Clinical Scenarios Emerging from Combined Immunophenotypic, Molecular and Morphologic Analysis of Pancreatic Cancer: The Good, the Bad and the Ugly Scenario

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Molecular Classification of Pancreatic Cancer
3. Morphology Matters: The Important Role of Tumor Budding
4. Heterogeneity of Immune Cell Composition in the TME of Pancreatic Cancer
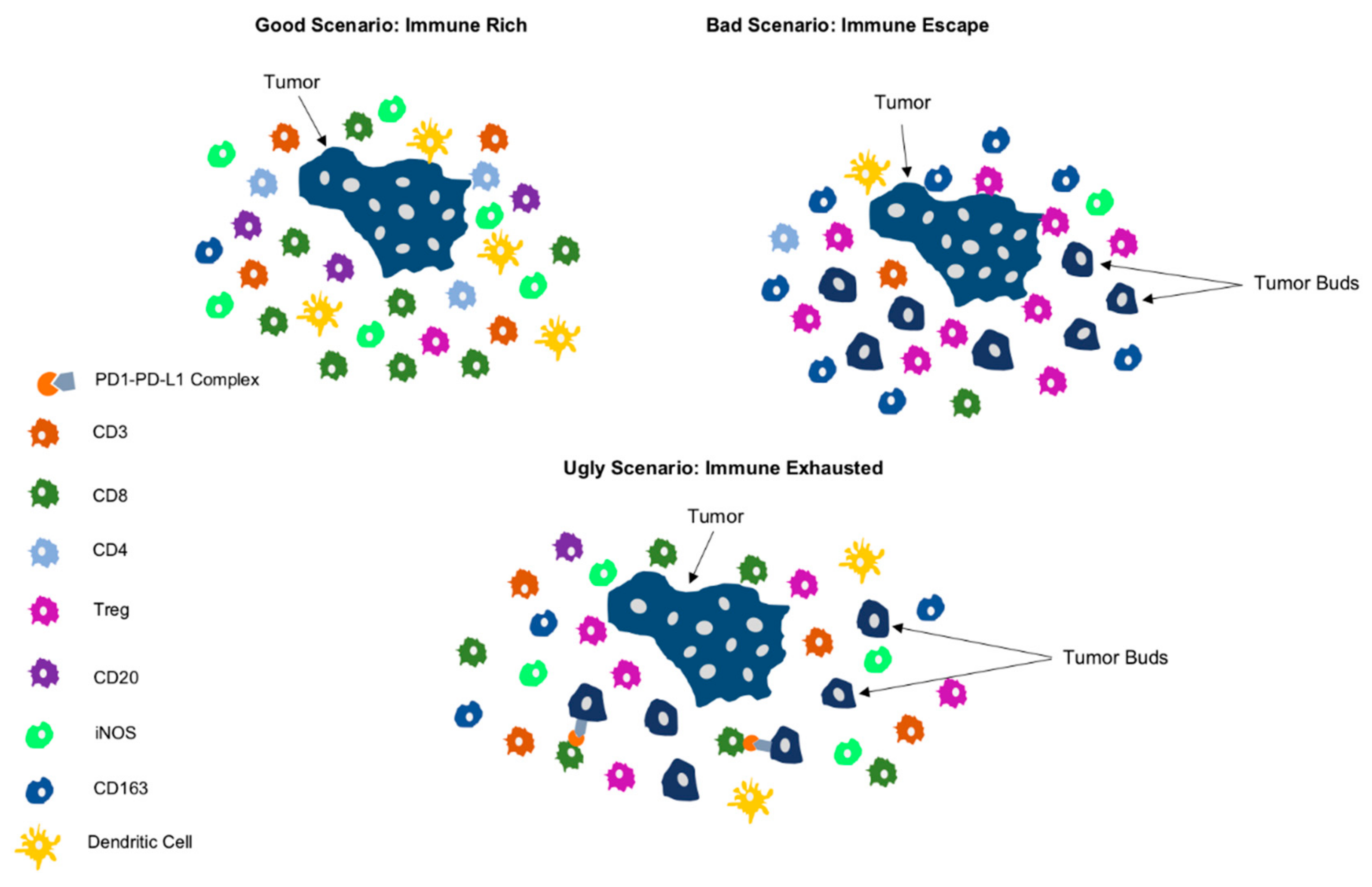
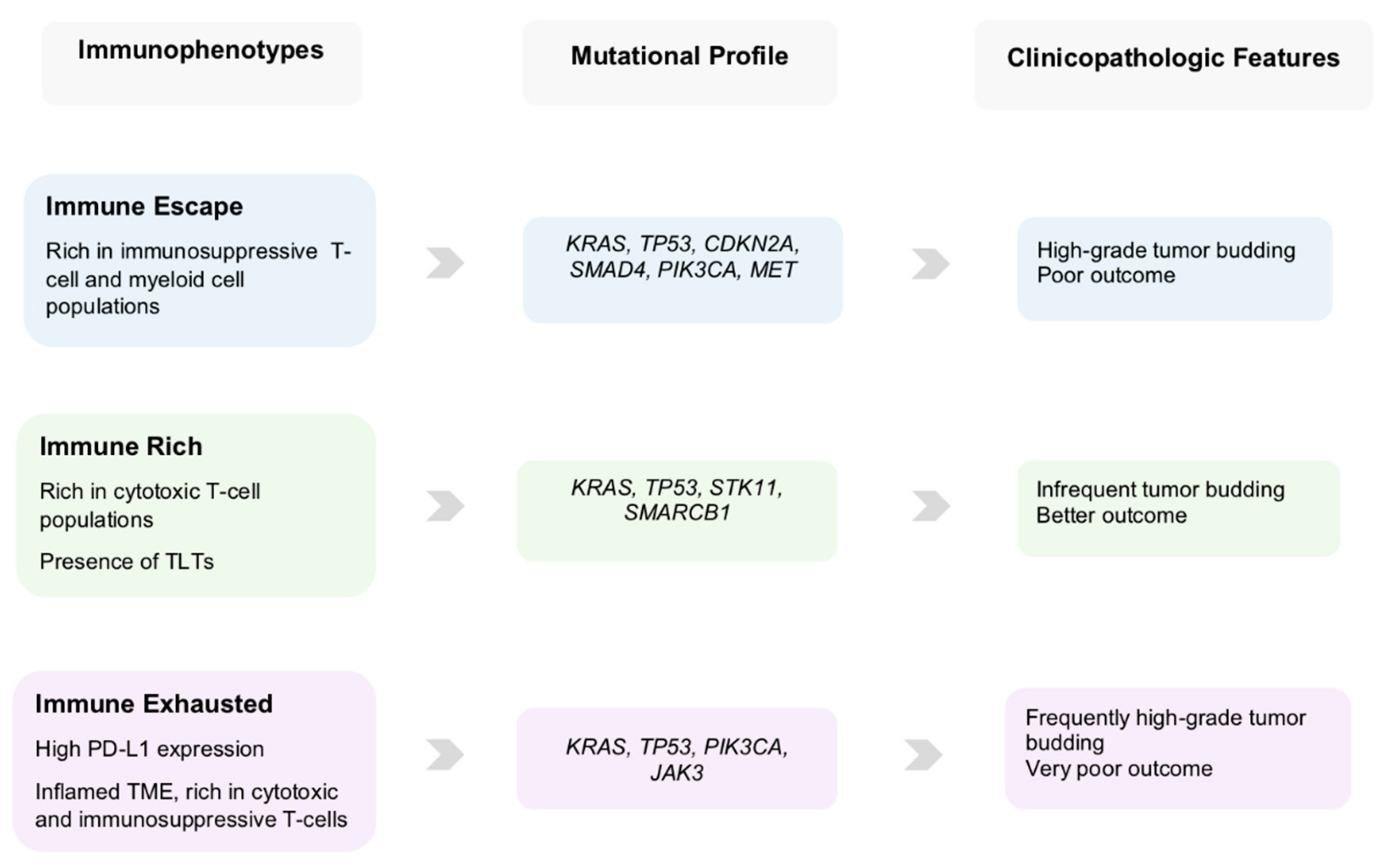
5. Clinical Scenarios
6. Emerging Opportunities and Challenges
7. Conclusions
Funding
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Goodman, A.M.; Piccioni, D.; Kato, S.; Boichard, A.; Wang, H.Y.; Frampton, G.; Lippman, S.M.; Connelly, C.; Fabrizio, D.; Miller, V.; et al. Prevalence of PDL1 Amplification and Preliminary Response to Immune Checkpoint Blockade in Solid Tumors. JAMA Oncol. 2018, 4, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Lupinacci, R.M.; Goloudina, A.; Buhard, O.; Bachet, J.B.; Maréchal, R.; Demetter, P.; Cros, J.; Bardier-Dupas, A.; Collura, A.; Cervera, P.; et al. Prevalence of Microsatellite Instability in Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2018, 154, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riquelme, E.; Maitra, A.; McAllister, F. Immunotherapy for Pancreatic Cancer: More Than Just a Gut Feeling. Cancer Discov. 2018, 8, 386–388. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, U.M.; Langhoff, E.; Goni, E.; Costello, E.; Greenhalf, W.; Halloran, C.; Ormanns, S.; Kruger, S.; Boeck, S.; Ribback, S.; et al. Immune Cell and Stromal Signature Associated with Progression-Free Survival of Patients with Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology 2018, 155, 1625–1639. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes with Outcomes of Patients with Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018, 4, e173420. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Maurer, H.C.; Holmstrom, S.R.; He, J.; Laise, P.; Su, T.; Ahmed, A.; Hibshoosh, H.; Chabot, J.A.; Oberstein, P.E.; Sepulveda, A.R.; et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 2019, 68, 1034–1043. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef]
- Karamitopoulou, E.; Zlobec, I.; Born, D.; Kondi-Pafiti, A.; Lykoudis, P.; Mellou, A.; Gennatas, K.; Gloor, B.; Lugli, A. Tumour budding is a strong and independent prognostic factor in pancreatic cancer. Eur. J. Cancer 2013, 49, 1032–1039. [Google Scholar] [CrossRef]
- Karamitopoulou, E.; Wartenberg, M.; Zlobec, I.; Cibin, S.; Worni, M.; Gloor, B.; Lugli, A. Tumour budding in pancreatic cancer revisited: Validation of the ITBCC scoring system. Histopathology 2018, 73, 137–146. [Google Scholar] [CrossRef]
- Kohler, I.; Bronsert, P.; Timme, S.; Werner, M.; Brabletz, T.; Hopt, U.T.; Schilling, O.; Bausch, D.; Keck, T.; Wellner, U.F. Detailed analysis of epithelial-mesenchymal transition and tumor budding identifies predictors of long-term survival in pancreatic ductal adenocarcinoma. J. Gastroenterol. Hepatol. 2015, 30, 78–84. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, K.; Li-Chang, H.H.; Kalloger, S.E.; Peixoto, R.D.; Webber, D.L.; Owen, D.A.; Driman, D.K.; Kirsch, R.; Serra, S.; Scudamore, C.H.; et al. Tumor budding is an independent adverse prognostic factor in pancreatic ductal adenocarcinoma. Am. J. Surg. Pathol. 2015, 4, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Lohneis, P.; Sinn, M.; Klein, F.; Bischoff, S.; Striefler, J.K.; Wislocka, L.; Sinn, B.V.; Pelzer, U.; Oettle, H.; Riess, H.; et al. Tumour buds determine prognosis in resected pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Galván, J.A.; Zlobec, I.; Wartenberg, M.; Lugli, A.; Gloor, B.; Perren, A.; Karamitopoulou, E. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br. J. Cancer 2015, 112, 1944–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigore, A.D.; Jolly, M.K.; Jia, D.; Farach-Carson, M.C.; Levine, H. Tumor budding: The name is EMT. Partial EMT. J. Clin. Med. 2016, 5, 51. [Google Scholar] [CrossRef]
- De Smedt, L.; Palmans, S.; Andel, D.; Govaere, O.; Boeckx, B.; Smeets, D.; Galle, E.; Wouters, J.; Barras, D.; Suffiotti, M. Expression profiling of budding cells in colorectal cancer reveals an EMT-like phenotype and molecular subtype switching. Br. J. Cancer 2017, 116, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Zlobec, I.; Lugli, A. Tumour budding in colorectal cancer: Molecular rationale for clinical translation. Nat. Rev. Cancer 2018, 18, 203–204. [Google Scholar] [CrossRef]
- Karamitopoulou, E.; Haemmig, S.; Baumgartner, U.; Schlup, C.; Wartenberg, M.; Vassella, E. MicroRNA dysregulation in the tumor microenvironment influences the phenotype of pancreatic cancer. Mod. Pathol. 2017, 30, 1116–1125. [Google Scholar] [CrossRef]
- Knudsen, K.N.; Lindebjerg, J.; Nielsen, B.S.; Hansen, T.F.; Sorensen, F.B. MicroRNA-200b is downregulated in colon cancer budding cells. PLoS ONE 2017, 12, e0178564. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M. Network of WNT and other regulatory signaling cascades in pluripotent stem cells and cancer stem cells. Curr. Pharm. Biotechnol. 2011, 12, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floor, S.; van Staveren, W.C.; Larsimont, D.; Dumont, J.E.; Maenhaut, C. Cancer cells in epithelial-to-mesenchymal transition and tumor-propagating-cancer stem cells: Distinct, overlapping or same populations. Oncogene 2011, 30, 4609–4621. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Vail, P.; Balaji, U.; Ngo, H.; Botros, I.W.; Makarov, V.; Riaz, N.; Balachandran, V.; Leach, S.; Thompson, D.M.; et al. Stratification of Pancreatic Ductal Adenocarcinoma: Combinatorial Genetic, Stromal, and Immunologic Markers. Clin. Cancer Res. 2017, 23, 4429–4440. [Google Scholar] [CrossRef]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef]
- Attri, K.S.; Mehla, K.; Singh, P.K. Evaluation of Macrophage Polarization in Pancreatic Cancer Microenvironment Under Hypoxia. Methods Mol. Biol. 2018, 1742, 265–276. [Google Scholar]
- Wartenberg, M.; Cibin, S.; Zlobec, I.; Vassella, E.; Eppenberger-Castori, S.M.M.; Terracciano, L.; Worni, M.; Gloor, B.; Perren, A.; Karamitopoulou, E. Integrated genomic and immunophenotypic classification of pancreatic cancer reveals three distinct subtypes with prognostic/predictive significance. Clin. Cancer Res. 2018, 24, 4444–4454. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castino, G.F.; Cortese, N.; Capretti, G.; Serio, S.; Di Caro, G.; Mineri, R.; Magrini, E.; Grizzi, F.; Cappello, P.; Novelli, F.; et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology 2016, 5, e1085147. [Google Scholar] [CrossRef] [PubMed]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yelensky, R.; Jooss, K.; Chan, T.A. Update on Tumor Neoantigens and Their Utility: Why It Is Good to Be Different. Trends Immunol. 2018, 39, 536–548. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Federico, L.; Li, Y.; Dai, H.; Lu, Y.; Mills, G.B.; Yi, S.; Lin, S.Y.; Sahni, N. Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat. Commun. 2018, 9, 1317. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures with Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef]
- Wartenberg, M.; Zlobec, I.; Perren, A.; Koelzer, V.H.; Lugli, A.; Karamitopoulou, E. Accumulation of FOXP3+T-cells in the tumor microenvironment is associated with an epithelial-mesenchymal-transition-type tumor budding phenotype and is an independent prognostic factor in surgically resected pancreatic ductal adenocarcinoma. Oncotarget 2015, 6, 4190–4201. [Google Scholar] [CrossRef] [Green Version]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef]
- Tamborero, D.; Rubio-Perez, C.; Muiños, F.; Sabarinathan, R.; Piulats, J.M.; Muntasell, A.; Dienstmann, R.; Lopez-Bigas, N.; Gonzalez-Perez, A. A Pan-cancer Landscape of Interactions between Solid Tumors and Infiltrating Immune Cell Populations. Clin. Cancer Res. 2018, 24, 3717–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danilova, L.; Ho, W.J.; Zhu, Q.; Vithayathil, T.T.; De Jesus-Acosta, A.; Azad, N.S.; Laheru, D.A.; Fertig, E.J.; Anders, R.; Jaffee, E.M.; et al. Programmed cell death ligand-1 (PD-L1) and CD8 expression profiling identify an immunologic subtype of pancreatic ductal adenocarcinomas with favorable survival. Cancer Immunol. Res. 2019, 7, 886–895. [Google Scholar] [CrossRef]
- De Silva, D.M.; Roy, A.; Kato, T.; Cecchi, F.; Lee, Y.H.; Matsumoto, K.; Bottaro, D.P. Targeting the hepatocyte growth factor/Met pathway in cancer. Biochem. Soc. Trans. 2017, 45, 855–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, I.H.; Lowery, M.A.; Stadler, Z.K.; Salo-Mullen, E.; Iacobuzio-Donahue, C.A.; Kelsen, D.P.; O’Reilly, E.M. Genomic instability in pancreatic adenocarcinoma: A new step towards precision medicine and novel therapeutic approaches. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. The immune revolution: A case for priming, not checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the impact of immunotherapy to pancreatic cancer: Challenges and opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karamitopoulou, E.; Gloor, B. Clinical Scenarios Emerging from Combined Immunophenotypic, Molecular and Morphologic Analysis of Pancreatic Cancer: The Good, the Bad and the Ugly Scenario. Cancers 2019, 11, 968. https://doi.org/10.3390/cancers11070968
Karamitopoulou E, Gloor B. Clinical Scenarios Emerging from Combined Immunophenotypic, Molecular and Morphologic Analysis of Pancreatic Cancer: The Good, the Bad and the Ugly Scenario. Cancers. 2019; 11(7):968. https://doi.org/10.3390/cancers11070968
Chicago/Turabian StyleKaramitopoulou, Eva, and Beat Gloor. 2019. "Clinical Scenarios Emerging from Combined Immunophenotypic, Molecular and Morphologic Analysis of Pancreatic Cancer: The Good, the Bad and the Ugly Scenario" Cancers 11, no. 7: 968. https://doi.org/10.3390/cancers11070968
APA StyleKaramitopoulou, E., & Gloor, B. (2019). Clinical Scenarios Emerging from Combined Immunophenotypic, Molecular and Morphologic Analysis of Pancreatic Cancer: The Good, the Bad and the Ugly Scenario. Cancers, 11(7), 968. https://doi.org/10.3390/cancers11070968