Cancer Stem Cells and Radioresistance: DNA Repair and Beyond




Abstract
:1. Introduction
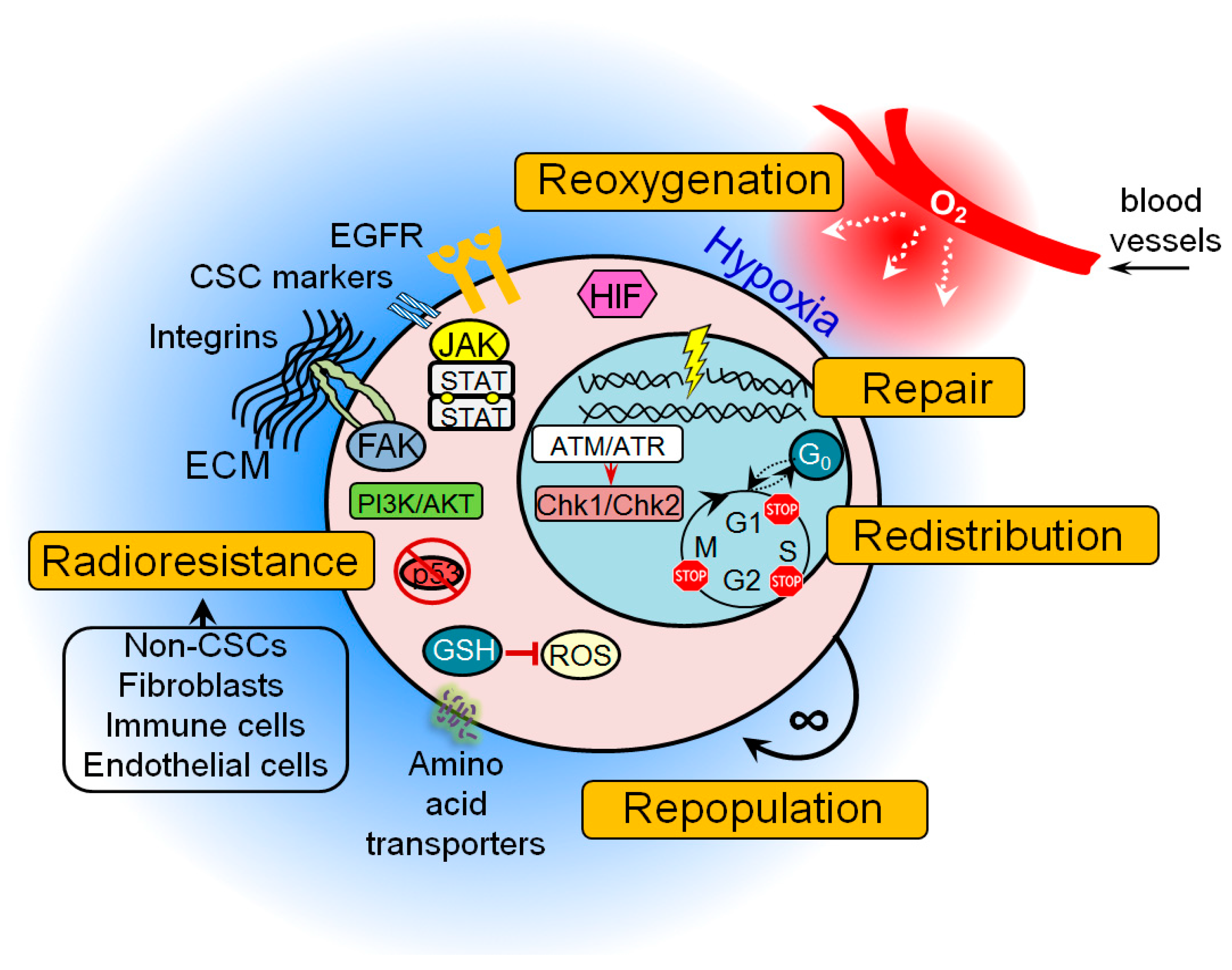
2. Cancer Stem Cells and 5Rs of Radiation Biology
3. Molecular Mechanisms of CSC Radioresistance
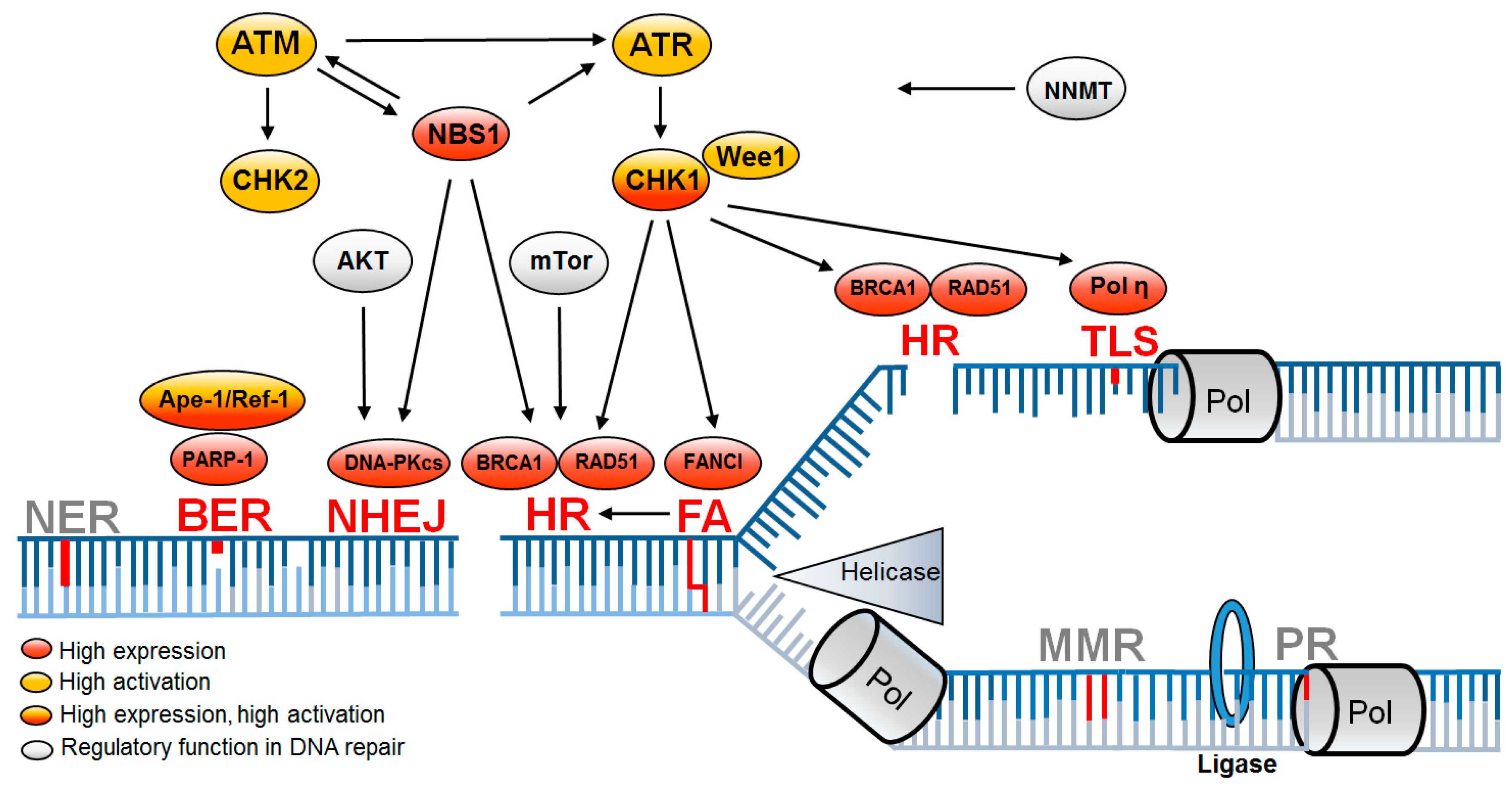
3.1. DNA Repair Factors and Pathways Upregulated in CSCs
3.2. Factors Indirectly Influencing DNA Repair Capacity of CSCs
4. CSC Heterogeneity and Plasticity
4.1. EMT and CSC Phenotype
4.2. CSC Induction by IR and ROS
4.3. Impact of Tumor Microenvironment (TME) on CSCs
4.4. The CSC Niche
4.5. Exosomes and microRNA
5. Targeting of DNA Repair Mechanisms in CSCs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. 2016, 7, 418–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global cancer transitions according to the Human Development Index (2008–2030): A population-based study. Lancet Oncol. 2012, 13, 790–801. [Google Scholar] [CrossRef]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Atun, R.; Jaffray, D.A.; Barton, M.B.; Bray, F.; Baumann, M.; Vikram, B.; Hanna, T.P.; Knaul, F.M.; Lievens, Y.; Lui, T.Y.; et al. Expanding global access to radiotherapy. Lancet Oncol. 2015, 16, 1153–1186. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M.; Overgaard, J.; Debus, J.; Bentzen, S.M.; Daartz, J.; Richter, C.; Zips, D.; Bortfeld, T. Radiation oncology in the era of precision medicine. Nat. Rev. Cancer 2016, 16, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Domina, E.A.; Philchenkov, A.; Dubrovska, A. Individual Response to Ionizing Radiation and Personalized Radiotherapy. Crit. Rev. Oncog. 2018, 23, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef]
- Butof, R.; Dubrovska, A.; Baumann, M. Clinical perspectives of cancer stem cell research in radiation oncology. Radiother. Oncol. 2013, 108, 388–396. [Google Scholar] [CrossRef]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Yaromina, A.; Eicheler, W.; Koch, U.; Baumann, M. Cancer stem cells: Targets and potential biomarkers for radiotherapy. Clin. Cancer Res. 2011, 17, 7224–7229. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Kurth, I.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother. Oncol. 2013, 108, 378–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.P.; Milas, L. The proportion of stem cells in murine tumors. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 513–518. [Google Scholar] [CrossRef]
- Baumann, M.; Dubois, W.; Suit, H.D. Response of human squamous cell carcinoma xenografts of different sizes to irradiation: Relationship of clonogenic cells, cellular radiation sensitivity in vivo, and tumor rescuing units. Radiat. Res. 1990, 123, 325–330. [Google Scholar] [CrossRef]
- Koch, U.; Krause, M.; Baumann, M. Cancer stem cells at the crossroads of current cancer therapy failures--radiation oncology perspective. Semin. Cancer Biol. 2010, 20, 116–124. [Google Scholar] [CrossRef]
- Dubben, H.H.; Thames, H.D.; Beck-Bornholdt, H.P. Tumor volume: A basic and specific response predictor in radiotherapy. Radiother. Oncol. 1998, 47, 167–174. [Google Scholar] [CrossRef]
- Linge, A.; Dubrovska, A.; Baumann, M.; Krause, M. The role of cancer stem cells in tumour radioresponse. In Strategies to Enhance the Therapeutic Ratio of Radiation as a Cancer Treatment; Anscher, M., Valerie, K., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 43–74. [Google Scholar]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef]
- Peitzsch, C.; Cojoc, M.; Hein, L.; Kurth, I.; Mabert, K.; Trautmann, F.; Klink, B.; Schrock, E.; Wirth, M.P.; Krause, M.; et al. An Epigenetic Reprogramming Strategy to Resensitize Radioresistant Prostate Cancer Cells. Cancer Res. 2016, 76, 2637–2651. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzouk, S. Translational genomics and head and neck cancer: Toward precision medicine. Clin. Genet. 2014, 86, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Steel, G.G.; McMillan, T.J.; Peacock, J.H. The 5Rs of radiobiology. Int. J. Radiat. Biol. 1989, 56, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Withers, H.R. The four R’s of radiotherapy. In Advances in Radiation Biology; Lett, J.T., Adler, H., Eds.; Academic Press: New York, NY, USA, 1975; Volume 5, pp. 241–271. [Google Scholar]
- Morgan, M.A.; Lawrence, T.S. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Hufnagl, A.; Herr, L.; Friedrich, T.; Durante, M.; Taucher-Scholz, G.; Scholz, M. The link between cell-cycle dependent radiosensitivity and repair pathways: A model based on the local, sister-chromatid conformation dependent switch between NHEJ and HR. Dna. Repair 2015, 27, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Mjelle, R.; Hegre, S.A.; Aas, P.A.; Slupphaug, G.; Drablos, F.; Saetrom, P.; Krokan, H.E. Cell cycle regulation of human DNA repair and chromatin remodeling genes. Dna. Repair 2015, 30, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 2012, 47, 320–329. [Google Scholar] [CrossRef]
- Borgmann, K.; Kocher, S.; Kriegs, M.; Mansour, W.Y.; Parplys, A.C.; Rieckmann, T.; Rothkamm, K. DNA Repair. In Molecular Radio-Oncology, Recent Results in Cancer Research; Springer: Berlin/Heidelberg, Germany, 2016; Volume 198, pp. 1–24. [Google Scholar]
- Moore, N.; Lyle, S. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J. Oncol. 2011, 2011, 396076. [Google Scholar] [CrossRef]
- Ambrosio, S.; Di Palo, G.; Napolitano, G.; Amente, S.; Dellino, G.I.; Faretta, M.; Pelicci, P.G.; Lania, L.; Majello, B. Cell cycle-dependent resolution of DNA double-strand breaks. Oncotarget 2016, 7, 4949–4960. [Google Scholar] [CrossRef]
- Chen, Y.; Li, D.; Wang, D.; Liu, X.; Yin, N.; Song, Y.; Lu, S.H.; Ju, Z.; Zhan, Q. Quiescence and attenuated DNA damage response promote survival of esophageal cancer stem cells. J. Cell. Biochem. 2012, 113, 3643–3652. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Deng, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Li, Y. Cancer stem cells and signaling pathways in radioresistance. Oncotarget 2016, 7, 11002–11017. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Hockel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef]
- Peitzsch, C.; Perrin, R.; Hill, R.P.; Dubrovska, A.; Kurth, I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int. J. Radiat. Biol. 2014, 90, 636–652. [Google Scholar] [CrossRef] [PubMed]
- Gallamini, A.; Zwarthoed, C.; Borra, A. Positron Emission Tomography (PET) in Oncology. Cancers 2014, 6, 1821–1889. [Google Scholar] [CrossRef] [Green Version]
- Abramyuk, A.; Appold, S.; Zophel, K.; Baumann, M.; Abolmaali, N. Modification of staging and treatment of head and neck cancer by FDG-PET/CT prior to radiotherapy. Strahlenther. Onkol. 2013, 189, 197–201. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Wang, P.; Lan, C.; Xiong, S.; Zhao, X.; Shan, Y.; Hu, R.; Wan, W.; Yu, S.; Liao, B.; Li, G.; et al. HIF1alpha regulates single differentiated glioma cell dedifferentiation to stem-like cell phenotypes with high tumorigenic potential under hypoxia. Oncotarget 2017, 8, 28074–28092. [Google Scholar]
- Luo, M.; Wicha, M.S. Targeting Cancer Stem Cell Redox Metabolism to Enhance Therapy Responses. Semin. Radiat. Oncol. 2019, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Liu, Y.; Lu, Y.; Liu, M.; Li, M.; Li, J.; Wu, L. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci. Rep. 2017, 7, 10592. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J. Bone Oncol. 2016, 5, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Han, Z.P.; Jing, Y.Y.; Yang, X.; Zhang, S.S.; Sun, K.; Hao, C.; Meng, Y.; Yu, F.H.; Liu, X.Q.; et al. Autophagy prevents irradiation injury and maintains stemness through decreasing ROS generation in mesenchymal stem cells. Cell Death Dis. 2013, 4, e844. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.C.; Ten Hoeve, J.J.; Grenman, R.; Wessels, L.F.; Kerkhoven, R.; Te Riele, H.; van den Brekel, M.W.; Verheij, M.; Begg, A.C. Pretreatment microRNA Expression Impacting on Epithelial-to-Mesenchymal Transition Predicts Intrinsic Radiosensitivity in Head and Neck Cancer Cell Lines and Patients. Clin. Cancer Res. 2015, 21, 5630–5638. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855. [Google Scholar] [CrossRef]
- Huang, T.; Wan, X.; Alvarez, A.A.; James, C.D.; Song, X.; Yang, Y.; Sastry, N.; Nakano, I.; Sulman, E.P.; Hu, B.; et al. MIR93 (microRNA-93) Regulates Tumorigenicity and Therapy Response of Glioblastoma by Targeting Autophagy. Autophagy 2019. [Google Scholar] [CrossRef]
- Chan, R.; Sethi, P.; Jyoti, A.; McGarry, R.; Upreti, M. Investigating the Radioresistant Properties of Lung Cancer Stem Cells in the Context of the Tumor Microenvironment. Radiat. Res. 2016, 185, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Butof, R.; Baumann, M. Time in radiation oncology—Keep it short! Radiother. Oncol. 2013, 106, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Blum, W.; Zhu, C.Q.; Yun, Z.; Pecze, L.; Kohno, M.; Chan, M.L.; Zhao, Y.; Felley-Bosco, E.; Schwaller, B.; et al. Putative cancer stem cells may be the key target to inhibit cancer cell repopulation between the intervals of chemoradiation in murine mesothelioma. BMC Cancer 2018, 18, 471. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Blanpain, C.; Rossi, D.J. DNA damage response in adult stem cells: Pathways and consequences. Nat. Rev. Mol. Cell Biol. 2011, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA Damage in Stem Cells. Mol. Cell 2017, 66, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Webb, B.; Gerson, S.L. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother. Oncol. 2014, 110, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Harding, A.; Kozlov, S.; Kijas, A.W.; Ensbey, K.S.; Walker, D.G.; Lavin, M.F. A role for homologous recombination and abnormal cell-cycle progression in radioresistance of glioma-initiating cells. Mol. Cancer Ther. 2012, 11, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Balbous, A.; Cortes, U.; Guilloteau, K.; Rivet, P.; Pinel, B.; Duchesne, M.; Godet, J.; Boissonnade, O.; Wager, M.; Bensadoun, R.J.; et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer 2016, 16, 604. [Google Scholar] [CrossRef] [PubMed]
- Mathews, L.A.; Cabarcas, S.M.; Hurt, E.M.; Zhang, X.; Jaffee, E.M.; Farrar, W.L. Increased expression of DNA repair genes in invasive human pancreatic cancer cells. Pancreas 2011, 40, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, H.; Liu, T.; Huang, M.; Butter, P.P.; Li, C.; Zhang, L.; Kao, G.D.; Gong, Y.; Maity, A.; et al. Temporal DNA-PK activation drives genomic instability and therapy resistance in glioma stem cells. JCI Insight 2018, 3, 98096. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Busheri, F.; Rasouli-Nia, A.; Mackey, J.R.; Weinfeld, M. Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast Cancer Res. 2010, 12, R31. [Google Scholar] [CrossRef] [PubMed]
- Gilabert, M.; Launay, S.; Ginestier, C.; Bertucci, F.; Audebert, S.; Pophillat, M.; Toiron, Y.; Baudelet, E.; Finetti, P.; Noguchi, T.; et al. Poly(ADP-ribose) polymerase 1 (PARP1) overexpression in human breast cancer stem cells and resistance to olaparib. PLoS ONE 2014, 9, e104302. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Han, C.; Zhao, R.; Cui, T.; Dai, Y.; Mao, C.; Zhao, W.; Zhang, X.; Yu, J.; Wang, Q.E. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4411–4416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, S.E.; De Witt Hamer, P.C.; Krawczyk, P.M.; Balaj, L.; Claes, A.; Niers, J.M.; Van Tilborg, A.A.; Zwinderman, A.H.; Geerts, D.; Kaspers, G.J.; et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell 2010, 18, 244–257. [Google Scholar] [CrossRef]
- Wu, J.; Mu, Q.; Thiviyanathan, V.; Annapragada, A.; Vigneswaran, N. Cancer stem cells are enriched in Fanconi anemia head and neck squamous cell carcinomas. Int. J. Oncol. 2014, 45, 2365–2372. [Google Scholar] [CrossRef]
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 4416–4428. [Google Scholar] [CrossRef] [Green Version]
- Gallmeier, E.; Hermann, P.C.; Mueller, M.T.; Machado, J.G.; Ziesch, A.; De Toni, E.N.; Palagyi, A.; Eisen, C.; Ellwart, J.W.; Rivera, J.; et al. Inhibition of ataxia telangiectasia- and Rad3-related function abrogates the in vitro and in vivo tumorigenicity of human colon cancer cells through depletion of the CD133(+) tumor-initiating cell fraction. Stem Cells 2011, 29, 418–429. [Google Scholar] [CrossRef]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, F.P.; Safwat, A.; Kassem, M.; Gautier, L.; Overgaard, J.; Horsman, M.R. Cancer stem cell overexpression of nicotinamide N-methyltransferase enhances cellular radiation resistance. Radiother. Oncol. 2011, 99, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E.; et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012, 19, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, Q.; Huang, Z.; Guryanova, O.A.; Huang, Q.; Shou, W.; Rich, J.N.; Bao, S. L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. EMBO J. 2011, 30, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef] [Green Version]
- Lundholm, L.; Haag, P.; Zong, D.; Juntti, T.; Mork, B.; Lewensohn, R.; Viktorsson, K. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013, 4, e478. [Google Scholar] [CrossRef]
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125–139. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; Chen, Z.; Medhurst, A.L.; Neal, J.A.; Bao, Z.; Mortusewicz, O.; McGouran, J.; Song, X.; Shen, H.; Hamdy, F.C.; et al. DNA-PKcs and PARP1 Bind to Unresected Stalled DNA Replication Forks Where They Recruit XRCC1 to Mediate Repair. Cancer Res. 2016, 76, 1078–1088. [Google Scholar] [CrossRef]
- McCord, A.M.; Jamal, M.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. CD133+ glioblastoma stem-like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin. Cancer Res. 2009, 15, 5145–5153. [Google Scholar] [CrossRef] [PubMed]
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; et al. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol. Cancer Res. 2009, 7, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchino, S.; Abdouh, M.; Chatoo, W.; Bernier, G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. 2010, 30, 10096–10111. [Google Scholar] [CrossRef] [PubMed]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Sacca, M.; Bartucci, M.; De Maria, R. DNA damage repair pathways in cancer stem cells. Mol. Cancer Ther. 2012, 11, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Ghisolfi, L.; Keates, A.C.; Hu, X.; Lee, D.K.; Li, C.J. Ionizing radiation induces stemness in cancer cells. PLoS ONE 2012, 7, e43628. [Google Scholar] [CrossRef]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bierie, B.; Li, A.G.; Pathania, S.; Toomire, K.; Dimitrov, S.D.; Liu, B.; Gelman, R.; Giobbie-Hurder, A.; Feunteun, J.; et al. BRCA1/FANCD2/BRG1-Driven DNA Repair Stabilizes the Differentiation State of Human Mammary Epithelial Cells. Mol. Cell 2016, 63, 277–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Jorg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 2016, 95, S20–S25. [Google Scholar] [CrossRef] [PubMed]
- Marie-Egyptienne, D.T.; Lohse, I.; Hill, R.P. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett. 2013, 341, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.P.; Marie-Egyptienne, D.T.; Hedley, D.W. Cancer stem cells, hypoxia and metastasis. Semin. Radiat. Oncol. 2009, 19, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Moncharmont, C.; Levy, A.; Guy, J.B.; Falk, A.T.; Guilbert, M.; Trone, J.C.; Alphonse, G.; Gilormini, M.; Ardail, D.; Toillon, R.A.; et al. Radiation-enhanced cell migration/invasion process: A review. Crit. Rev. Oncol. Hematol. 2014, 92, 133–142. [Google Scholar] [CrossRef]
- Wang, S.S.; Jiang, J.; Liang, X.H.; Tang, Y.L. Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets Ther. 2015, 8, 2973–2980. [Google Scholar]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Ansieau, S.; Collin, G.; Hill, L. EMT or EMT-Promoting Transcription Factors, Where to Focus the Light? Front. Oncol. 2014, 4, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Mahabir, R.; Tanino, M.; Elmansuri, A.; Wang, L.; Kimura, T.; Itoh, T.; Ohba, Y.; Nishihara, H.; Shirato, H.; Tsuda, M.; et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014, 16, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Yang, M.H.; Tsai, M.L.; Lan, H.Y.; Su, S.H.; Chang, S.C.; Teng, H.W.; Yang, S.H.; Lan, Y.T.; Chiou, S.H.; et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 2011, 141, 279–291.e5. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Gouaze-Andersson, V.; Gherardi, M.J.; Lemarie, A.; Gilhodes, J.; Lubrano, V.; Arnauduc, F.; Cohen-Jonathan Moyal, E.; Toulas, C. FGFR1/FOXM1 pathway: A key regulator of glioblastoma stem cells radioresistance and a prognosis biomarker. Oncotarget 2018, 9, 31637–31649. [Google Scholar] [CrossRef]
- Konge, J.; Leteurtre, F.; Goislard, M.; Biard, D.; Morel-Altmeyer, S.; Vaurijoux, A.; Gruel, G.; Chevillard, S.; Lebeau, J. Breast cancer stem cell-like cells generated during TGFbeta-induced EMT are radioresistant. Oncotarget 2018, 9, 23519–23531. [Google Scholar]
- Martin, M.; Vozenin, M.C.; Gault, N.; Crechet, F.; Pfarr, C.M.; Lefaix, J.L. Coactivation of AP-1 activity and TGF-beta1 gene expression in the stress response of normal skin cells to ionizing radiation. Oncogene 1997, 15, 981–989. [Google Scholar] [CrossRef]
- Jobling, M.F.; Mott, J.D.; Finnegan, M.T.; Jurukovski, V.; Erickson, A.C.; Walian, P.J.; Taylor, S.E.; Ledbetter, S.; Lawrence, C.M.; Rifkin, D.B.; et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 2006, 166, 839–848. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, J.G.; Kim, N.D.; Yang, K.; Shim, J.W.; Heo, K. Estradiol, TGF-beta1 and hypoxia promote breast cancer stemness and EMT-mediated breast cancer migration. Oncol. Lett. 2016, 11, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, G.; Tirino, V.; Camerlingo, R.; Franco, R.; La Rocca, A.; Liguori, E.; Martucci, N.; Paino, F.; Normanno, N.; Rocco, G. Epithelial to mesenchymal transition by TGFbeta-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS ONE 2011, 6, e21548. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yi, J.; Tao, L.; Huang, G.; Chu, X.; Song, H.; Chen, L. Wnt signaling induces radioresistance through upregulating HMGB1 in esophageal squamous cell carcinoma. Cell Death Dis. 2018, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Lapouge, G.; Rorive, S.; Drogat, B.; Desaedelaere, K.; Delafaille, S.; Dubois, C.; Salmon, I.; Willekens, K.; Marine, J.C.; et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 2015, 16, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Celia-Terrassa, T.; Meca-Cortes, O.; Mateo, F.; Martinez de Paz, A.; Rubio, N.; Arnal-Estape, A.; Ell, B.J.; Bermudo, R.; Diaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schafer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambach, D.M.; Sodi, V.L.; Lelkes, P.I.; Azizkhan-Clifford, J.; Reginato, M.J. ErbB2, FoxM1 and 14-3-3zeta prime breast cancer cells for invasion in response to ionizing radiation. Oncogene 2014, 33, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, D.; Melo, T.; Deng, Z.; Almeida, C.; Zhao, W. ERK/GSK3beta/Snail signaling mediates radiation-induced alveolar epithelial-to-mesenchymal transition. Free Radic. Biol. Med. 2012, 52, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Kam, W.W.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015, 2015, 750798. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, I.G.; Lee, S.H.; Kwak, M.K. Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance. Oxid. Med. Cell. Longev. 2016, 2016, 2428153. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A.; Stehr, H.; Newman, A.M.; Gentles, A.J.; Kong, W.; Truong, D.; Martin, S.; Chaudhuri, A.; et al. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; He, Y.; Ji, J.; Yao, Y.; Shen, W.; Luo, J.; Zhu, W.; Cao, H.; Geng, Y.; Xu, J.; et al. Hypoxia-inducible factor 1alpha (HIF-1alpha) and reactive oxygen species (ROS) mediates radiation-induced invasiveness through the SDF-1alpha/CXCR4 pathway in non-small cell lung carcinoma cells. Oncotarget 2015, 6, 10893–10907. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.E.; Nor, J.E. Perivascular stem cell niche in head and neck cancer. Cancer Lett. 2013, 338, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Liang, D.; Liu, J.; Axcrona, K.; Kvalheim, G.; Stokke, T.; Nesland, J.M.; Suo, Z. Prostate cancer cell lines under hypoxia exhibit greater stem-like properties. PLoS ONE 2011, 6, e29170. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra373. [Google Scholar] [CrossRef] [PubMed]
- Chiblak, S.; Tang, Z.; Lemke, D.; Knoll, M.; Dokic, I.; Warta, R.; Moustafa, M.; Mier, W.; Brons, S.; Rapp, C.; et al. Carbon irradiation overcomes glioma radioresistance by eradicating stem cells and forming an antiangiogenic and immunopermissive niche. JCI Insight 2019, 4, 123837. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.C.; Chaudhary, V.; Bartscherer, K.; Boutros, M. Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 2012, 14, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Tooi, M.; Komaki, M.; Morioka, C.; Honda, I.; Iwasaki, K.; Yokoyama, N.; Ayame, H.; Izumi, Y.; Morita, I. Placenta Mesenchymal Stem Cell Derived Exosomes Confer Plasticity on Fibroblasts. J. Cell. Biochem. 2016, 117, 1658–1670. [Google Scholar] [CrossRef] [PubMed]
- de Araujo Farias, V.; O’Valle, F.; Serrano-Saenz, S.; Anderson, P.; Andres, E.; Lopez-Penalver, J.; Tovar, I.; Nieto, A.; Santos, A.; Martin, F.; et al. Exosomes derived from mesenchymal stem cells enhance radiotherapy-induced cell death in tumor and metastatic tumor foci. Mol. Cancer 2018, 17, 122. [Google Scholar] [CrossRef]
- Mutschelknaus, L.; Azimzadeh, O.; Heider, T.; Winkler, K.; Vetter, M.; Kell, R.; Tapio, S.; Merl-Pham, J.; Huber, S.M.; Edalat, L.; et al. Radiation alters the cargo of exosomes released from squamous head and neck cancer cells to promote migration of recipient cells. Sci. Rep. 2017, 7, 12423. [Google Scholar] [CrossRef]
- Sanchez, C.A.; Andahur, E.I.; Valenzuela, R.; Castellon, E.A.; Fulla, J.A.; Ramos, C.G.; Trivino, J.C. Exosomes from bulk and stem cells from human prostate cancer have a differential microRNA content that contributes cooperatively over local and pre-metastatic niche. Oncotarget 2016, 7, 3993–4008. [Google Scholar] [CrossRef]
- Bell, E.H.; Kirste, S.; Fleming, J.L.; Stegmaier, P.; Drendel, V.; Mo, X.; Ling, S.; Fabian, D.; Manring, I.; Jilg, C.A.; et al. A novel miRNA-based predictive model for biochemical failure following post-prostatectomy salvage radiation therapy. PLoS ONE 2015, 10, e0118745. [Google Scholar] [CrossRef] [PubMed]
- Hatano, K.; Kumar, B.; Zhang, Y.; Coulter, J.B.; Hedayati, M.; Mears, B.; Ni, X.; Kudrolli, T.A.; Chowdhury, W.H.; Rodriguez, R.; et al. A functional screen identifies miRNAs that inhibit DNA repair and sensitize prostate cancer cells to ionizing radiation. Nucleic Acids Res. 2015, 43, 4075–4086. [Google Scholar] [CrossRef]
- Gong, P.; Zhang, T.; He, D.; Hsieh, J.T. MicroRNA-145 Modulates Tumor Sensitivity to Radiation in Prostate Cancer. Radiat. Res. 2015, 184, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.P.; Chien, Y.; Chiou, G.Y.; Cherng, J.Y.; Wang, M.L.; Lo, W.L.; Chang, Y.L.; Huang, P.I.; Chen, Y.W.; Shih, Y.H.; et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials 2012, 33, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Fuse, M.; Nohata, N.; Kojima, S.; Sakamoto, S.; Chiyomaru, T.; Kawakami, K.; Enokida, H.; Nakagawa, M.; Naya, Y.; Ichikawa, T.; et al. Restoration of miR-145 expression suppresses cell proliferation, migration and invasion in prostate cancer by targeting FSCN1. Int. J. Oncol. 2011, 38, 1093–1101. [Google Scholar] [PubMed]
- Josson, S.; Sung, S.Y.; Lao, K.; Chung, L.W.; Johnstone, P.A. Radiation modulation of microRNA in prostate cancer cell lines. Prostate 2008, 68, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Venere, M.; Hamerlik, P.; Wu, Q.; Rasmussen, R.D.; Song, L.A.; Vasanji, A.; Tenley, N.; Flavahan, W.A.; Hjelmeland, A.B.; Bartek, J.; et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 258–269. [Google Scholar] [CrossRef]
- Al-Ejeh, F.; Pajic, M.; Shi, W.; Kalimutho, M.; Miranda, M.; Nagrial, A.M.; Chou, A.; Biankin, A.V.; Grimmond, S.M.; Australian Pancreatic Cancer Genome, I.; et al. Gemcitabine and CHK1 inhibition potentiate EGFR-directed radioimmunotherapy against pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2014, 20, 3187–3197. [Google Scholar] [CrossRef]
- Signore, M.; Buccarelli, M.; Pilozzi, E.; De Luca, G.; Cappellari, M.; Fanciulli, M.; Goeman, F.; Melucci, E.; Biffoni, M.; Ricci-Vitiani, L. UCN-01 enhances cytotoxicity of irinotecan in colorectal cancer stem-like cells by impairing DNA damage response. Oncotarget 2016, 7, 44113–44128. [Google Scholar] [CrossRef] [Green Version]
- Tachon, G.; Cortes, U.; Guichet, P.O.; Rivet, P.; Balbous, A.; Masliantsev, K.; Berger, A.; Boissonnade, O.; Wager, M.; Karayan-Tapon, L. Cell Cycle Changes after Glioblastoma Stem Cell Irradiation: The Major Role of RAD51. Int. J. Mol. Sci. 2018, 19, 3018. [Google Scholar] [CrossRef]
- Meng, E.; Mitra, A.; Tripathi, K.; Finan, M.A.; Scalici, J.; McClellan, S.; Madeira da Silva, L.; Reed, E.; Shevde, L.A.; Palle, K.; et al. ALDH1A1 maintains ovarian cancer stem cell-like properties by altered regulation of cell cycle checkpoint and DNA repair network signaling. PLoS ONE 2014, 9, e107142. [Google Scholar] [CrossRef]
- Kahn, J.; Hayman, T.J.; Jamal, M.; Rath, B.H.; Kramp, T.; Camphausen, K.; Tofilon, P.J. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro Oncol. 2014, 16, 29–37. [Google Scholar] [CrossRef]
- Zhang, M.; Atkinson, R.L.; Rosen, J.M. Selective targeting of radiation-resistant tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3522–3527. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhang, Y.; Chen, S.; Kmieciak, M.; Leng, Y.; Lin, H.; Rizzo, K.A.; Dumur, C.I.; Ferreira-Gonzalez, A.; Dai, Y.; et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia 2015, 29, 807–818. [Google Scholar] [CrossRef]
- Fulton, B.; Short, S.C.; James, A.; Nowicki, S.; McBain, C.; Jefferies, S.; Kelly, C.; Stobo, J.; Morris, A.; Williamson, A.; et al. PARADIGM-2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin. Transl. Radiat. Oncol. 2018, 8, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Lesueur, P.; Chevalier, F.; El-Habr, E.A.; Junier, M.P.; Chneiweiss, H.; Castera, L.; Muller, E.; Stefan, D.; Saintigny, Y. Radiosensitization Effect of Talazoparib, a Parp Inhibitor, on Glioblastoma Stem Cells Exposed to Low and High Linear Energy Transfer Radiation. Sci. Rep. 2018, 8, 3664. [Google Scholar] [CrossRef]
- Chang, C.W.; Chen, Y.S.; Chou, S.H.; Han, C.L.; Chen, Y.J.; Yang, C.C.; Huang, C.Y.; Lo, J.F. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 2014, 74, 6291–6305. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Resistance Mechanism | Therapeutic Approach | Tumor | Citation |
|---|---|---|---|---|
| IR | ATM, ATR, CHK1, PARP-1 upregulation | ATR i + PARP1 i | Glioblastoma | [82] |
| IR | Replication stress + DDR activation | ATR i + PARP1 i | Glioblastoma | [84] |
| IR | SSB repair | PARP1 i | Glioblastoma | [160] |
| IR | DNA-PK activation | DNA-PK i | Glioblastoma | [76] |
| IR | DNA-PKCS activation | DNA-PKCS i | Glioblastoma | [99] |
| IR | DDR activation | ATM i | Glioblastoma | [87] |
| IR | DDR activation | CHK1 i | Glioblastoma | [69] |
| IR | ZEB1-mediated CHK1 stabilization | ZEB1 depletion by siRNA | Breast | [104] |
| IR | RAD51 overexpression | RAD51 i | NSCLC | [163] |
| IR | WEE1 overexpression | WEE1 i | Glioblastoma | [80] |
| IR | Akt signalling | Akt i | Breast | [166] |
| IR | mTor signalling | mTorC1/2 inhibition | Glioblastoma | [165] |
| Cisplatin/paclitaxel | CHK1 activation | CHK1 i | NSCLC | [86] |
| Radionuclide antibody | CHK1 activation | CHK1 i | Pancreatic | [161] |
| Irinotecan | CHK1 activation | CHK1 i | Colon | [162] |
| ICL | ATR/CHK1 activation | ATR depletion by siRNA | Colon | [83] |
| Olaparib | RAD51 overexpression | RAD51 depletion by shRNA | Breast | [73] |
| Cisplatin | TLS activity | Pol η depletion by siRNA | Ovary | [79] |
| Cisplatin | Overexpression of ROS scavengers | ROS scavenger i by 2-ME, 3AT | HNSCC | [170] |
| Vorinostat | DDR activation and G2 Checkpoint | HDACsi + WEE1 i | Leukemia | [167] |
| Carboplatin | ALDH1 overexpression | ALDH1 i depletion by siRNA | Ovary | [164] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers 2019, 11, 862. https://doi.org/10.3390/cancers11060862
Schulz A, Meyer F, Dubrovska A, Borgmann K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers. 2019; 11(6):862. https://doi.org/10.3390/cancers11060862
Chicago/Turabian StyleSchulz, Alexander, Felix Meyer, Anna Dubrovska, and Kerstin Borgmann. 2019. "Cancer Stem Cells and Radioresistance: DNA Repair and Beyond" Cancers 11, no. 6: 862. https://doi.org/10.3390/cancers11060862
APA StyleSchulz, A., Meyer, F., Dubrovska, A., & Borgmann, K. (2019). Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers, 11(6), 862. https://doi.org/10.3390/cancers11060862