Deciphering the Molecular Basis of Melatonin Protective Effects on Breast Cells Treated with Doxorubicin: TWIST1 a Transcription Factor Involved in EMT and Metastasis, a Novel Target of Melatonin

 ,
,  , ,
, , 

Abstract
:1. Introduction
2. Results
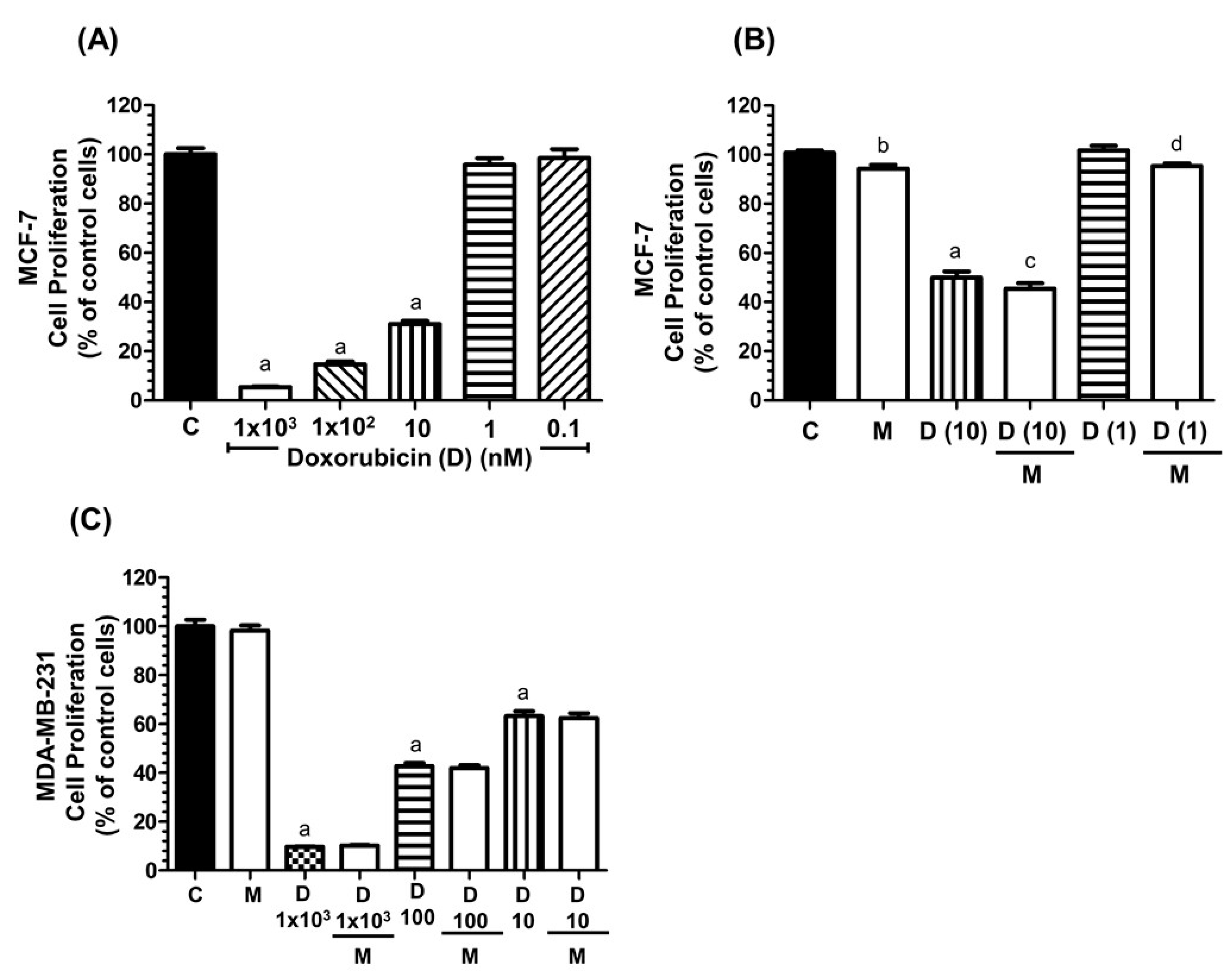
2.1. Effects of Melatonin and Doxorubicin on the Proliferation of MCF-7 and MDA-MB-231 Cells
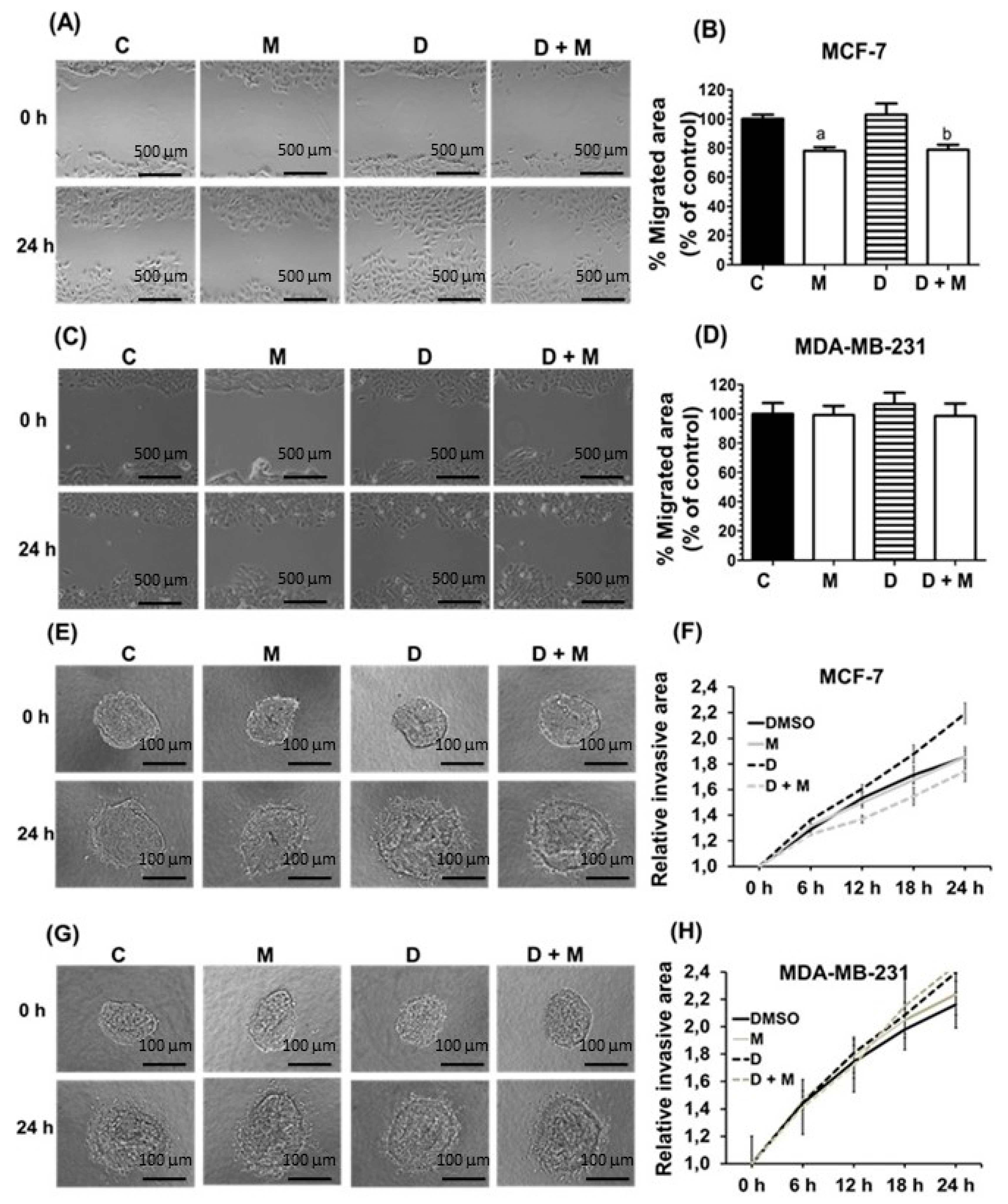
2.2. Effects of Melatonin and Doxorubicin on Cell Migration and Invasion in MDA-MB-231 and MCF-7 Cells
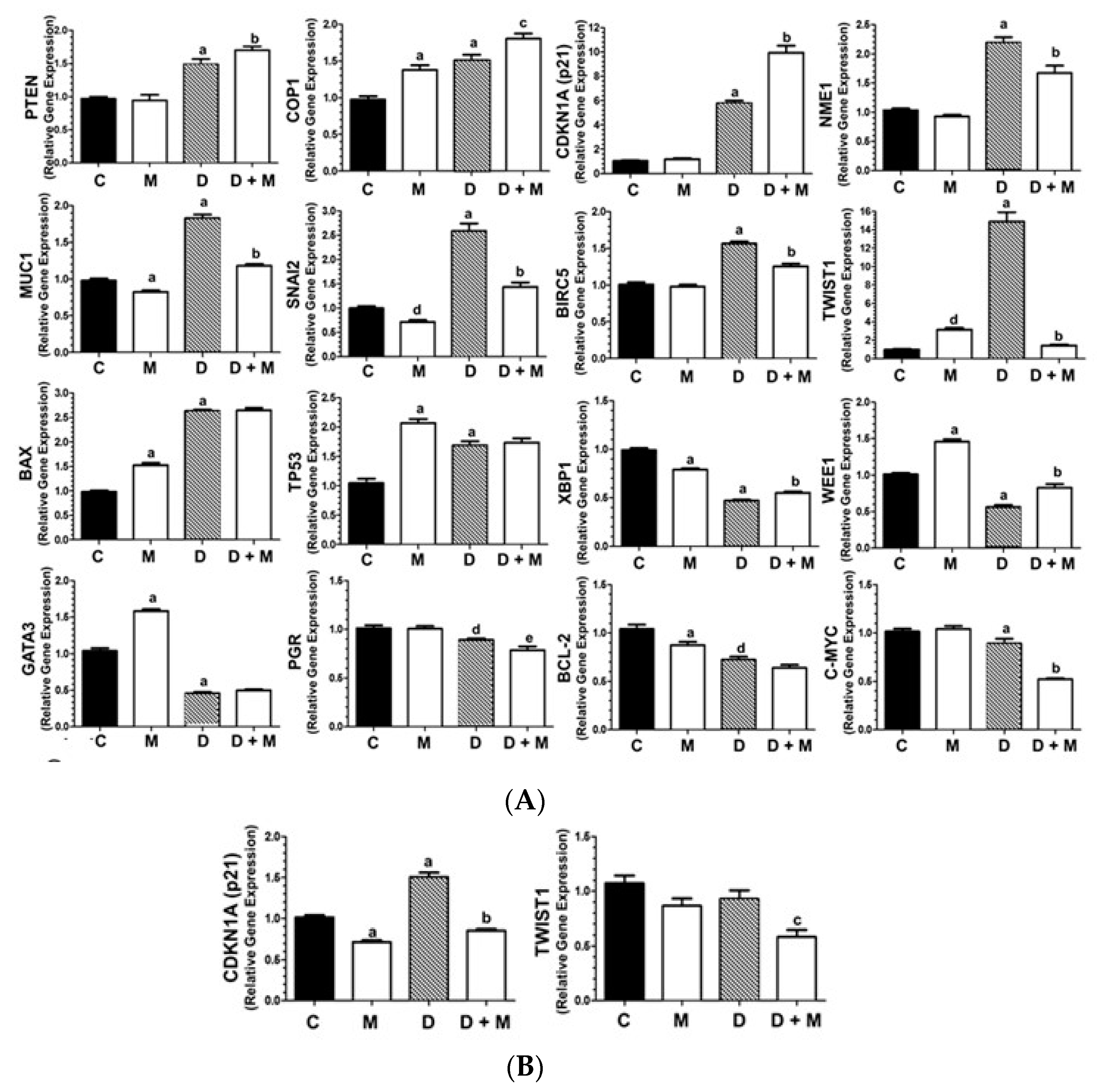
2.3. Effects of Doxorubicin and Melatonin on the Expression of Cancer-Related Genes
2.4. TWIST1 Protein Levels Are Upregulated by Doxorubicin and Inhibited by Melatonin in MCF-7 Cells
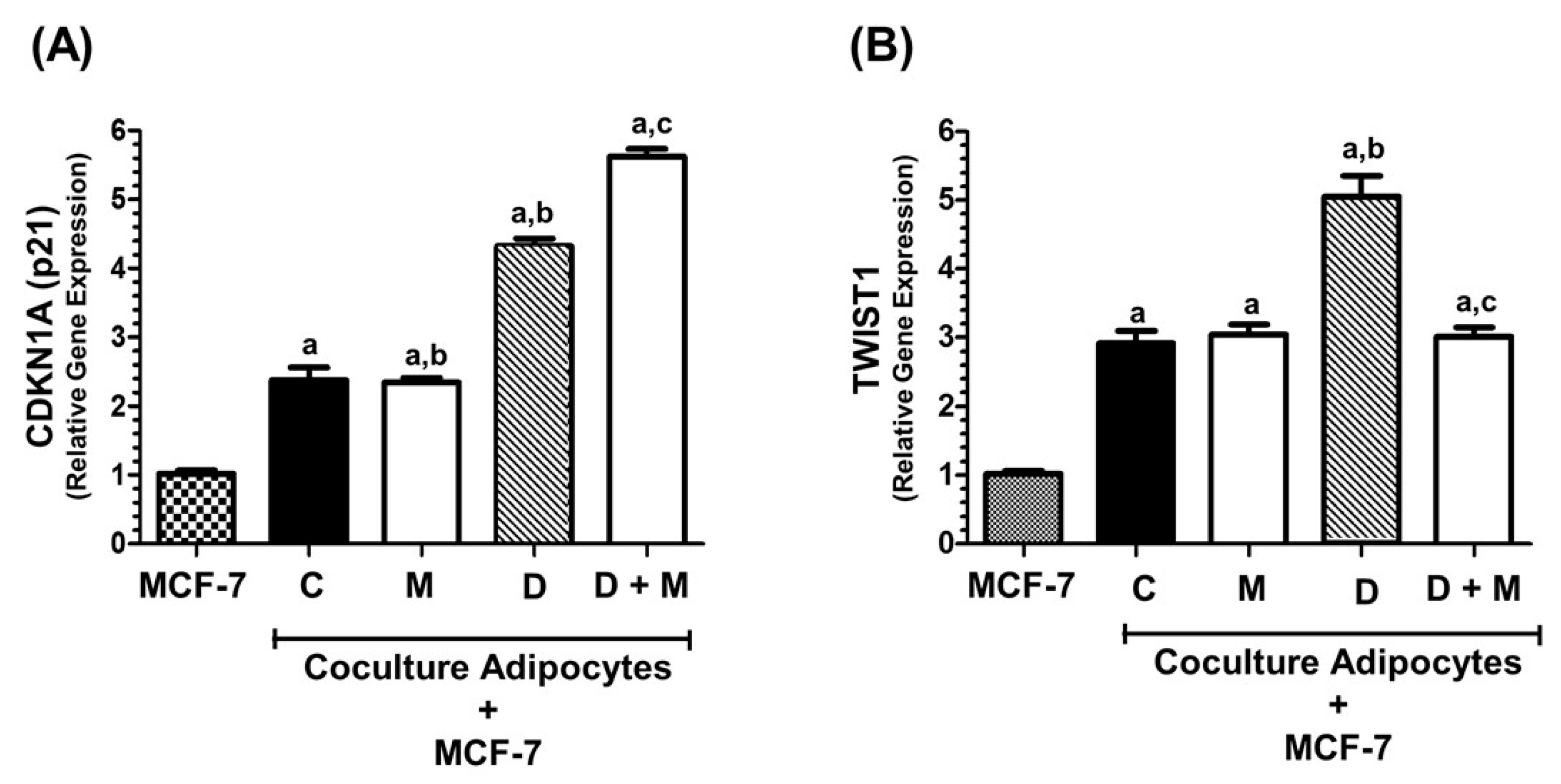
2.5. TWIST1 Gene Expression Is Regulated by Co-Culture of MCF-7 with Adipocytes
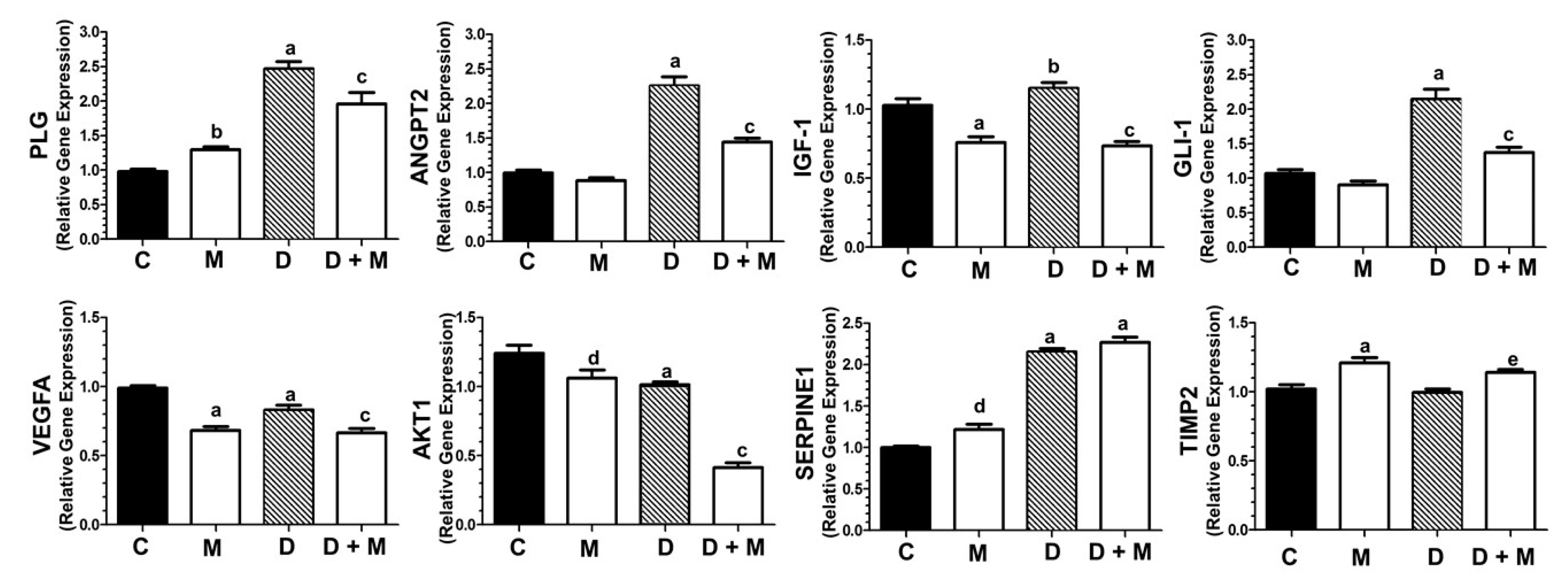
2.6. Effects of Doxorubicin and Melatonin on the Expression of Angiogenesis-Related Genes
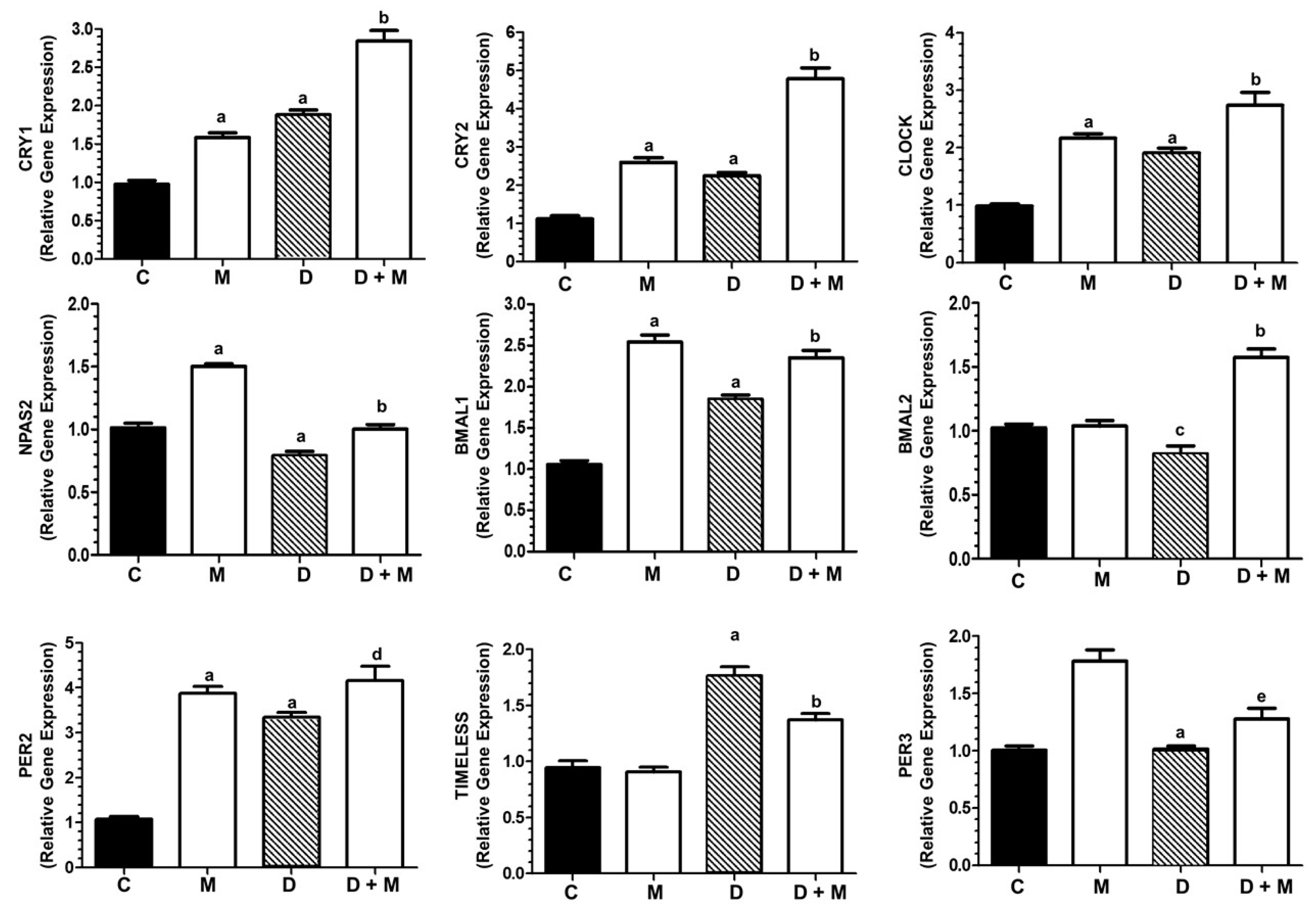
2.7. Effects of Doxorubicin and Melatonin on the Expression of Clock Genes
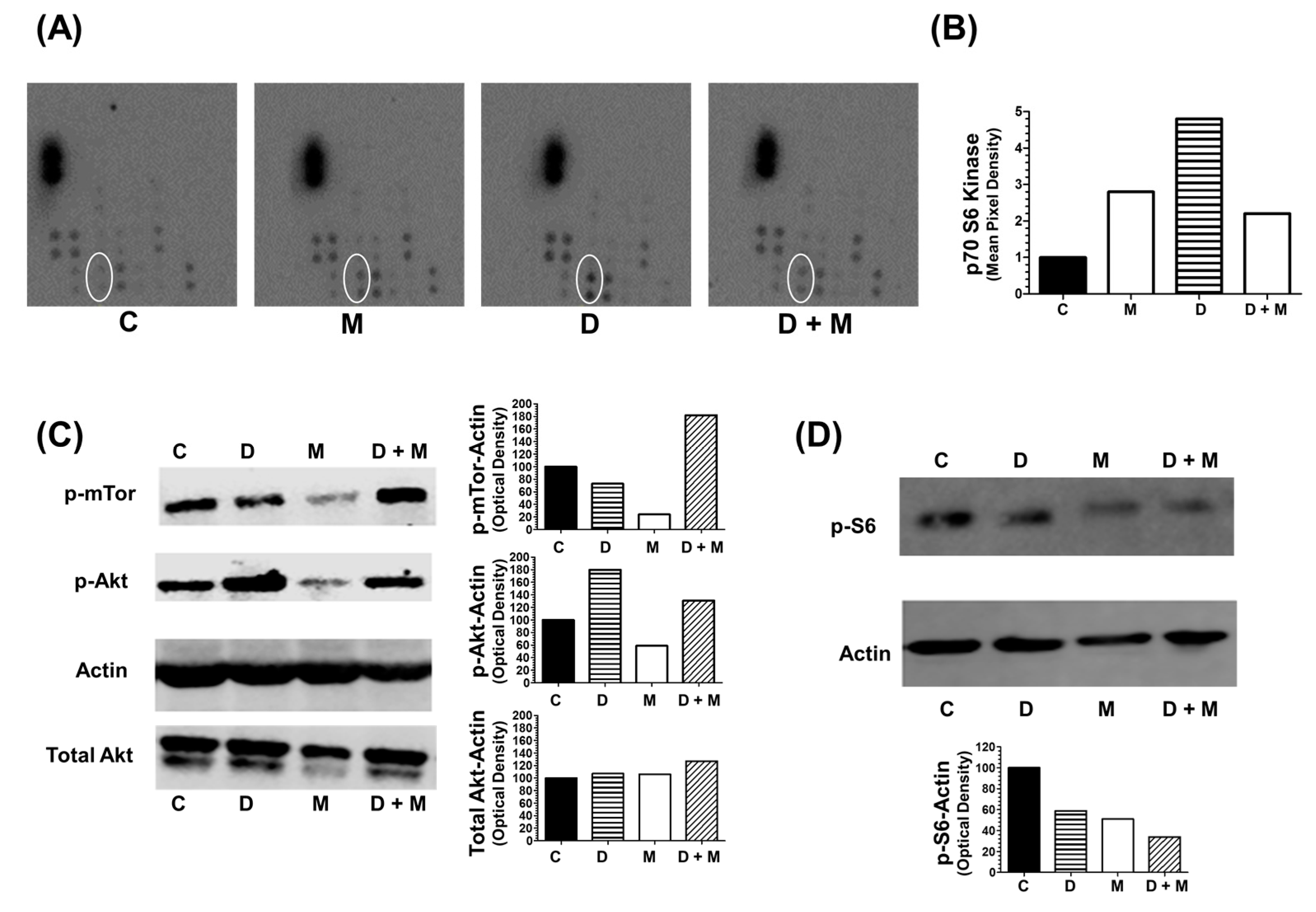
2.8. Combination of Melatonin and Doxorubicin on Kinase Intracellular Regulators
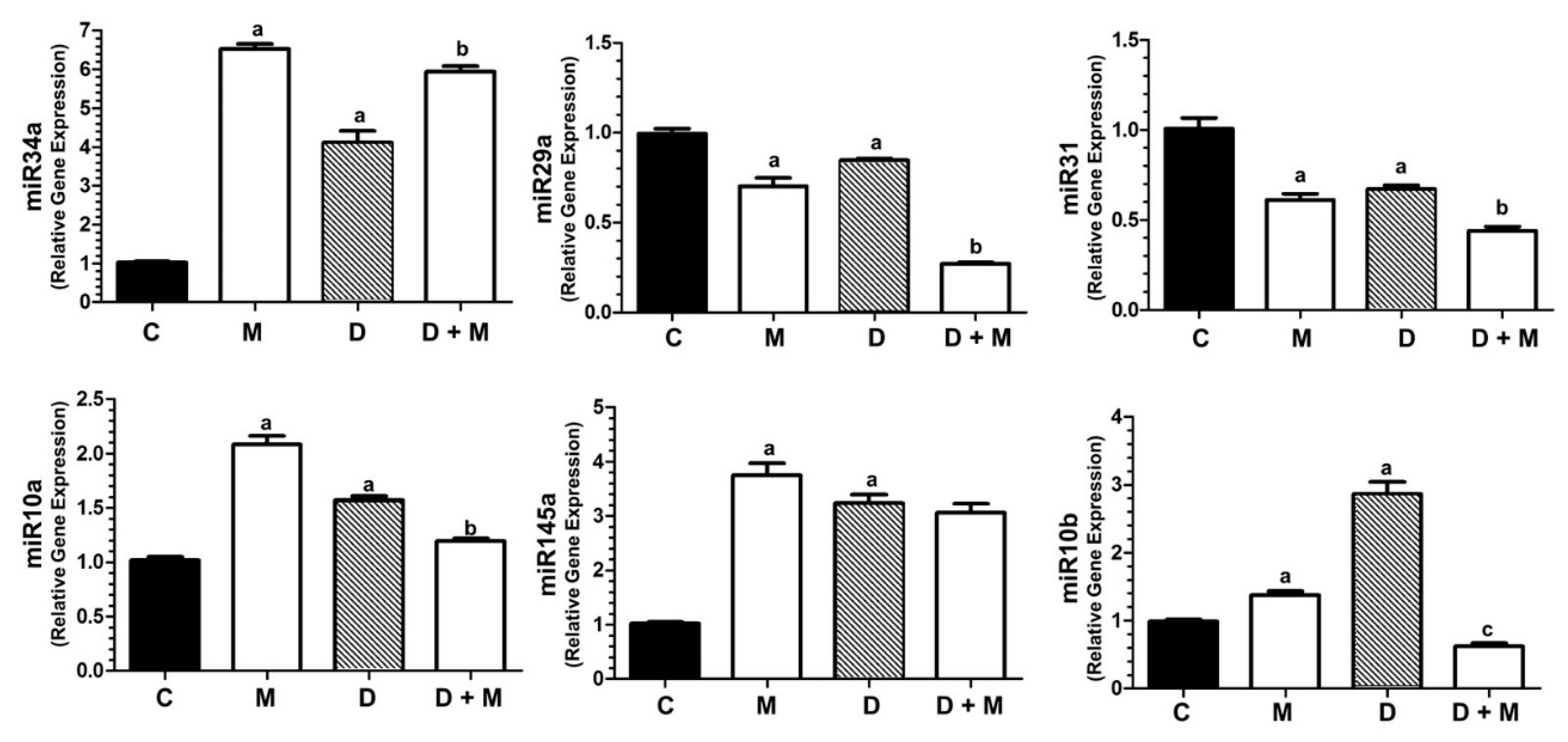
2.9. Doxorubicin and Melatonin Modulate the Expression of miR-34a, miR-29a, miR-31, miR-10a, miR145 and miR-10b in MCF-7 Cells
2.10. TWIST1 Blockade Abolishes the Stimulatory Effect of Doxorubicin on the Expression of miR-10b, miR-10a and VEGFA
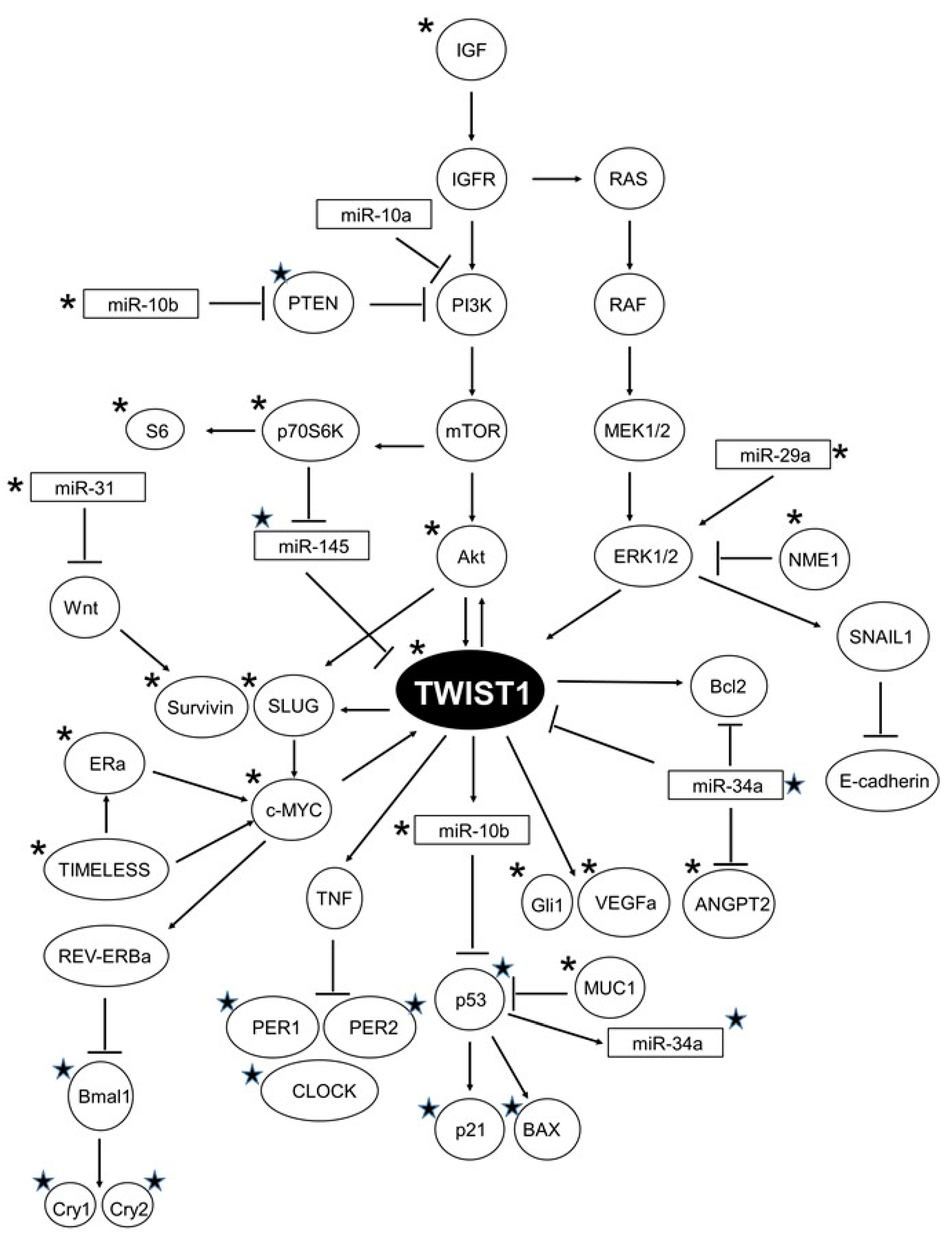
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Co-Culture of Breast Adipocytes and MCF-7 Cells as an In Vitro Breast Cancer Model
4.3. Cell Proliferation Assay
4.4. Cell Migration Assay: Wound Healing Assay
4.5. Three-Dimensional Spheroid Invasion Assays
4.6. RNA Isolation and cDNA Synthesis
4.7. RT2 ProfilerTM PCR Array
4.8. Measurement of Specific mRNA Gene Expression
4.9. Real-Time RT-PCR Data Analysis
4.10. Quantification of miRNAs Expression
4.11. Phosphokinase Screening
4.12. Antibodies
4.13. Western Blotting
4.14. Immunofluorescence Staining
4.15. Small Interfering RNA (siRNA)
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Reiter, R.J.; Tan, D.X.; Fuentes-Broto, L. Melatonin: A multitasking molecule. Prog. Brain. Res. 2010, 181, 127–151. [Google Scholar] [PubMed]
- Majidinia, M.; Reiter, R.J.; Shakouri, S.K.; Yousefi, B. The role of melatonin, a multitasking molecule, in retarding the processes of ageing. Ageing Res. Rev. 2018, 47, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Zisapel, N. New perspectives on the role of melatonin in human sleep, circadian rhythms and their regulation. Br. J. Pharmacol. 2018, 10, 3190–3199. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.F.; Xu, K. Melatonin, a full service anti-cancer agent: Inhibition of initiation, progression and metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef] [PubMed]
- Favero, G.; Moretti, E.; Bonomini, F.; Reiter, R.J.; Rodella, L.F.; Rezzani, R. Promising antineoplastic actions of melatonin. Front. Pharmacol. 2018, 9, 1086. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Salehi, E.; Farhood, B.; Nashtaei, M.S.; Hashemi Goradel, N.; Khanlarkhani, N.; Namjoo, Z. Adjuvant chemotherapy with melatonin for targeting human cancers: A review. J. Cell. Physiol. 2018, 7, 2356–2372. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Campa, C.; Menéndez-Menéndez, J.; Alonso-González, C.; González, A.; Álvarez-García, V.; Cos, S. What is known about melatonin, chemotherapy and altered gene expression in breast cancer. Oncol. Lett. 2017, 13, 2003–2014. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Menéndez, J.; Martínez-Campa, C. Melatonin: An anti-tumor agent in hormone-dependent cancers. Int. J. Endocrinol. 2018, 2018, 3271948. [Google Scholar] [CrossRef]
- Cos, S.; González, A.; Martínez-Campa, C.; Mediavilla, M.D.; Alonso-González, C.; Sánchez-Barceló, E.J. Melatonin as a selective estrogen enzyme modulator. Curr. Cancer Drug Targets 2008, 8, 691–702. [Google Scholar] [CrossRef]
- Rato, A.G.; Pedrero, J.G.; Martínez, M.A.; del Río, B.; Lazo, P.S.; Ramos, S. Melatonin blocks the activation of estrogen receptor for DNA binding. FASEB J. 1999, 13, 857–868. [Google Scholar] [CrossRef]
- Bouhoute, A.; Leclerq, G. Calmodulin decreases the estrogen binding capacity of the estrogen receptor. Biochem. Biophys. Res. Commun. 1996, 227, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Tancini, G.; Paolorossi, F.; Mandalà, M.; Ardizzoia, A.; Malugani, F.; Giani, L.; Barni, S. Chemoneuroendocrine therapy of metastatic breast cancer with persistent thrombocytopenia with weekly low-dose epirubicin plus melatonin: A phase II study. J. Pineal Res. 1999, 26, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Kisková, T.; Ekmekcioglu, C.; Garajová, M.; Orendás, P.; Bojková, B.; Bobrov, N.; Jäger, W.; Kassayová, M.; Thalhammer, T. A combination of resveratrol and melatonin exerts chemopreventive effects in N-methyl-N-nitrosourea-induced rat mammary carcinogenesis. Eur. J. Cancer Prev. 2012, 21, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Orendás, P.; Kubatka, P.; Bojková, B.; Kassayová, M.; Kajo, K.; Výbohová, D.; Kruzliak, P.; Péc, M.; Adamkov, M.; Kapinová, A.; et al. Melatonin potentiates the anti-tumour effect of pravastatin in rat mammary gland carcinoma model. Int. J. Exp. Pathol. 2014, 95, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Alonso-González, C.; Menéndez-Menéndez, J.; González-González, A.; González, A.; Cos, S.; Martínez-Campa, C. Melatonin enhances the apoptotic effects and modulates the changes in gene expression induced by docetaxel in MCF-7 human breast cancer cells. Int. J. Oncol. 2018, 52, 560–570. [Google Scholar]
- Granzotto, M.; Rapozzi, V.; Decorti, G.; Giraldi, T. Effects of melatonin on doxorubicin cytotoxicity in sensitive and pleiotropically resistant tumor cells. J. Pineal Res. 2001, 31, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, S.; Dauchy, R.T.; Hauch, A.; Mao, L.; Yuan, L.; Wren, M.A.; Belancio, V.P.; Mondal, D.; Frasch, T.; Blask, D.E.; et al. Doxorubicin resistance in breast cancer is driven by light at night-induced disruption of the circadian melatonin signal. J. Pineal Res. 2015, 59, 60–69. [Google Scholar] [CrossRef]
- Ma, C.; Li, L.X.; Zhang, Y.; Xiang, C.; Ma, T.; Ma, Z.Q.; Zhang, Z.P. Protective and sensitive effects of melatonin combined with adraimycin on ER+ (estrogen receptor) breast cancer. Eur. J. Gynaecol. Oncol. 2015, 36, 197–202. [Google Scholar]
- Kosar, P.A.; Naziroglu, M.; Övey, I.S.; Cig, B. Synergic effects of doxorubicin and melatonin on apoptosis and mitochondrial oxidative stress in MCF-7 breast cancer cells: Involvement of TRPV1 channels. J. Membr. Biol. 2016, 249, 129–140. [Google Scholar] [CrossRef]
- Zhu, Q.Q.; Ma, C.; Wang, Q.; Song, Y.; Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumour Biol. 2016, 37, 185–197. [Google Scholar] [CrossRef]
- Tang, H.; Massi, D.; Hemmings, B.A.; Mandalà, M.; Hu, Z.; Wicki, A.; Xue, G. AKT-ions with a TWIST between EMT and MET. Oncotarget 2016, 7, 62767–62777. [Google Scholar] [CrossRef]
- Wushou, A.; Hou, J.; Zhao, Y.J.; Shao, Z.M. Twist-1 up-regulation in carcinoma correlates to poor survival. Int. J. Mol. Sci. 2014, 15, 21621–21630. [Google Scholar] [CrossRef]
- Abdelmaksoud-Dammak, R.; Chamtouri, N.; Triki, M.; Saadallah-Kallel, A.; Ayadi, W.; Charfi, S.; Khabir, A.; Ayadi, L.; Sallemi-Boudawara, T.; Mokdad-Gargouri, R. Overexpression of miR-10b in colorectal cancer patients: Correlation with TWIST-1 and E-cadherin expression. Tumour Biol. 2017, 39, 1010428317695916. [Google Scholar] [CrossRef]
- Shen, X.; Fang, J.; Lv, X.; Pei, Z.; Wang, Y.; Jiang, S.; Ding, K. Heparin impairs angiogenesis through inhibition of microRNA-10b. J. Biol. Chem. 2011, 286, 26616–26627. [Google Scholar] [CrossRef]
- M, J.R.; S, V. BMI1 and PTEN are key determinants of breast cancer therapy: A plausible therapeutic target in breast cancer. Gene 2018, 678, 302–311. [Google Scholar] [CrossRef]
- Zohny, S.F.; Al-Malki, A.L.; Zamzami, M.A.; Choudhry, H. p21 Waf1/Cip1: Its paradoxical effect in the regulation of breast cancer. Breast Cancer 2019, 26, 131–137. [Google Scholar] [CrossRef]
- Shao, J.; Teng, Y.; Padia, R.; Hong, S.; Noh, H.; Xie, X.; Mumm, J.S.; Dong, Z.; Ding, H.F.; Cowell, J.; et al. COP1 and GSK3β cooperate to promote c-Jun degradation and inhibit breast cancer cell tumorigenesis. Neoplasia 2013, 15, 1075–1085. [Google Scholar] [CrossRef]
- McCorkle, J.R.; Leonard, M.K.; Kraner, S.D.; Blalock, E.M.; Ma, D.; Zimmer, S.G.; Kaetzel, D.M. The metastasis suppressor NME1 regulates expression of genes linked to metastasis and patient outcome in melanoma and breast carcinoma. Cancer Genom. Proteom. 2014, 11, 175–194. [Google Scholar]
- Wei, X.; Xu, H.; Kufe, D. Human mucin 1 oncoprotein represses transcription of the p53 tumor suppressor gene. Cancer Res. 2007, 67, 1853–1858. [Google Scholar] [CrossRef]
- Wang, W.; Hind, T.; Lam, B.W.S.; Herr, D.R. Sphingosine 1-phosphate signaling induces SNAI2 expression to promote cell invasion in breast cancer. FASEB J. 2019, 7, 7180–7191. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, K.; Yagüe, E. miR-218 targets survivin and regulates resistance to chemotherapeutics in breast cancer. Breast Cancer Res. Treat. 2015, 151, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef]
- Li, Q.Q.; Xu, J.D.; Wang, W.J.; Cao, X.X.; Chen, Q.; Tang, F.; Chen, Z.Q.; Liu, X.P.; Xu, Z.D. Twist1-mediated Adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer Res. 2009, 15, 2657–2665. [Google Scholar] [CrossRef]
- Basu, A.; Haldar, S. The relationship between Bcl2, Bax and p53: Consequences for cell cycle progression and cell death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, F.; Pan, Y.; Chen, X.; Chen, J.; Wang, Y.; Zheng, X.; Zhang, J. XBP1 promotes tumor invasion and is associated with poor prognosis in oral squamous cell carcinoma. Oncol. Rep. 2018, 40, 988–998. [Google Scholar] [CrossRef]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 kinase in cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- Asch-Kendrick, R.; Cimino-Mathews, A. The role of GATA3 in breast carcinomas: A review. Hum. Pathol. 2016, 48, 37–47. [Google Scholar] [CrossRef]
- Yip, C.H.; Rhodes, A. Estrogen and progesterone receptors in breast cancer. Future Oncol. 2014, 10, 2293–2301. [Google Scholar] [CrossRef] [Green Version]
- Merino, D.; Lok, S.W.; Visvader, J.E.; Lindeman, G.J. Targeting BCL-2 to enhance vulnerability to therapy in estrogen receptor-positive breast cancer. Oncogene 2016, 35, 1877–1887. [Google Scholar] [CrossRef]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-driven pathways in breast cancer subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]
- Molis, T.M.; Spriggs, L.L.; Jupiter, Y.; Hill, S.M. Melatonin modulation of estrogen-regulated proteins, growth factors, and proto-oncogenes in human breast cancer. J. Pineal Res. 1995, 18, 93–103. [Google Scholar] [CrossRef]
- Shirley, S.H.; Rundhaug, J.E.; Tian, J.; Cullinan-Ammann, N.; Lambertz, I.; Conti, C.J.; Fuchs-Young, R. Transcriptional regulation of estrogen receptor-alpha by p53 in human breast cancer cells. Cancer Res. 2009, 69, 3405–3413. [Google Scholar] [CrossRef]
- Vesuna, F.; Lisok, A.; Kimble, B.; Domek, J.; Kato, Y.; van der Groep, P.; Artemov, D.; Kowalski, J.; Carraway, H.; van Diest, P.; et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-α. Oncogene 2012, 31, 3223–3234. [Google Scholar] [CrossRef]
- Han, K.Y.; Chen, P.N.; Hong, M.C.; Hseu, Y.C.; Chen, K.M.; Hsu, L.S.; Chen, W.J. Naringenin attenuated prostate cancer invasion via reversal of epithelial-to-mesenchymal transition and inhibited uPA activity. Anticancer Res. 2018, 38, 6753–6758. [Google Scholar] [CrossRef]
- Avraham, H.K.; Jiang, S.; Fu, Y.; Nakshatri, H.; Ovadia, H.; Avraham, S. Angiopoietin-2 medaites blood-brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J. Pathol. 2014, 232, 369–381. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, Q.; Wang, B.; Sun, Q.; He, Z.; Hong, J.; Kuehn, F.; Liu, E.; Zhang, Z. IGF-1 induces the epithelial-mesenchymal transition via Stat5 in hepatocellular carcinoma. Oncotarget 2017, 8, 111922–111930. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Cao, Y.; Yu, Y.; Chen, X.; Gao, X.; Hu, Q. Upregulation of twist is involved in Gli1 induced migration and invasion of hepatocarcinoma cells. Biol. Chem. 2018, 399, 911–919. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, R.; Zhao, Y.; Dong, S.; Zhang, J.; Luo, Y.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; et al. MACC-1 promotes endothelium-dependent angiogenesis in gastric cancer by activating TWIST1/VEGFA signal pathway. PLoS ONE 2016, 11, e0157137. [Google Scholar]
- Xue, G.; Restuccia, D.F.; Lan, Q.; Hynx, D.; Dirnhofer, S.; Hess, D.; Rüegg, C.; Hemmings, B.A. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov. 2012, 2, 248–259. [Google Scholar] [CrossRef]
- Cardenas, C.; van de Sandt, L.; Edlund, K.; Lohr, M.; Hellwig, B.; Marchan, R.; Schmidt, M.; Rahnenführer, J.; Oster, H.; Hengstler, J.G. Loss of circadian clock gene expression is associated with tumor progression in breast cancer. Cell Cycle 2014, 13, 3282–3291. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society. Cancer Facts and Figures 2017. Available online: www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2017.html (accessed on 17 May 2018).
- Jawad, B.; Poudel, L.; Podgornik, R.; Steinmetz, N.F.; Ching, W.Y. Molecular mechanism and binding free energy of doxorubicin intercalation in DNA. Phys. Chem. Chem. Phys. 2019, 21, 3877–3893. [Google Scholar] [CrossRef]
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharmacother. 2019, 114, 108800. [Google Scholar] [CrossRef]
- Lumachi, F.; Chiara, G.B.; Foltran, L.; Basso, S.M. Proteomics as a guide for personalized adjuvant chemotherapy in patients with early breast cancer. Cancer Genom. Proteom. 2015, 12, 385–390. [Google Scholar]
- Kim, C.; Kim, N.; Joo, H.; Youm, J.B.; Park, W.S.; Cuong, D.V.; Park, Y.S.; Kim, E.; Min, C.K.; Han, J. Modulation by melatonin of the cardiotoxic and antitumor activities of adriamycin. J. Cardiovasc. Pharmacol. 2005, 46, 200–210. [Google Scholar] [CrossRef]
- Rapozzi, V.; Zorzet, S.; Comelli, M.; Mavelli, I.; Perissin, L.; Giraldi, T. Melatonin decreases bone marrow and lymphatic toxicity of Adriamycin in mice bearing TLX5 lymphoma. Life Sci. 1998, 63, 1701–1713. [Google Scholar] [CrossRef]
- Fic, M.; Podhorska-Okolow, M.; Dziegiel, P.; Gebarowska, E.; Wysocka, T.; Drag-Zalesinska, M.; Zabel, M. Effect of melatonin on cytotoxicity of doxorubicin toward selected cell lines (human keratinocytes, lung cancer cell line A-549, laryngeal cancer cell line Hep-2). In Vivo 2007, 21, 513–518. [Google Scholar]
- Pariente, R.; Pariente, J.A.; Rodríguez, A.B.; Espino, J. Melatonin sensitizes human cervical cancer HeLa cells to cisplatin-induced cytotoxicity and apoptosis: Effects on oxidative stress and DNA fragmentation. J. Pineal Res. 2016, 60, 55–64. [Google Scholar] [CrossRef]
- Wan, L.; Tian, Y.; Zhang, R.; Peng, Z.; Sun, J.; Zhang, W. MicroRNA-103 confers the resistance to long-treatment of Adriamycin to human leukemia cells by regulation of COP1. J. Cell. Biochem. 2018, 119, 3843–3852. [Google Scholar] [CrossRef]
- Thisse, B.; el Messal, M.; Perrin-Schmitt, F. The twist gene: Isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Res. 1987, 15, 3439–3453. [Google Scholar] [CrossRef]
- Wang, S.M.; Coljee, V.W.; Pignolo, R.J.; Rotenberg, M.O.; Cristofalo, V.J.; Sierra, F. Cloning of the human twist gene: Its expression is retained in adult mesodermally-derived tissues. Gene 1997, 187, 83–92. [Google Scholar] [CrossRef]
- Maestro, R.; Dei Tos, A.P.; Hamamori, Y.; Krasnokutsy, S.; Sartorelli, V.; Kedes, L.; Doglioni, C.; Beach, D.H.; Hannon, G.J. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999, 13, 2207–2217. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, D.Q.; Chen, M.Y.; Ji, K.Y.; Ma, D.X.; Zhou, L.F. Endothelial cells by inactivation of VHL gene direct angiogenesis, not vasculogenesis via Twist1 accumulation associated with hemangioblastoma neovascularization. Sci. Rep. 2017, 7, 5463. [Google Scholar] [CrossRef]
- Mironchik, Y.; Winnard, P.T., Jr.; Vesuna, F.; Kato, Y.; Wildes, F.; Pathak, A.P.; Kominsky, S.; Artemov, D.; Bhujwalla, Z.; Van Diest, P.; et al. Twist overexpression induces in vivo angiogenesis and correlates with chromosomal instability in breast cancer. Cancer Res. 2005, 65, 10801–10809. [Google Scholar] [CrossRef]
- Zhou, M.; Hou, Y.; Yang, G.; Zhang, H.; Tu, G.; Du, Y.E.; Wen, S.; Xu, L.; Tang, X.; Tang, S.; et al. LncRNA-Hh strengthen cancer stem cells generation in twist-positive breast cancer via activation of Hedgehog signaling pathway. Stem Cells 2016, 34, 55–66. [Google Scholar] [CrossRef]
- Mier, D.; Lopez, M.; Franken, P.; Fontana, A. Twist1 is a TNF-inducible inhibitor of Clock mediated activation of period genes. PLoS ONE 2015, 10, e0137229. [Google Scholar] [CrossRef]
- Hwang-Verslues, W.W.; Chang, P.H.; Jeng, Y.M.; Kuo, W.H.; Chiang, P.H.; Chang, Y.C.; Hsieh, T.H.; Su, F.Y.; Lin, L.C.; Abbondante, S.; et al. Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proc. Natl. Acad. Sci. USA 2013, 110, 12331–12336. [Google Scholar] [CrossRef] [Green Version]
- Samulin Erdem, J.; Skare, O.; Petersen-Overleir, M.; Noto, H.O.; Lie, J.S.; Reszka, E.; Peplonska, B.; Zienolddiny, S. Mechanisms of breast cancer in shift workers: DNA methylation in five core circadian genes in nurses working night shifts. J. Cancer 2017, 8, 2876–2884. [Google Scholar] [CrossRef]
- Chi, L.; Zou, Y.; Qin, L.; Ma, W.; Hao, Y.; Tang, Y.; Luo, R.; Wu, Z. TIMELESS contributes to the progression of breast cancer through activation of MYC. Breast Cancer Res. 2017, 19, 53. [Google Scholar] [CrossRef] [Green Version]
- Magne Nde, C.B.; Casas Gimeno, G.; Docanto, M.; Knower, K.C.; Young, M.J.; Buehn, J.; Sayed, E.; Clyne, C.D. Timeless is a novel estrogen receptor co-activator involved in multiple signaling pathways in MCF-7 cells. J. Mol. Biol. 2018, 430, 1531–1543. [Google Scholar] [CrossRef]
- Álvarez-García, V.; González, A.; Alonso-González, C.; Martínez-Campa, C.; Cos, S. Regulation of vascular endothelial growth factor by melatonin in human breast cancer cells. J. Pineal Res. 2013, 54, 373–380. [Google Scholar] [CrossRef]
- Proietti, S.; Cucina, A.; D’Anselmi, F.; Dinicola, S.; Pasqualato, A.; Lisi, E.; Bizzarri, M. Melatonin and vitamin D3 synergistically down-regulate Akt and MDM2 leading to TGFβ-1-dependent growth inhibition of breast cancer cells. J. Pineal Res. 2011, 50, 150–158. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, X.; Zhang, Y.; Shi, D.; Chen, W.; Fu, L.; Liu, L.; Xie, F.; Kang, T.; Huang, W.; et al. Simultaneous modulation of COX-2, p300, Akt, and Apaf-1 signaling by melatonin to inhibit proliferation and induce apoptosis in breast cancer cells. J. Pineal Res. 2012, 53, 77–90. [Google Scholar] [CrossRef]
- Wu, Y.H.; Huang, Y.F.; Chen, C.C.; Chou, C.Y. Akt inhibitor SC66 promotes cell sensitivity to cisplatin in chemoresistant ovarian cancer cells through inhibition of COL11A1 expression. Cell Death Dis. 2019, 10, 322. [Google Scholar] [CrossRef]
- Cicenas, J.; Urban, P.; Vuaroqueaux, V.; Labuhn, M.; Küng, W.; Wight, E.; Mayhew, M.; Eppenberger, U.; Eppenberger-Castori, S. Increased level of phosphorylated akt measured by chemiluminescence-linked immunosorbent assay is a predictor of poor prognosis in primary breast cancer overexpressing ErbB-2. Breast Cancer Res. 2005, 7, R394–R401. [Google Scholar] [CrossRef]
- Yndestad, S.; Austreid, E.; Svanberg, I.R.; Knappskog, S.; Lonning, P.E.; Eikesdal, H.P. Activation of Akt characterizes estrogen receptor positive human breast cancer which respond to anthracyclines. Oncotarget 2017, 8, 41227–41241. [Google Scholar] [CrossRef]
- Steelman, L.S.; Navolanic, P.M.; Sokolosky, M.L.; Taylor, J.R.; Lehmann, B.D.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Stadelman, K.M.; Terrain, D.M.; et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene 2008, 27, 4086–4095. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Cheng, J.; Sun, G.; Wu, S.; Li, M.; Gao, Z.; Zhai, S.; Li, P.; Su, D.; Wang, X. p70S6K promotes IL-6-induced epithelial-mesenchymal transition and metastasis of head and neck squamous cell carcinoma. Oncotarget 2016, 7, 36539–36550. [Google Scholar] [CrossRef] [Green Version]
- Bostner, J.; Karlsson, E.; Eding, C.B.; Perez-Tenorio, G.; Franzén, H.; Konstantinell, A.; Fornander, T.; Nordenskjöld, B.; Stal, O. S6 kinase signaling: Tamoxifen response and prognostic indication in two breast cancer cohorts. Endocr. Relat. Cancer 2015, 22, 331–343. [Google Scholar] [CrossRef]
- Lee, Y.; Jung, W.H.; Koo, J.S. Adipocytes can induce epithelial-mesenchymal transition in breast cancer cells. Breast Cancer Res. Treat. 2015, 153, 323–335. [Google Scholar] [CrossRef]
- Yao-Borengasser, A.; Monzavi-Karbassi, B.; Hedges, R.A.; Rogers, L.J.; Kadlubar, S.A.; Kieber-Emmons, T. Adipocyte hypoxia promotes epithelial-mesenchymal transition-related gene expression and estrogen receptor-negative phenotype in breast cancer cells. Oncol. Rep. 2015, 33, 2689–2694. [Google Scholar] [CrossRef]
- Shiota, M.; Izumi, H.; Onitsuka, T.; Miyamoto, N.; Kashiwagi, E.; Kidani, A.; Hirano, G.; Takahashi, M.; Naito, S.; Kohno, K. Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene 2008, 27, 5543–5553. [Google Scholar] [CrossRef]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Rui, X.; Zhao, H.; Xiao, X.; Wang, L.; Mo, L.; Yao, Y. MicroRNA-34a suppresses breast cancer cell proliferation and invasion by targeting Notch1. Exp. Ther. Med. 2018, 16, 4387–4392. [Google Scholar] [CrossRef]
- Imani, S.; Wei, C.; Cheng, J.; Khan, M.A.; Fu, S.; Yang, L.; Tania, M.; Zhang, X.; Xiao, X.; Zhang, X.; et al. MicroRNA-34a targets epithelial to mesenchymal transition-inducing transcription factors (EMT-TFs) and inhibits breast cancer cell migration and invasion. Oncotarget 2017, 8, 21362–21379. [Google Scholar] [CrossRef] [Green Version]
- Rokavec, M.; Öner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.H.; Xiong, Q.Y.; Xu, L.; Duan, P.; Yang, Q.O.; Zhou, P.; Tu, J.H. miR-29a regulated ER-positive breast cancer cell growth and invasion and is involved in the insulin signaling pathway. Oncotarget 2017, 8, 32566–32575. [Google Scholar] [CrossRef] [Green Version]
- Khamisipour, G.; Mansourabadi, E.; Naeimi, B.; Moazzeni, A.; Tahmasebi, R.; Hasanpour, M.; Mohammadi, M.M.; Mansourabadi, Z.; Shamsian, S. Knockdown of microRNA-29a regulates the expression of apoptosis-related genes in MCF-7 breast carcinoma cells. Mol. Clin. Oncol. 2018, 8, 362–369. [Google Scholar] [CrossRef]
- Soheilyfar, S.; Velashjerdi, Z.; Sayed Hajizadeh, Y.; Fathi Maroufi, N.; Amini, Z.; Khorrami, A.; Haj Azimian, S.; Isazadeh, A.; Taefehshokr, S.; Taefehshokr, N. In vivo and in vitro impact of miR-31 and miR143 on the suppression of metastasis and invasion in breast cancer. J. BOUN 2018, 23, 1290–1296. [Google Scholar]
- Lv, C.; Li, F.; Li, X.; Tian, Y.; Zhang, Y.; Sheng, X.; Song, Y.; Meng, Q.; Yuan, S.; Luan, L.; et al. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nat. Commun. 2017, 8, 1036. [Google Scholar] [CrossRef]
- Ke, K.; Lou, T. MicroRNA-10a suppresses breast cancer progression via PI3K/Akt/mTOR pathway. Oncol. Lett. 2017, 14, 5994–6000. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.S.; Ip, C.K.; Mak, A.S.; Wong, A.S. A novel p70 S6 kinase-microRNA biogenesis axis mediates multicellular spheroid formation in ovarian cancer progression. Oncotarget 2016, 7, 38064–38077. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Y.; Li, X.Y.; Chen, W.Q.; Hu, H.; Luo, B.; Shi, Y.X.; Wu, T.W.; Li, Y.; Kong, Q.Z.; Lu, H.D.; et al. Demethylation of the MIR145 promoter suppresses migration and invasion in breast cancer. Oncotarget 2017, 8, 61731–61741. [Google Scholar] [CrossRef]
- Liu, X.; Guan, Y.; Wang, L.; Niu, Y. MicroRNA-10b expression in node-negative breast cancer-correlation with metastasis and angiogenesis. Oncol. Lett. 2017, 14, 5845–5852. [Google Scholar] [CrossRef] [Green Version]
- Tsukerman, P.; Yamin, R.; Seidel, E.; Khawaled, S.; Schmiedel, D.; Bar-Mag, T.; Mandelboim, O. MiR-520d-5p directly targets TWIST1 and downregulates the metastamiR miR-10b. Oncotarget 2014, 5, 12141–12150. [Google Scholar] [CrossRef] [Green Version]
- Croset, M.; Goehrig, D.; Frackowiak, A.; Bonnelye, E.; Ansieau, S.; Puisieux, A.; Clézardin, P. TWIST1 expression in breast cancer cells facilitates bone metastasis formation. J. Bone Miner. Res. 2014, 29, 1886–1899. [Google Scholar] [CrossRef]
- Chao, C.C.; Chen, P.C.; Chiou, P.C.; Hsu, C.J.; Liu, P.I.; Yang, Y.C.; Reiter, R.J.; Yang, S.F.; Tang, C.H. Melatonin suppresses lung cancer metastasis by inhibition of epithelial-mesenchymal transition through targeting to Twist. Clin. Sci. 2019, 133, 709–722. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breast Cancer Array | D vs. C | D + M vs. D | M vs. C | |||
|---|---|---|---|---|---|---|
| Up | Down | Up | Down | Up | Down | |
| Angiogenesis, cell adhesion and proteases | 5 | 3 | 2 | 1 | 0 | 2 |
| Breast cancer classification markers | 2 | 1 | 1 | 4 | 1 | 5 |
| Signal transduction | 8 | 3 | 3 | 5 | 3 | 6 |
| Cell cycle | 5 | 1 | 0 | 7 | 4 | 3 |
| Apoptosis and DNA damage and repair | 7 | 2 | 1 | 6 | 7 | 3 |
| Transcription factors | 0 | 7 | 2 | 3 | 2 | 3 |
| Angiogenesis Array | D vs. C | D + M vs. D | M vs. C | |||
|---|---|---|---|---|---|---|
| Up | Down | Up | Down | Up | Down | |
| Adhesion molecules | 4 | 0 | 0 | 2 | 0 | 1 |
| Proteases, inhibitors and matrix proteins | 4 | 2 | 1 | 5 | 2 | 1 |
| Cytokines and other factors | 0 | 3 | 1 | 3 | 0 | 0 |
| Growth factors and receptors | 8 | 3 | 3 | 4 | 2 | 0 |
| Gene | Forward | Reverse |
|---|---|---|
| PTEN | AGGTTTCCTCTGGTCCTGGT | CGACGGGAAGACAAGTTCAT |
| COP1 | CTGGAAGCAGAATCACATGC | TGTGCTATCCTCACTGACAGG |
| CDKN1A | CAGCATGACAGATTTCTACC | CAGGGTATGTACATGAGGAG |
| NME1 | CAGAAGTCTCCACGGATGGT | AGAAAGGATTCCGCCTTGTT |
| MUC1 | GCAAGAGCACTCCATTCTCAATT | TGGCATCAGTCTTGGTGCTATG |
| SNAI2 | CAGTGATTATTTCCCCGTATC | CCCCAAAGATGAGGAGTATC |
| BIRC5 | TCTCCGCAGTTTCCTCAAAT | GGACCACCGCATCTCTACAT |
| TWIST1 | CTAGATGTCATTGTTTCCAGAG | CCCTGTTTCTTTCAATTTGG |
| BAX | AACTGGACAGTAACATGGAG | TTGCTGGCAAAGTAGAAAAG |
| TP53 | CCTATGCTTGTATGGCTAAC | TAGATCCATGCCTTCTTCTTC |
| XBP1 | ACTGGGTCCAAGTTGTCCAG | TCACCCCTCCAGAACATCTC |
| WEE1 | AGTGCCATTGCTGAAGGTCT | ACCTCGGATACCACAAGTGC |
| GATA3 | CGGTCCAGCACAGGCAGGGAGT | GAGCCCACAGGCATTGCAGACA |
| PGR | GAGAGCTCATCAAGGCAATTGG | CACCATCCCTGCCAATATCTTG |
| BCL2 | CCTTTGGAATGGAAGCTTAG | GAGGGAATGTTTTCTCCTTG |
| c-MYC | TGAGGAGGAACAAGAAGATG | ATCCAGACTCTGACCTTTTG |
| PLG | TAGATTCTCACCTGCTACAC | CGCAGTAGTCATATCTCTTTTC |
| ANGPT2 | AAGAGAAAGATCAGCTACAGG | CCTTAGAGTTTGATGTGGAC |
| IGF1 | ATAGAGCCTGCGCAATGGAA | GAGATGGGAGATGTTGAGAGCA |
| GLI1 | CTCGTAGCTTTCATCAACT | TTTTTGGTGATTCATCTGGG |
| VEGFA | TGGTGATGTTGGACTCCTCA | GGGCAGAATCATCACGAAGT |
| AKT1 | AAGTACTCTTTCCAGACCC | TTCTCCAGCTTGAGGTC |
| SERPINE1 | ATCCACAGCTGTCATAGTC | CACTTGGCCCATGAAAAG |
| TIMP2 | GGCCTGAGAAGGATATAGAG | CTTTCCTGCAATGAGATATTCC |
| CRY1 | TTTTGCAGGGAAGCCTCTTA | CTGCTATTGCCCTGTTGGTT |
| CRY2 | GCGAAAGCTGCTGGTAAATC | TACCTGCCCAAATTGAAAGC |
| CLOCK | TATCATGCGTGTCCGTTGTT | ACAAGGCATGTCCCAGTTTC |
| NPAS2 | AAGGCTTCCAGTCTTGCTGA | CGGGACCAGTTCAATGTTCT |
| BMAL1 | GACGAGGCAGCTGAGGTTAC | CCACAGCACAGGCTATTTGA |
| BMAL2 | AATCCAACTGTGCACCATCA | GCTACCAGGCAAAACCAGAG |
| PER2 | AGTGGGACTGGAAAATGCTG | CACACACAGAAGGAGGAGCA |
| TIMELESS | CACTGGCGTCATCATCAATC | GCAGGAGGAAGACAACTTGC |
| PER3 | TCTTTGGGTCCAGTTGTTCC | TCCTGGCGTCTTCTCACTTT |
| ACTIN | TAGCACAGCCTGGATAGCAA | AAATCTGGCACCACACCTTC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menéndez-Menéndez, J.; Hermida-Prado, F.; Granda-Díaz, R.; González, A.; García-Pedrero, J.M.; Del-Río-Ibisate, N.; González-González, A.; Cos, S.; Alonso-González, C.; Martínez-Campa, C. Deciphering the Molecular Basis of Melatonin Protective Effects on Breast Cells Treated with Doxorubicin: TWIST1 a Transcription Factor Involved in EMT and Metastasis, a Novel Target of Melatonin. Cancers 2019, 11, 1011. https://doi.org/10.3390/cancers11071011
Menéndez-Menéndez J, Hermida-Prado F, Granda-Díaz R, González A, García-Pedrero JM, Del-Río-Ibisate N, González-González A, Cos S, Alonso-González C, Martínez-Campa C. Deciphering the Molecular Basis of Melatonin Protective Effects on Breast Cells Treated with Doxorubicin: TWIST1 a Transcription Factor Involved in EMT and Metastasis, a Novel Target of Melatonin. Cancers. 2019; 11(7):1011. https://doi.org/10.3390/cancers11071011
Chicago/Turabian StyleMenéndez-Menéndez, Javier, Francisco Hermida-Prado, Rocío Granda-Díaz, Alicia González, Juana María García-Pedrero, Nagore Del-Río-Ibisate, Alicia González-González, Samuel Cos, Carolina Alonso-González, and Carlos Martínez-Campa. 2019. "Deciphering the Molecular Basis of Melatonin Protective Effects on Breast Cells Treated with Doxorubicin: TWIST1 a Transcription Factor Involved in EMT and Metastasis, a Novel Target of Melatonin" Cancers 11, no. 7: 1011. https://doi.org/10.3390/cancers11071011
APA StyleMenéndez-Menéndez, J., Hermida-Prado, F., Granda-Díaz, R., González, A., García-Pedrero, J. M., Del-Río-Ibisate, N., González-González, A., Cos, S., Alonso-González, C., & Martínez-Campa, C. (2019). Deciphering the Molecular Basis of Melatonin Protective Effects on Breast Cells Treated with Doxorubicin: TWIST1 a Transcription Factor Involved in EMT and Metastasis, a Novel Target of Melatonin. Cancers, 11(7), 1011. https://doi.org/10.3390/cancers11071011