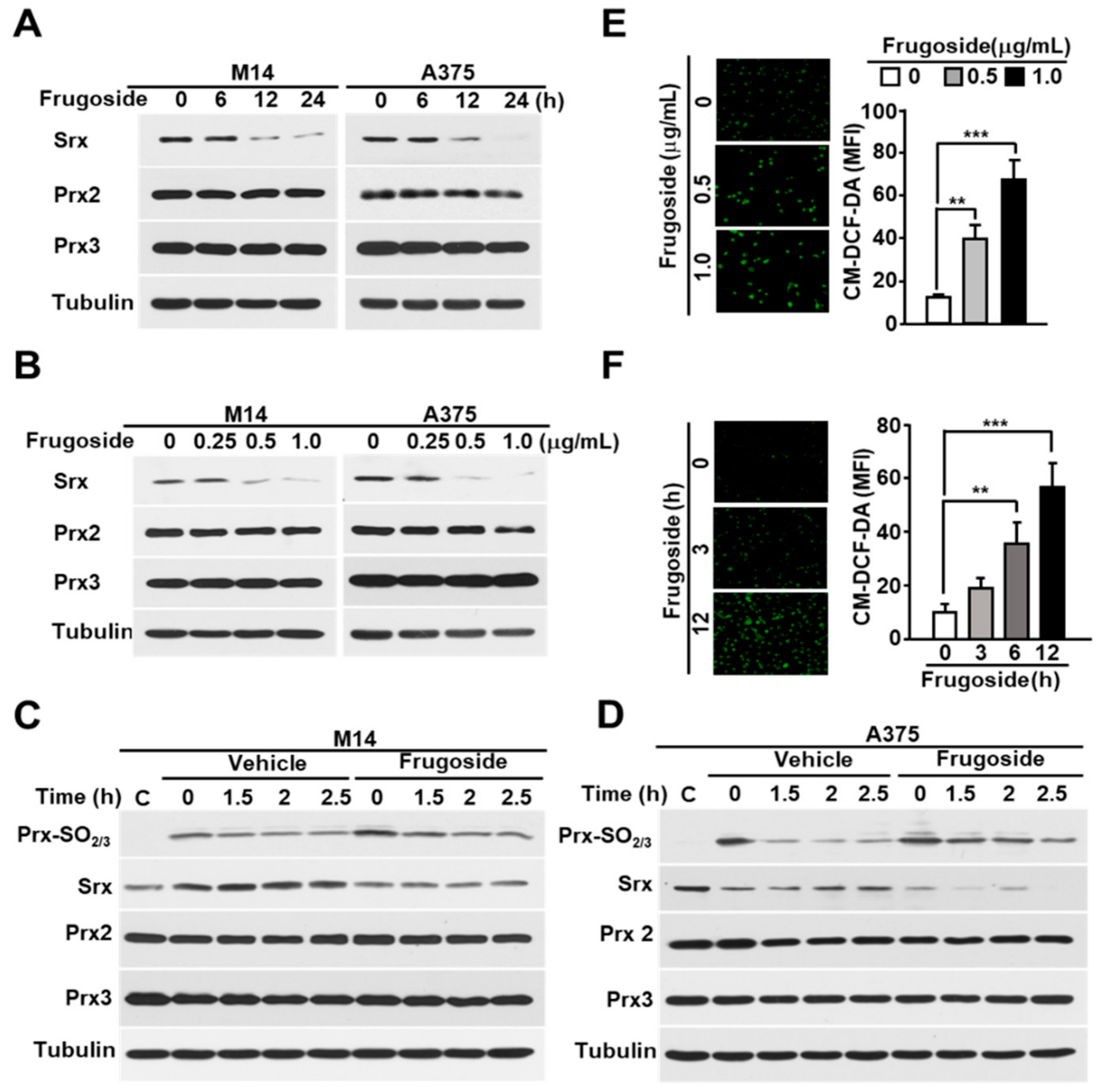
2.1. Frugoside Leads to Attenuated Srx Expression and Subsequently Delays Reduction of Hyperoxidized Prxs in Melanoma Cells
2.2. Frugoside Induces Mitochondria-Mediated Apoptotic Cell Death in Melanoma Cells
2.3. Frugoside Induces Oxidative Mitochondrial Damage
2.4. Frugoside Induces Cell Death via p38 MAPK Activation and ROS Accumulation
2.5. Frugoside-Mediated Cytotoxic Effects Are Reduced by Srx Overexpression
2.6. Frugoside-Mediated Srx Deficiency Impairs Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Antibodies, and Chemicals
4.2. Extraction and Isolation of Frugoside
4.3. In Vitro Cell Death Assays
4.4. Quantitative Reverse Transcriptase Polymerase Chain Reaction
4.5. Protein Isolation and Western Blotting
4.6. Measurement of Mitochondrial Activity
4.7. Clonogenic Assay and Colony-Forming Assay
4.8. Subcellular Fractionation
4.9. Evaluation of Tumorigenicity and Toxicity
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hambright, H.G.; Meng, P.; Kumar, A.P.; Ghosh, R. Inhibition of PI3K/AKT/mTOR axis disrupts oxidative stress-mediated survival of melanoma cells. Oncotarget 2015, 6, 7195–7208. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Zhang, Z. Gambogic Acid Inhibits Malignant Melanoma Cell Proliferation Through Mitochondrial p66shc/ROS-p53/Bax-Mediated Apoptosis. Cell Physiol. Biochem. 2016, 38, 1618–1630. [Google Scholar] [CrossRef] [PubMed]
- Cummins, D.L.; Cummins, J.M.; Pantle, H.; Silverman, M.A.; Leonard, A.L.; Chanmugam, A. Cutaneous malignant melanoma. Mayo Clin. Proc. 2006, 81, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Arcidiacono, P.; Ragonese, F.; Stabile, A.; Pistilli, A.; Kuligina, E.; Rende, M.; Bottoni, U.; Calvieri, S.; Crisanti, A.; Spaccapelo, R. Antitumor activity and expression profiles of genes induced by sulforaphane in human melanoma cells. Eur. J. Nutr. 2017, 1, 017–1527. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Grob, J.J.; Aaronson, N.; Fierlbeck, G.; Tilgen, W.; Seiter, S.; Gore, M.; Aamdal, S.; Cebon, J.; Coates, A.; et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J. Clin. Oncol. 2000, 18, 158–166. [Google Scholar] [CrossRef]
- Garbe, C.; Peris, K.; Hauschild, A.; Saiag, P.; Middleton, M.; Bastholt, L.; Grob, J.J.; Malvehy, J.; Newton-Bishop, J.; Stratigos, A.J.; et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline-Update 2016. Eur. J. Cancer 2016, 63, 201–217. [Google Scholar] [CrossRef]
- Wong, D.J.; Ribas, A. Targeted Therapy for Melanoma. Cancer Treat Res 2016, 167, 251–262. [Google Scholar]
- Kwong, L.N.; Davies, M.A. Targeted therapy for melanoma: Rational combinatorial approaches. Oncogene 2014, 33, 1–9. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Khan, I.A.; Ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef] [PubMed]
- Maiuthed, A.; Chantarawong, W.; Chanvorachote, P. Lung Cancer Stem Cells and Cancer Stem Cell-targeting Natural Compounds. Anticancer Res. 2018, 38, 3797–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlQathama, A.; Prieto, J.M. Natural products with therapeutic potential in melanoma metastasis. Nat. Prod. Rep. 2015, 32, 1170–1182. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, A.B.; Lopes, R.M.; Schwartsmann, G. Natural products in anticancer therapy. Curr. Opin. Pharmacol. 2001, 1, 364–369. [Google Scholar] [CrossRef]
- Ibrahim, S.R.; Mohamed, G.A.; Shaala, L.A.; Moreno, L.; Banuls, Y.; Kiss, R.; Youssef, D.T. Proceraside A, a new cardiac glycoside from the root barks of Calotropis procera with in vitro anticancer effects. Nat. Prod. Res. 2014, 28, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Gurung, A.B.; Ali, M.A.; Bhattacharjee, A.; AbulFarah, M.; Al-Hemaid, F.; Abou-Tarboush, F.M.; Al-Anazi, K.M.; Al-Anazi, F.S.; Lee, J. Molecular docking of the anticancer bioactive compound proceraside with macromolecules involved in the cell cycle and DNA replication. Genet. Mol. Res. 2016, 15, 15027829. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi, F.; Fukao, Y.; Maruyama, T.; Obata, T.; Tanaka, M.; Sasaki, T.; Mikage, M.; Haque, M.E.; Tsuda, Y. Cytotoxic principles of a Bangladeshi crude drug, akond mul (roots of Calotropis gigantea L.). Chem. Pharm. Bull. (Tokyo) 1998, 46, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Seeka, C.; Sutthivaiyakit, S. Cytotoxic cardenolides from the leaves of Calotropis gigantea. Chem. Pharm. Bull. (Tokyo) 2010, 58, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Nakamura, T.; Kado, Y.; Yamaguchi, T.; Matsumura, H.; Ishikawa, K.; Inoue, T. Crystal structure of peroxiredoxin from Aeropyrum pernix K1 complexed with its substrate, hydrogen peroxide. J. Biochem. 2010, 147, 109–115. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends. Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [PubMed] [Green Version]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001, 52, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Kang, S.W.; Woo, H.A.; Hwang, S.C.; Chae, H.Z.; Kim, K.; Rhee, S.G. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J. Biol. Chem. 2002, 277, 38029–38036. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Jiang, H.; Matthews, C.P.; Colburn, N.H. Sulfiredoxin is an AP-1 target gene that is required for transformation and shows elevated expression in human skin malignancies. Proc. Natl. Acad. Sci. USA 2008, 105, 19738–19743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Lee, H.L.; Lee, K.B.; Park, J.H.; Chung, W.Y.; Lee, K.S.; Sheen, S.S.; Park, K.J.; Hwang, S.C. Nuclear factor E2-related factor 2 dependent overexpression of sulfiredoxin and peroxiredoxin III in human lung cancer. Korean J. Intern. Med. 2011, 26, 304–313. [Google Scholar] [CrossRef]
- Kim, J.; Lee, G.R.; Kim, H.; Jo, Y.J.; Hong, S.E.; Lee, J.; Lee, H.I.; Jang, Y.S.; Oh, S.H.; Lee, H.J.; et al. Effective killing of cancer cells and regression of tumor growth by K27 targeting sulfiredoxin. Free Radic. Biol. Med. 2016, 101, 384–392. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar]
- Loesch, M.; Chen, G. The p38 MAPK stress pathway as a tumor suppressor or more? Front. Biosci. 2008, 13, 3581–3593. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Leiter, U. Melanoma epidemiology and trends. Clin. Dermatol. 2009, 27, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Massaoka, M.H.; Matsuo, A.L.; Figueiredo, C.R.; Farias, C.F.; Girola, N.; Arruda, D.C.; Scutti, J.A.; Romoff, P.; Favero, O.A.; Ferreira, M.J.; et al. Jacaranone induces apoptosis in melanoma cells via ROS-mediated downregulation of Akt and p38 MAPK activation and displays antitumor activity in vivo. PLoS ONE 2012, 7, e38698. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Badana, A.K.; G, M.M.; G, S.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re308. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Nguyen, F.D.; Noory, M.A.; Sharma, A. Current State of Animal (Mouse) Modeling in Melanoma Research. Cancer Growth Metastasis 2015, 8, 81–94. [Google Scholar] [CrossRef]
- Perez-Guijarro, E.; Day, C.P.; Merlino, G.; Zaidi, M.R. Genetically engineered mouse models of melanoma. Cancer 2017, 123, 2089–2103. [Google Scholar] [CrossRef] [Green Version]
- Mishra, M.; Jiang, H.; Wu, L.; Chawsheen, H.A.; Wei, Q. The sulfiredoxin-peroxiredoxin (Srx-Prx) axis in cell signal transduction and cancer development. Cancer Lett. 2015, 366, 150–159. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kang, D. The Role of Peroxiredoxins in the Transduction of H2O2 Signals. Antioxid. Redox Signal. 2018, 28, 537–557. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, Y.; Cooper, J.A. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. J. Biol. Chem. 1998, 273, 17477–17482. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Elgamal, M.H.A.; Hanna, A.G.; Morsy, N.A.M.; Duddeck, H.; Simon, A.; Gáti, T.; Tóth, G. Complete 1H and 13C signal assignments of 5α-cardenolides isolated from Calotropis procera R. BR. J. Mol. Struct. 1999, 477, 201–208. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, I.-S.; Jeong, Y.J.; Kim, J.E.; Shin, J.; Jang, S.-W. Frugoside Induces Mitochondria-Mediated Apoptotic Cell Death through Inhibition of Sulfiredoxin Expression in Melanoma Cells. Cancers 2019, 11, 854. https://doi.org/10.3390/cancers11060854
Song I-S, Jeong YJ, Kim JE, Shin J, Jang S-W. Frugoside Induces Mitochondria-Mediated Apoptotic Cell Death through Inhibition of Sulfiredoxin Expression in Melanoma Cells. Cancers. 2019; 11(6):854. https://doi.org/10.3390/cancers11060854
Chicago/Turabian StyleSong, In-Sung, Yu Jeong Jeong, Ji Eun Kim, Jimin Shin, and Sung-Wuk Jang. 2019. "Frugoside Induces Mitochondria-Mediated Apoptotic Cell Death through Inhibition of Sulfiredoxin Expression in Melanoma Cells" Cancers 11, no. 6: 854. https://doi.org/10.3390/cancers11060854
APA StyleSong, I. -S., Jeong, Y. J., Kim, J. E., Shin, J., & Jang, S. -W. (2019). Frugoside Induces Mitochondria-Mediated Apoptotic Cell Death through Inhibition of Sulfiredoxin Expression in Melanoma Cells. Cancers, 11(6), 854. https://doi.org/10.3390/cancers11060854