Synthesis and Enhanced Cellular Uptake In Vitro of Anti-HER2 Multifunctional Gold Nanoparticles


Abstract
:1. Introduction
2. Results
2.1. Nanoparticle Design
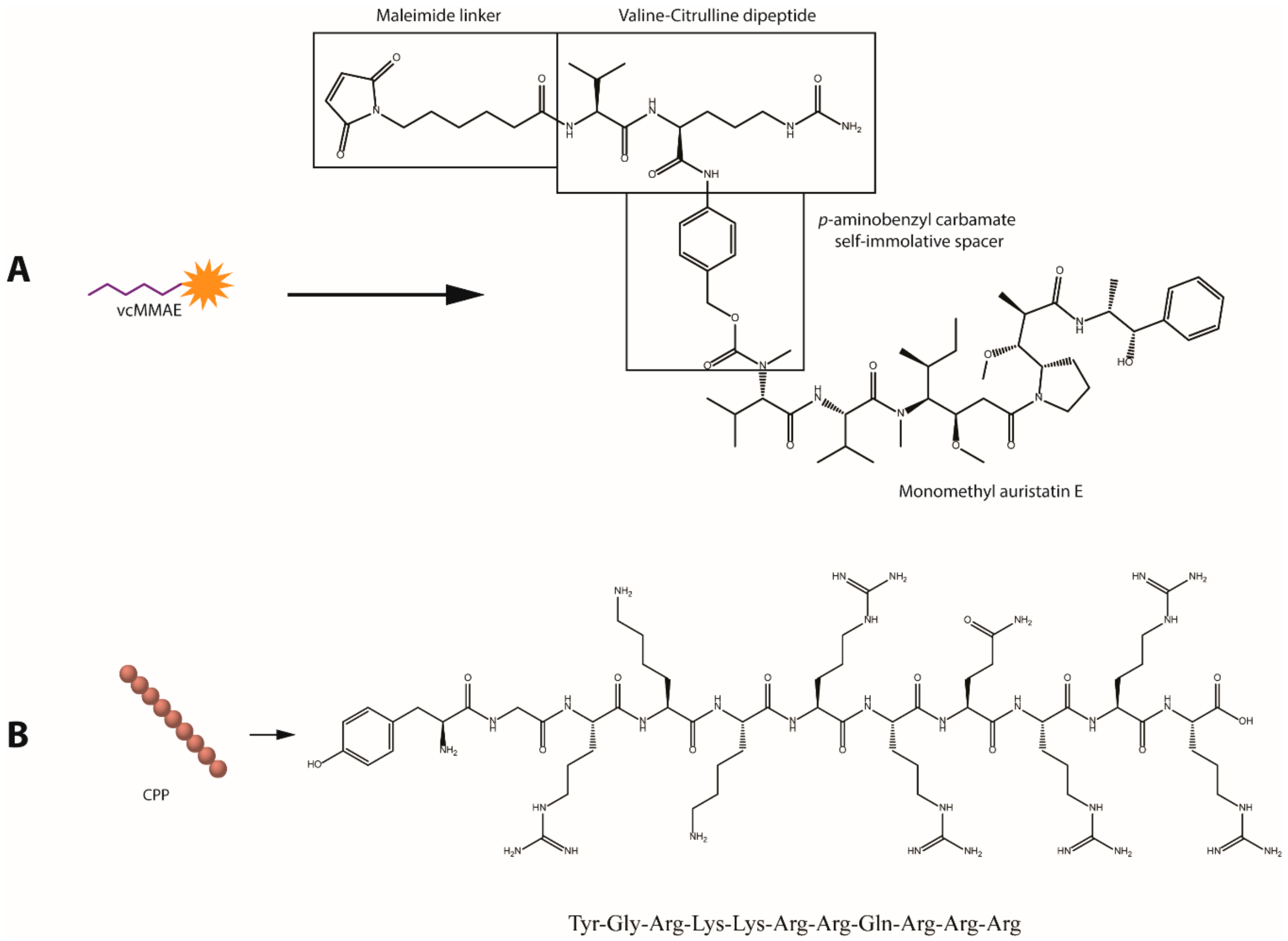
2.2. Antibody-Drug Conjugate (Tmab-vcMMAE) Synthesis
2.3. Antibody and CPP PEGylation
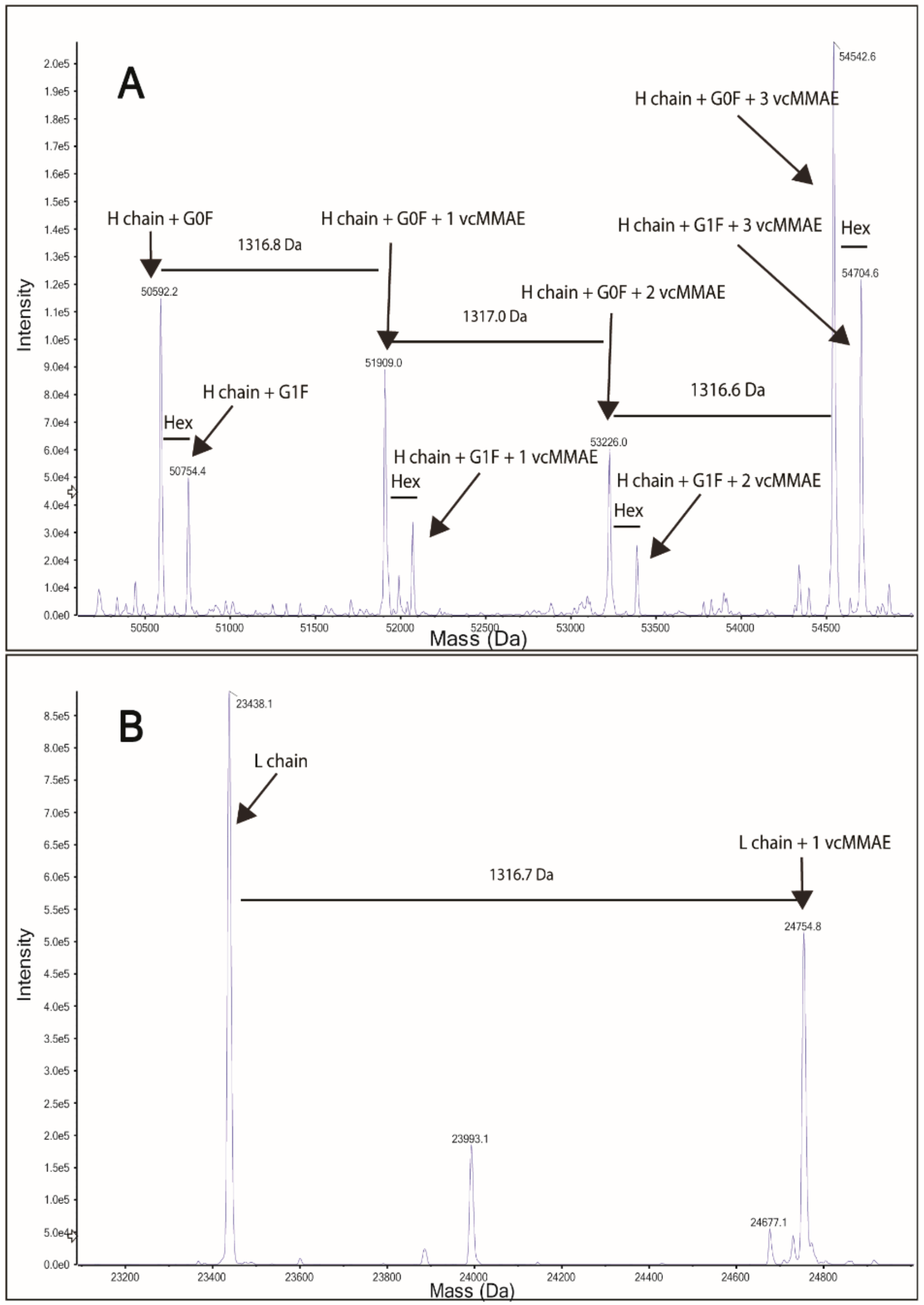
2.3.1. Structural Characterization
2.3.2. Binding Kinetics of Functionalized Trastuzumab
2.4. Gold Nanoparticle Surface Functionalization
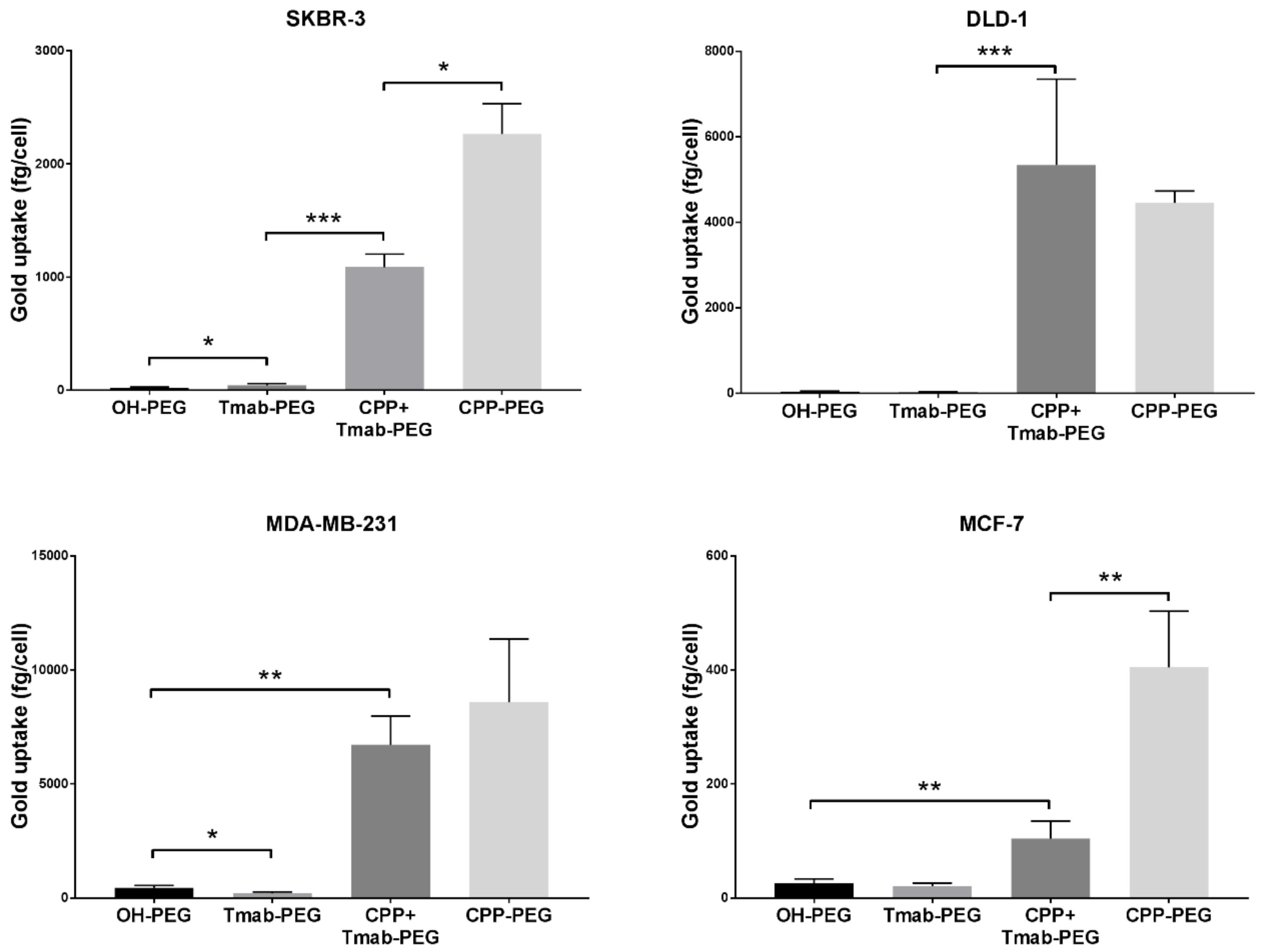
2.5. Cellular Uptake in Various Breast Cancer Cell Lines
2.5.1. Active Targeting in HER2-Positive SBKR-3 Cells
2.5.2. CPP-Driven Enhanced Internalization
2.6. In Vitro Cytotoxicity of ADC-PEG-AuNP in HER2 Overexpressing Cancer Cell Lines
3. Discussion
3.1. Trastuzumab and HIV-TAT PEGylation
3.2. ADC Construction
3.3. Gold Nanoparticle Surface Functionalization
3.4. Active Targeting and Cellular Uptake
3.5. Cellular Uptake Enhancement with HIV-TAT
3.6. In Vitro Cytotoxicity of ADC-PEG-AuNP in HER2 Overexpressing Cancer Cell Lines
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Spherical Citrate-Capped Gold Nanoparticles
4.3. Tmab PEGylation (Tmab-PEG-SH)
4.4. HIV-TAT Cell Penetrating Peptide (CPP) PEGylation (CPP-PEG-SH)
4.5. Tmab-vcMMAE Conjugate Synthesis
4.5.1. Antibody Partial Reduction
4.5.2. Conjugate Synthesis
4.5.3. Intact Mass Analysis
4.6. Binding Kinetics to Recombinant HER2 through Surface Plasmon Resonance
4.7. Gold Nanoparticle Surface Functionalization
4.8. UV-Vis Spectroscopy
4.9. Size-Exclusion High-Performance Liquid Chromatography (SE-HPLC)
4.10. DLS and Zeta Potential Measurements
4.11. Cellular Uptake Quantification through Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
4.12. Cellular Uptake Evaluation by Transmission Electron Microscopy (TEM)
4.13. Cell Cytotoxicity Evaluation
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure-an obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Netti, P.A.; Baxter, L.T.; Boucher, Y.; Skalak, R.; Jain, R.K. Time-dependent behavior of interstitial fluid pressure in solid tumors: Implications for drug delivery. Cancer Res. 1995, 55, 5451–5458. [Google Scholar] [PubMed]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Tran, S.; DeGiovanni, P.-J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef]
- Jin, S.; Leach, J.C.; Ye, K. Nanoparticle-mediated gene delivery. Methods Mol. Biol. 2009, 544, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Manthe, R.L.; Foy, S.P.; Krishnamurthy, N.; Sharma, B.; Labhasetwar, V. Tumor ablation and nanotechnology. Mol. Pharm. 2010, 7, 1880–1898. [Google Scholar] [CrossRef]
- Will, O.; Purkayastha, S.; Chan, C.; Athanasiou, T.; Darzi, A.W.; Gedroyc, W.; Tekkis, P.P. Diagnostic precision of nanoparticle-enhanced MRI for lymph-node metastases: A meta-analysis. Lancet Oncol. 2006, 7, 52–60. [Google Scholar] [CrossRef]
- Lukianova-Hleb, E.Y.; Ren, X.; Sawant, R.R.; Wu, X.; Torchilin, V.P.; Lapotko, D.O. On-demand intracellular amplification of chemoradiation with cancer-specific plasmonic nanobubbles. Nat. Med. 2014, 20, 778. [Google Scholar] [CrossRef]
- Kyriazi, M.-E.; Giust, D.; El-Sagheer, A.H.; Lackie, P.M.; Muskens, O.L.; Brown, T.; Kanaras, A.G. Multiplexed mRNA Sensing and Combinatorial-Targeted Drug Delivery Using DNA-Gold Nanoparticle Dimers. ACS Nano 2018, 12, 3333–3340. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Li, X.; Li, H.; Zhang, W. Quantifying thiol–gold interactions towards the efficient strength control. Nat. Commu. 2014, 5, 4348. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Alkilany, A.M.; Murphy, C.J. Toxicity and cellular uptake of gold nanoparticles: What we have learned so far? J. Nanopart. Res. Interdiscip. Forum Nanoscale Sci. Technol. 2010, 12, 2313–2333. [Google Scholar] [CrossRef] [PubMed]
- Goel, R.; Shah, N.; Visaria, R.; Paciotti, G.F.; Bischof, J.C. Biodistribution of TNF-alpha-coated gold nanoparticles in an in vivo model system. Nanomedicine 2009, 4, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commu. 2018, 9, 1410. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Fléjou, J.F.; Paraf, F.; Muzeau, F.; Fékété, F.; Hénin, D.; Jothy, S.; Potet, F. Expression of c-erbB-2 oncogene product in Barrett’s adenocarcinoma: Pathological and prognostic correlations. J. Clin. Pathol. 1994, 47, 23. [Google Scholar] [CrossRef]
- Berchuck, A.; Kamel, A.; Whitaker, R.; Kerns, B.; Olt, G.; Kinney, R.; Soper, J.T.; Dodge, R.; Clarke-Pearson, D.L.; Marks, P.; et al. Overexpression of HER-2/neu Is Associated with Poor Survival in Advanced Epithelial Ovarian Cancer. Cancer Res. 1990, 50, 4087. [Google Scholar]
- Rolitsky, C.D.; Theil, K.S.; McGaughy, V.R.; Copeland, L.J.; Niemann, T.H. HER-2/neu amplification and overexpression in endometrial carcinoma. Int. J. Gynecol. Pathol. 1999, 18, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Sawada, H.; Yamada, Y.; Watanabe, A.; Tatsumi, M.; Yamashita, J.; Matsuda, M.; Sakaguchi, T.; Hirao, T.; Nakano, H. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer 1999, 85, 1894–1902. [Google Scholar] [CrossRef]
- Santin, A.D.; Bellone, S.; Van Stedum, S.; Bushen, W.; Palmieri, M.; Siegel, E.R.; De Las Casas, L.E.; Roman, J.J.; Burnett, A.; Pecorelli, S. Amplification of c-erbB2 oncogene. Cancer 2005, 104, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, Y.; Ninomiya, I.; Yamaguchi, A.; Fushida, S.; Kimura, H.; Ohoyama, S.; Miyazaki, I.; Endou, Y.; Tanaka, M.; Sasaki, T. Evaluation of Immunoreactivity for erbB-2 Protein as a Marker of Poor Short Term Prognosis in Gastric Cancer. Cancer Res. 1991, 51, 1034. [Google Scholar] [PubMed]
- Hudis, C.A. Trastuzumab--mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, Y.J.; Zheng, H.; Zhong, X.R.; Wang, Y.; Wang, Z.; Wang, Y.G.; Wang, Y.P. Membrane-bound complement regulatory proteins are prognostic factors of operable breast cancer treated with adjuvant trastuzumab: A retrospective study. Oncol. Rep. 2014, 32, 2619–2627. [Google Scholar] [CrossRef] [Green Version]
- Elgundi, Z.; Reslan, M.; Cruz, E.; Sifniotis, V.; Kayser, V. The state-of-play and future of antibody therapeutics. Adv. Drug Deliv. Rev. 2017, 122, 2–19. [Google Scholar] [CrossRef]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.M.; Tannock, I.F. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer 2010, 10, 255. [Google Scholar] [CrossRef]
- Hermanson, G.T. Chapter 18-PEGylation and Synthetic Polymer Modification. In Bioconjugate Techniques, 3rd ed.; Hermanson, G.T., Ed.; Academic Press: Boston, MA, USA, 2013; 2p. [Google Scholar]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef]
- Haiss, W.; Thanh, N.T.K.; Aveyard, J.; Fernig, D.G. Determination of Size and Concentration of Gold Nanoparticles from UV−Vis Spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Libutti, S.K.; Paciotti, G.F.; Byrnes, A.A.; Alexander, H.R., Jr.; Gannon, W.E.; Walker, M.; Seidel, G.D.; Yuldasheva, N.; Tamarkin, L. Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin. Cancer Res. 2010, 16, 6139–6149. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef]
- Adem, Y.T.; Schwarz, K.A.; Duenas, E.; Patapoff, T.W.; Galush, W.J.; Esue, O. Auristatin Antibody Drug Conjugate Physical Instability and the Role of Drug Payload. Bioconj. Chem. 2014, 25, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Kim, B.Y.S.; Rutka, J.T.; Chan, W.C.W. Nanoparticle-mediated cellular response is size-dependent. Nat. Nanotechnol. 2008, 3, 145. [Google Scholar] [CrossRef]
- Cho, E.C.; Xie, J.; Wurm, P.A.; Xia, Y. Understanding the Role of Surface Charges in Cellular Adsorption versus Internalization by Selectively Removing Gold Nanoparticles on the Cell Surface with a I2/KI Etchant. Nano Lett. 2009, 9, 1080–1084. [Google Scholar] [CrossRef]
- Ma, X.; Wu, Y.; Jin, S.; Tian, Y.; Zhang, X.; Zhao, Y.; Yu, L.; Liang, X.-J. Gold Nanoparticles Induce Autophagosome Accumulation through Size-Dependent Nanoparticle Uptake and Lysosome Impairment. ACS Nano 2011, 5, 8629–8639. [Google Scholar] [CrossRef]
- Tkachenko, A.G.; Xie, H.; Liu, Y.; Coleman, D.; Ryan, J.; Glomm, W.R.; Shipton, M.K.; Franzen, S.; Feldheim, D.L. Cellular Trajectories of Peptide-Modified Gold Particle Complexes: Comparison of Nuclear Localization Signals and Peptide Transduction Domains. Bioconj. Chem. 2004, 15, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Baulida, J.; Kraus, M.H.; Alimandi, M.; Di Fiore, P.P.; Carpenter, G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J. Biol. Chem. 1996, 271, 5251–5257. [Google Scholar] [PubMed]
- Kim, B.; Han, G.; Toley, B.J.; Kim, C.-k.; Rotello, V.M.; Forbes, N.S. Tuning payload delivery in tumour cylindroids using gold nanoparticles. Nat. Nanotechnol. 2010, 5, 465. [Google Scholar] [CrossRef] [PubMed]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. The Formation of Colloidal Gold. J. Phys. Chem. 1953, 57, 670–673. [Google Scholar] [CrossRef]
- Frens, G. Controlled Nucleation for the Regulation of the Particle Size in Monodisperse Gold Suspensions. Nat. Phys. Sci. 1973, 241, 20. [Google Scholar] [CrossRef]
- Kelly, S.T.; Zydney, A.L. Effects of intermolecular thiol–disulfide interchange reactions on bsa fouling during microfiltration. Biotechnol. Bioeng. 1994, 44, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.V.; Laurence, J.S.; Siahaan, T.J. The role of thiols and disulfides on protein stability. Curr. Protein Pept. Sci. 2009, 10, 614–625. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trastuzumab Variant | Ka (× 106) M−1·s−1 | Kd (× 105) s−1 | KD (pM) |
|---|---|---|---|
| Tmab | 3.24 ± 0.15 | 1.98 ± 0.50 | 6.07 ± 1.27 |
| Tmab-PEG-SH 2× | 3.53 ± 0.15 | 2.47 ± 0.24 | 6.83 ± 0.68 |
| Tmab-PEG-SH 5× | 2.86 ± 0.03 | 1.97 ± 0.20 | 6.91 ± 0.78 |
| Tmab-PEG-SH 10× | 2.87 ± 0.11 | 1.98 ± 0.14 | 6.89 ± 0.21 |
| Tmab-PEG-SH 25× | 2.05 ± 0.03 | 1.12 ± 0.11 | 5.46 ± 0.44 |
| ADC | 2.25 ± 0.01 | 1.58 ± 0.09 | 7.05 ± 0.41 |
| ADC-PEG-SH | 7.45 ± 0.07 | 61.80 ± 0.05 | 85.01 ± 10.92 |
| NP | Z-ave (nm) | PDI | ζ (mV) | λ max (nm) | TEM (nm) |
|---|---|---|---|---|---|
| Cit-AuNP | 60.62 ± 0.19 | 0.29 | −34.60 ± 0.91 | 530.5 | 48.29 ± 5.58 |
| OH-PEG-AuNP | 86.61 ± 0.12 | 0.17 | −14.37 ± 0.12 | 532.4 | |
| Tmab-PEG-AuNP | 87.35 ± 0.41 | 0.17 | −1.10 ± 0.46 | 534.2 | |
| CPP+Tmab-PEG-AuNP | 83.42 ± 2.14 | 0.20 | 1.5 ± 0.46 | 533.8 | |
| CPP-PEG-AuNP | 81.22 ± 0.39 | 0.17 | 6.17 ± 0.71 | 533.2 | |
| ADC-PEG-AuNP | 85.45 ± 1.34 | 0.19 | −2.3 ± 0.37 | 534.1 |
| Sample | SKBR-3 | SKOV-3 | ||||
|---|---|---|---|---|---|---|
| Agent | GR50 (nM) | GR50 95% CI | R2 | SKOV-3 | GR50 95% CI | R2 |
| Free MMAE | 0.33 | (0.28–0.37) | 0.9986 | 0.14 | (0.11–0.17) | 0.9851 |
| ADC | 34.91 | (29.04–41.02) | 0.9847 | 4.81 | (3.56–6.32) | 0.9636 |
| ADC-PEG-AuNP | 19.45 | (16.52–22.80) | 0.9913 | 10.14 | (8.55–11.83) | 0.9878 |
| Tmab | 2118.36 | (1849.27–2426.61) | 0.9931 | N.D. | N.D. | N.D. |
| Sample | 248 nm | 280 nm |
|---|---|---|
| Trastuzumab | 7.75 × 104 | 2.25 × 105 |
| MMAE | 1.59 × 104 | 1.50 × 103 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, E.; Kayser, V. Synthesis and Enhanced Cellular Uptake In Vitro of Anti-HER2 Multifunctional Gold Nanoparticles. Cancers 2019, 11, 870. https://doi.org/10.3390/cancers11060870
Cruz E, Kayser V. Synthesis and Enhanced Cellular Uptake In Vitro of Anti-HER2 Multifunctional Gold Nanoparticles. Cancers. 2019; 11(6):870. https://doi.org/10.3390/cancers11060870
Chicago/Turabian StyleCruz, Esteban, and Veysel Kayser. 2019. "Synthesis and Enhanced Cellular Uptake In Vitro of Anti-HER2 Multifunctional Gold Nanoparticles" Cancers 11, no. 6: 870. https://doi.org/10.3390/cancers11060870
APA StyleCruz, E., & Kayser, V. (2019). Synthesis and Enhanced Cellular Uptake In Vitro of Anti-HER2 Multifunctional Gold Nanoparticles. Cancers, 11(6), 870. https://doi.org/10.3390/cancers11060870