Context-Dependent Role of NF-κB Signaling in Primary Liver Cancer—from Tumor Development to Therapeutic Implications



{kind=link}
Abstract
:1. Introduction
2. NF-κB Signaling in Liver Homeostasis, Inflammation and Fibrogenesis
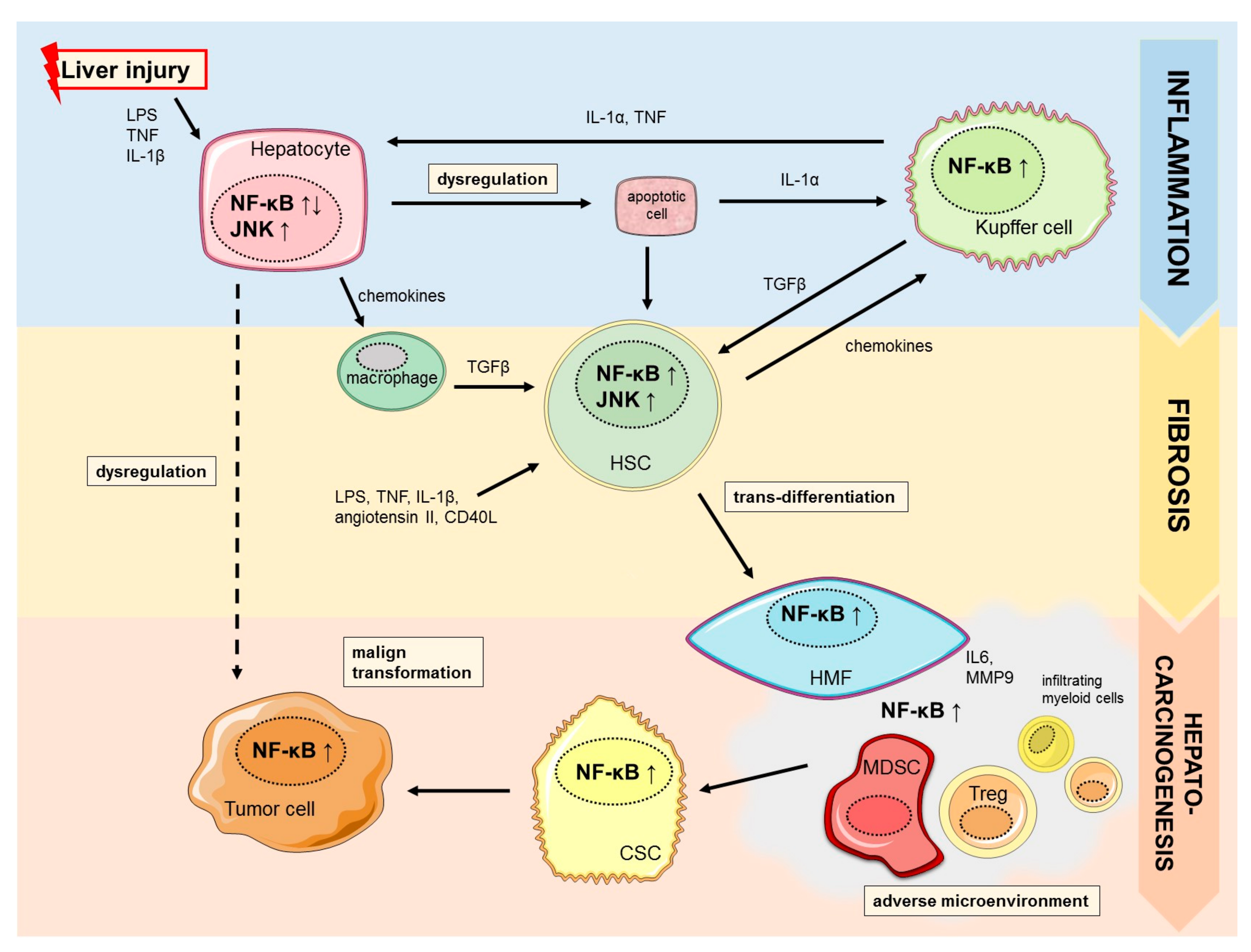
2.1. Hepato-Protective Effects of NF-κB
2.2. NF-κB in Liver Fibrogenesis
3. NF-κB in Hepatocarcinogenesis
3.1. NF-κB in Cancer Stemness
3.2. Immune Cells and NF-κB Signaling
4. NF-κB as a Target for Cancer Prevention and Therapy
5. NF-κB Signaling and Cholangiocarcinoma
Targeting NF-κB in Cholangiocarcinoma
6. Outlook
Funding
Conflicts of Interest
References
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Galle, P.R.; Teufel, A. Molecular diagnosis and therapy of hepatocellular carcinoma (HCC): An emerging field for advanced technologies. J. Hepatol. 2012, 56, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiang, Y.; Yuan, H.; Fang, Q.; Cai, N.; Suo, C.; Jin, L.; Zhang, T.; Chen, X. The trends in incidence of primary liver cancer caused by specific etiologies: Results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J. Hepatol. 2019, 70, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer risk: Role of environment-response. Science 2015, 347, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Karin, M. The Fire within: Cell-Autonomous Mechanisms in Inflammation-Driven Cancer. Cancer Cell 2019, 35, 714–720. [Google Scholar] [CrossRef]
- Seton-Rogers, S. Field effect. Nat. Rev. Cancer 2012, 12, 508–509. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-kappaB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.J.; Lerner, J.; et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Castven, D.; Fischer, M.; Becker, D.; Heinrich, S.; Andersen, J.B.; Strand, D.; Sprinzl, M.F.; Strand, S.; Czauderna, C.; Heilmann-Heimbach, S.; et al. Adverse genomic alterations and stemness features are induced by field cancerization in the microenvironment of hepatocellular carcinomas. Oncotarget 2017, 8, 48688–48700. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef]
- Elsharkawy, A.M.; Mann, D.A. Nuclear factor-kappaB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 2007, 46, 590–597. [Google Scholar] [CrossRef]
- Li, Z.W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. IkappaB kinases: Key regulators of the NF-kappaB pathway. Trends Biochem. Sci. 2004, 29, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Baltimore, D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Fuentes, M.E.; Yamaguchi, K.; Durnin, M.H.; Dalrymple, S.A.; Hardy, K.L.; Goeddel, D.V. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity 1999, 10, 421–429. [Google Scholar] [CrossRef]
- Li, Q.; Van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, M.; Metcalf, D.; Merryfull, J.; Beg, A.; Baltimore, D.; Gerondakis, S. The combined absence of the transcription factors Rel and RelA leads to multiple hemopoietic cell defects. Proc. Natl. Acad. Sci. USA 1999, 96, 11848–11853. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, D.; Yeh, W.C.; Wakeham, A.; Rudolph, B.; Nallainathan, D.; Potter, J.; Elia, A.J.; Mak, T.W. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000, 14, 854–862. [Google Scholar]
- Luedde, T.; Trautwein, C. Intracellular survival pathways in the liver. Liver Int. 2006, 26, 1163–1174. [Google Scholar] [CrossRef]
- Luedde, T.; Heinrichsdorff, J.; de Lorenzi, R.; De Vos, R.; Roskams, T.; Pasparakis, M. IKK1 and IKK2 cooperate to maintain bile duct integrity in the liver. Proc. Natl. Acad. Sci. USA 2008, 105, 9733–9738. [Google Scholar] [CrossRef] [Green Version]
- Heinrichsdorff, J.; Luedde, T.; Perdiguero, E.; Nebreda, A.R.; Pasparakis, M. p38 alpha MAPK inhibits JNK activation and collaborates with IkappaB kinase 2 to prevent endotoxin-induced liver failure. EMBO Rep. 2008, 9, 1048–1054. [Google Scholar] [CrossRef]
- Tang, G.; Minemoto, Y.; Dibling, B.; Purcell, N.H.; Li, Z.; Karin, M.; Lin, A. Inhibition of JNK activation through NF-kappaB target genes. Nature 2001, 414, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Ehlken, H.; Krishna-Subramanian, S.; Ochoa-Callejero, L.; Kondylis, V.; Nadi, N.E.; Straub, B.K.; Schirmacher, P.; Walczak, H.; Kollias, G.; Pasparakis, M. Death receptor-independent FADD signalling triggers hepatitis and hepatocellular carcinoma in mice with liver parenchymal cell-specific NEMO knockout. Cell Death Differ. 2014, 21, 1721–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubero, F.J.; Singh, A.; Borkham-Kamphorst, E.; Nevzorova, Y.A.; Al Masaoudi, M.; Haas, U.; Boekschoten, M.V.; Gassler, N.; Weiskirchen, R.; Muller, M.; et al. TNFR1 determines progression of chronic liver injury in the IKKgamma/Nemo genetic model. Cell Death Differ. 2013, 20, 1580–1592. [Google Scholar] [CrossRef]
- Liedtke, C.; Bangen, J.M.; Freimuth, J.; Beraza, N.; Lambertz, D.; Cubero, F.J.; Hatting, M.; Karlmark, K.R.; Streetz, K.L.; Krombach, G.A.; et al. Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology 2011, 141, 2176–2187. [Google Scholar] [CrossRef] [PubMed]
- Kondylis, V.; Polykratis, A.; Ehlken, H.; Ochoa-Callejero, L.; Straub, B.K.; Krishna-Subramanian, S.; Van, T.M.; Curth, H.M.; Heise, N.; Weih, F.; et al. NEMO Prevents Steatohepatitis and Hepatocellular Carcinoma by Inhibiting RIPK1 Kinase Activity-Mediated Hepatocyte Apoptosis. Cancer Cell 2015, 28, 582–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsin, I.F.; Montano, E.; Seki, E. Finding a new role for NEMO: A key player in preventing hepatocyte apoptosis and liver tumorigenesis by inhibiting RIPK1. Hepatology 2016, 64, 295–297. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How useful are they? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef]
- Njei, B.; Rotman, Y.; Ditah, I.; Lim, J.K. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015, 61, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, I.; Sutti, S.; Vacchiano, M.; Bozzola, C.; Albano, E. NF-kappaB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clin. Sci. 2013, 124, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bullard, J.; Kalra, M.; Assefa, S.; Kaul, A.K.; Vonfeldt, K.; Strom, S.C.; Conrad, R.S.; Sharp, H.L.; Kaul, R. Status of bacterial colonization, Toll-like receptor expression and nuclear factor-kappa B activation in normal and diseased human livers. Clin. Immunol. 2011, 138, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Leithauser, F.; Gul, S.; Fiedler, K.; Guldiken, N.; Espenlaub, S.; Holzmann, K.H.; Hipp, N.; Sindrilaru, A.; Luedde, T.; et al. Hepatic activation of IKK/NFkappaB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology 2012, 56, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Son, G.; Iimuro, Y.; Seki, E.; Hirano, T.; Kaneda, Y.; Fujimoto, J. Selective inactivation of NF-kappaB in the liver using NF-kappaB decoy suppresses CCl4-induced liver injury and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G631–G639. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Wang, B.B.; Cheng, J.Y.; Gao, H.H.; Zhang, Y.; Chen, Z.N.; Bian, H. Hepatic stellate cells in inflammation-fibrosis-carcinoma axis. Anat. Rec. 2010, 293, 1492–1496. [Google Scholar] [CrossRef]
- Kluwe, J.; Pradere, J.P.; Gwak, G.Y.; Mencin, A.; De Minicis, S.; Osterreicher, C.H.; Colmenero, J.; Bataller, R.; Schwabe, R.F. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 2010, 138, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Moles, A.; Sanchez, A.M.; Banks, P.S.; Murphy, L.B.; Luli, S.; Borthwick, L.; Fisher, A.; O’Reilly, S.; van Laar, J.M.; White, S.A.; et al. Inhibition of RelA-Ser536 phosphorylation by a competing peptide reduces mouse liver fibrosis without blocking the innate immune response. Hepatology 2013, 57, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Oakley, F.; Meso, M.; Iredale, J.P.; Green, K.; Marek, C.J.; Zhou, X.; May, M.J.; Millward-Sadler, H.; Wright, M.C.; Mann, D.A. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology 2005, 128, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 2006, 103, 10544–10551. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Aigelsreiter, A.; Haybaeck, J.; Schauer, S.; Kiesslich, T.; Bettermann, K.; Griessbacher, A.; Stojakovic, T.; Bauernhofer, T.; Samonigg, H.; Kornprat, P.; et al. NEMO expression in human hepatocellular carcinoma and its association with clinical outcome. Hum. Pathol. 2012, 43, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Dong, X.; Zhang, D.; Chen, X.; Zhu, X. High expression of Snail and NF-kappaB predicts poor survival in Chinese hepatocellular carcinoma patients. Oncotarget 2017, 8, 4543–4548. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Gerstenlauer, M.; Chan, L.K.; Leithauser, F.; Yeh, M.M.; Wirth, T.; Maier, H.J. Block of NF-kB signaling accelerates MYC-driven hepatocellular carcinogenesis and modifies the tumor phenotype towards combined hepatocellular cholangiocarcinoma. Cancer Lett. 2019, 458, 113–122. [Google Scholar] [CrossRef]
- Schneider, A.T.; Gautheron, J.; Feoktistova, M.; Roderburg, C.; Loosen, S.H.; Roy, S.; Benz, F.; Schemmer, P.; Buchler, M.W.; Nachbur, U.; et al. RIPK1 Suppresses a TRAF2-Dependent Pathway to Liver Cancer. Cancer Cell 2017, 31, 94–109. [Google Scholar] [CrossRef]
- Sunami, Y.; Ringelhan, M.; Kokai, E.; Lu, M.; O’Connor, T.; Lorentzen, A.; Weber, A.; Rodewald, A.K.; Mullhaupt, B.; Terracciano, L.; et al. Canonical NF-kappaB signaling in hepatocytes acts as a tumor-suppressor in hepatitis B virus surface antigen-driven hepatocellular carcinoma by controlling the unfolded protein response. Hepatology 2016, 63, 1592–1607. [Google Scholar] [CrossRef] [PubMed]
- Bettermann, K.; Vucur, M.; Haybaeck, J.; Koppe, C.; Janssen, J.; Heymann, F.; Weber, A.; Weiskirchen, R.; Liedtke, C.; Gassler, N.; et al. TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell 2010, 17, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Haybaeck, J.; Zeller, N.; Wolf, M.J.; Weber, A.; Wagner, U.; Kurrer, M.O.; Bremer, J.; Iezzi, G.; Graf, R.; Clavien, P.A.; et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell 2009, 16, 295–308. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Karin, M. NF-kappaB and STAT3-key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Finkin, S.; Yuan, D.; Stein, I.; Taniguchi, K.; Weber, A.; Unger, K.; Browning, J.L.; Goossens, N.; Nakagawa, S.; Gunasekaran, G.; et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 2015, 16, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, J.; Stein, I.; Haag, D.; Riehl, A.; Longerich, T.; Horwitz, E.; Breuhahn, K.; Gebhardt, C.; Schirmacher, P.; Hahn, M.; et al. S100A8 and S100A9 are novel nuclear factor kappa B target genes during malignant progression of murine and human liver carcinogenesis. Hepatology 2009, 50, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.S.; Park, S.; Park, S.H.; Park, E.R.; Cha, P.H.; Kim, B.Y.; Chung, Y.M.; Woo, S.R.; Han, C.J.; Kim, S.B.; et al. Overexpression of Romo1 promotes production of reactive oxygen species and invasiveness of hepatic tumor cells. Gastroenterology 2012, 143, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, Y.H.; Chung, J.S.; Yoo, Y.D. Romo1 and the NF-kappaB pathway are involved in oxidative stress-induced tumor cell invasion. Int. J. Oncol. 2015, 46, 2021–2028. [Google Scholar] [CrossRef]
- Wang, F.; Yang, J.L.; Yu, K.K.; Xu, M.; Xu, Y.Z.; Chen, L.; Lu, Y.M.; Fang, H.S.; Wang, X.Y.; Hu, Z.Q.; et al. Activation of the NF-kappaB pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol. Cancer 2015, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Zhang, B.; Hua, R.; Tai, W.C.; Zeng, Z.; Xie, B.; Huang, C.; Xue, J.; Xiong, S.; Yang, J.; et al. URG4/URGCP enhances the angiogenic capacity of human hepatocellular carcinoma cells in vitro via activation of the NF-kappaB signaling pathway. BMC Cancer 2015, 15, 368. [Google Scholar] [CrossRef] [PubMed]
- Iyer, J.K.; Kalra, M.; Kaul, A.; Payton, M.E.; Kaul, R. Estrogen receptor expression in chronic hepatitis C and hepatocellular carcinoma pathogenesis. World J. Gastroenterol. 2017, 23, 6802–6816. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Fan, X.; Jing, W.; Liang, Y.; Chen, R.; Liu, Y.; Zhu, M.; Jia, R.; Wang, H.; Zhang, X.; et al. Osteopontin promotes a cancer stem cell-like phenotype in hepatocellular carcinoma cells via an integrin-NF-kappaB-HIF-1alpha pathway. Oncotarget 2015, 6, 6627–6640. [Google Scholar] [CrossRef] [PubMed]
- You, N.; Zheng, L.; Liu, W.; Zhong, X.; Wang, W.; Li, J. Proliferation inhibition and differentiation induction of hepatic cancer stem cells by knockdown of BC047440: A potential therapeutic target of stem cell treatment for hepatocellular carcinoma. Oncol. Rep. 2014, 31, 1911–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; You, N.; Huang, X.; Gu, H.; Wu, K.; Mi, N.; Li, J. COMMD7 Regulates NF-kappaB Signaling Pathway in Hepatocellular Carcinoma Stem-like Cells. Mol. Ther. Oncolytics 2019, 12, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Castven, D.; Czauderna, C.; Marquardt, J.U. Contribution of the Cancer Stem Cell Phenotype to Hepatocellular Carcinoma Resistance. In Resistance to Molecular Therapies for Hepatocellular Carcinoma; Villanueva, A., Ed.; Springer International Publishing: Cham, Switzerland, 2017; Volume 13, pp. 65–91. [Google Scholar] [CrossRef]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Gomez-Quiroz, L.; Arreguin Camacho, L.O.; Pinna, F.; Lee, Y.H.; Kitade, M.; Dominguez, M.P.; Castven, D.; Breuhahn, K.; Conner, E.A.; et al. Curcumin effectively inhibits oncogenic NF-kappaB signaling and restrains stemness features in liver cancer. J. Hepatol. 2015, 63, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Fusilli, C.; Rappa, F.; Van Haele, M.; Douet, J.; Pindjakova, J.; Rocha, S.W.; Pata, I.; Valcikova, B.; Uldrijan, S.; et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology 2017. [Google Scholar] [CrossRef]
- Zhao, J.; Dong, L.; Lu, B.; Wu, G.; Xu, D.; Chen, J.; Li, K.; Tong, X.; Dai, J.; Yao, S.; et al. Down-regulation of osteopontin suppresses growth and metastasis of hepatocellular carcinoma via induction of apoptosis. Gastroenterology 2008, 135, 956–968. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.; Czauderna, C.; Marquardt, J.U. Immunotherapy of Hepatocellular Carcinoma. Oncol. Res. Treat. 2018, 41, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Thimme, R. Role of Immunity in Pathogenesis and Treatment of Hepatocellular Carcinoma. Dig. Dis. 2016, 34, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, J.; Lichtenheld, M.G.; Meadows, G.G. A role for NF-kappa B activation in perforin expression of NK cells upon IL-2 receptor signaling. J. Immunol. 2002, 169, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Bi, E.; Hu, Y.; Deng, W.; Tian, Z.; Dong, C.; Hu, Y.; Sun, B. A novel NF-kappaB binding site controls human granzyme B gene transcription. J. Immunol. 2006, 176, 4173–4181. [Google Scholar] [CrossRef] [PubMed]
- Evaristo, C.; Spranger, S.; Barnes, S.E.; Miller, M.L.; Molinero, L.L.; Locke, F.L.; Gajewski, T.F.; Alegre, M.L. Cutting Edge: Engineering Active IKKbeta in T Cells Drives Tumor Rejection. J. Immunol. 2016, 196, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Karyampudi, L.; Lamichhane, P.; Krempski, J.; Kalli, K.R.; Behrens, M.D.; Vargas, D.M.; Hartmann, L.C.; Janco, J.M.; Dong, H.; Hedin, K.E.; et al. PD-1 Blunts the Function of Ovarian Tumor-Infiltrating Dendritic Cells by Inactivating NF-kappaB. Cancer Res. 2016, 76, 239–250. [Google Scholar] [CrossRef]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NF-kappaB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 2017, 47, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-kappaB c-Rel is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Chiang, I.T.; Liu, Y.C.; Wang, W.H.; Hsu, F.T.; Chen, H.W.; Lin, W.J.; Chang, W.Y.; Hwang, J.J. Sorafenib inhibits TPA-induced MMP-9 and VEGF expression via suppression of ERK/NF-kappaB pathway in hepatocellular carcinoma cells. In Vivo 2012, 26, 671–681. [Google Scholar] [PubMed]
- Hsu, F.T.; Liu, Y.C.; Chiang, I.T.; Liu, R.S.; Wang, H.E.; Lin, W.J.; Hwang, J.J. Sorafenib increases efficacy of vorinostat against human hepatocellular carcinoma through transduction inhibition of vorinostat-induced ERK/NF-kappaB signaling. Int. J. Oncol. 2014, 45, 177–188. [Google Scholar] [CrossRef]
- Tsai, J.J.; Pan, P.J.; Hsu, F.T. Regorafenib induces extrinsic and intrinsic apoptosis through inhibition of ERK/NF-kappaB activation in hepatocellular carcinoma cells. Oncol. Rep. 2017, 37, 1036–1044. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Meng, L.; Liu, K.; Ji, B. Targeting the PD-L1/DNMT1 axis in acquired resistance to sorafenib in human hepatocellular carcinoma. Oncol. Rep. 2017, 38, 899–907. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, E.; Khosroushahi, A.Y.; Eftekhari, A.; Farajnia, S.; Babaei, H.; Eghbal, M.A. Novel angiotensin receptor blocker, azilsartan induces oxidative stress and NFkB-mediated apoptosis in hepatocellular carcinoma cell line HepG2. Biomed. Pharmacother. 2018, 99, 939–946. [Google Scholar] [CrossRef]
- Hsieh, S.C.; Tsai, J.P.; Yang, S.F.; Tang, M.J.; Hsieh, Y.H. Metformin inhibits the invasion of human hepatocellular carcinoma cells and enhances the chemosensitivity to sorafenib through a downregulation of the ERK/JNK-mediated NF-kappaB-dependent pathway that reduces uPA and MMP-9 expression. Amino Acids 2014, 46, 2809–2822. [Google Scholar] [CrossRef]
- Saber, S.; Mahmoud, A.A.A.; Goda, R.; Helal, N.S.; El-Ahwany, E.; Abdelghany, R.H. Perindopril, fosinopril and losartan inhibited the progression of diethylnitrosamine-induced hepatocellular carcinoma in mice via the inactivation of nuclear transcription factor kappa-B. Toxicol. Lett. 2018, 295, 32–40. [Google Scholar] [CrossRef]
- Mehmood, T.; Maryam, A.; Zhang, H.; Li, Y.; Khan, M.; Ma, T. Deoxyelephantopin induces apoptosis in HepG2 cells via oxidative stress, NF-kappaB inhibition and mitochondrial dysfunction. Biofactors 2017, 43, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pan, C.; Zhao, S.; Wang, Z.; Zhang, H.; Wu, W. Resveratrol inhibits tumor necrosis factor-alpha-mediated matrix metalloproteinase-9 expression and invasion of human hepatocellular carcinoma cells. Biomed. Pharmacother. 2008, 62, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Li, Z.; Li, Y.; Zhang, W.; Han, X. Sulforaphene enhances radiosensitivity of hepatocellular carcinoma through suppression of the NF-kappaB pathway. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.T.; Liu, Y.C.; Liu, T.T.; Hwang, J.J. Curcumin Sensitizes Hepatocellular Carcinoma Cells to Radiation via Suppression of Radiation-Induced NF-kappaB Activity. Biomed. Res. Int. 2015, 2015, 363671. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D.; Ishikawa, H.; Matsumoto, K.; Shibata, H.; Motoyoshi, Y.; Fukuta, M.; Kawashimo, H.; Goto, T.; Taura, N.; Ichikawa, T.; et al. DHMEQ, a novel NF-kappaB inhibitor, induces apoptosis and cell-cycle arrest in human hepatoma cells. Int. J. Oncol. 2006, 29, 713–719. [Google Scholar] [PubMed]
- Poma, P.; Notarbartolo, M.; Labbozzetta, M.; Sanguedolce, R.; Alaimo, A.; Carina, V.; Maurici, A.; Cusimano, A.; Cervello, M.; D’Alessandro, N. Antitumor effects of the novel NF-kappaB inhibitor dehydroxymethyl-epoxyquinomicin on human hepatic cancer cells: Analysis of synergy with cisplatin and of possible correlation with inhibition of pro-survival genes and IL-6 production. Int. J. Oncol. 2006, 28, 923–930. [Google Scholar] [PubMed]
- Lampiasi, N.; Azzolina, A.; D’Alessandro, N.; Umezawa, K.; McCubrey, J.A.; Montalto, G.; Cervello, M. Antitumor effects of dehydroxymethylepoxyquinomicin, a novel nuclear factor-kappaB inhibitor, in human liver cancer cells are mediated through a reactive oxygen species-dependent mechanism. Mol. Pharmacol. 2009, 76, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Lampiasi, N.; Azzolina, A.; Umezawa, K.; Montalto, G.; McCubrey, J.A.; Cervello, M. The novel NF-kappaB inhibitor DHMEQ synergizes with celecoxib to exert antitumor effects on human liver cancer cells by a ROS-dependent mechanism. Cancer Lett. 2012, 322, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Haruki, K.; Shiba, H.; Fujiwara, Y.; Furukawa, K.; Iwase, R.; Uwagawa, T.; Misawa, T.; Ohashi, T.; Yanaga, K. Inhibition of nuclear factor-kappaB enhances the antitumor effect of tumor necrosis factor-alpha gene therapy for hepatocellular carcinoma in mice. Surgery 2013, 154, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Aishima, S.; Oda, Y. Pathogenesis and classification of intrahepatic cholangiocarcinoma: Different characters of perihilar large duct type versus peripheral small duct type. J. Hepato-Biliary-Pancreat. Sci. 2015, 22, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Thorgeirsson, S.S. Genetic profiling of intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2012, 28, 266–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012, 142, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, Y.; Kokuryo, T.; Tsunoda, N.; Yamazaki, Y.; Oda, K.; Nimura, Y.; Naing Mon, N.; Huang, P.; Nakanuma, Y.; Chen, M.F.; et al. Tumor necrosis factor alpha promotes invasiveness of cholangiocarcinoma cells via its receptor, TNFR2. Cancer Lett. 2005, 219, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Seubwai, W.; Wongkham, C.; Puapairoj, A.; Khuntikeo, N.; Pugkhem, A.; Hahnvajanawong, C.; Chaiyagool, J.; Umezawa, K.; Okada, S.; Wongkham, S. Aberrant expression of NF-kappaB in liver fluke associated cholangiocarcinoma: Implications for targeted therapy. PLoS ONE 2014, 9, e106056. [Google Scholar] [CrossRef]
- Liu, B.; Yan, S.; Jia, Y.; Ma, J.; Wu, S.; Xu, Y.; Shang, M.; Mao, A. TLR2 promotes human intrahepatic cholangiocarcinoma cell migration and invasion by modulating NF-kappaB pathway-mediated inflammatory responses. FEBS J. 2016, 283, 3839–3850. [Google Scholar] [CrossRef]
- Phoomak, C.; Silsirivanit, A.; Wongkham, C.; Sripa, B.; Puapairoj, A.; Wongkham, S. Overexpression of O-GlcNAc-transferase associates with aggressiveness of mass-forming cholangiocarcinoma. Asian Pac. J. Cancer Prev. 2012, 13 (Suppl. 1), 101–105. [Google Scholar]
- Phoomak, C.; Vaeteewoottacharn, K.; Sawanyawisuth, K.; Seubwai, W.; Wongkham, C.; Silsirivanit, A.; Wongkham, S. Mechanistic insights of O-GlcNAcylation that promote progression of cholangiocarcinoma cells via nuclear translocation of NF-kappaB. Sci. Rep. 2016, 6, 27853. [Google Scholar] [CrossRef]
- Oishi, N.; Kumar, M.R.; Roessler, S.; Ji, J.; Forgues, M.; Budhu, A.; Zhao, X.; Andersen, J.B.; Ye, Q.H.; Jia, H.L.; et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology 2012, 56, 1792–1803. [Google Scholar] [CrossRef]
- Prakobwong, S.; Gupta, S.C.; Kim, J.H.; Sung, B.; Pinlaor, P.; Hiraku, Y.; Wongkham, S.; Sripa, B.; Pinlaor, S.; Aggarwal, B.B. Curcumin suppresses proliferation and induces apoptosis in human biliary cancer cells through modulation of multiple cell signaling pathways. Carcinogenesis 2011, 32, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.L.; Liang, Y.J.; Zheng, T.S.; Song, R.P.; Wang, J.B.; Sun, B.S.; Pan, S.H.; Qu, L.D.; Liu, J.R.; Jiang, H.C.; et al. EF24 inhibits tumor growth and metastasis via suppressing NF-kappaB dependent pathways in human cholangiocarcinoma. Sci. Rep. 2016, 6, 32167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.Y.; Wang, X.; Zheng, S.S.; Gao, X.M.; Jia, Q.A.; Zhu, W.W.; Lu, L.; Jia, H.L.; Chen, J.H.; Dong, Q.Z.; et al. RA190, a Proteasome Subunit ADRM1 Inhibitor, Suppresses Intrahepatic Cholangiocarcinoma by Inducing NF-KB-Mediated Cell Apoptosis. Cell. Physiol. Biochem. 2018, 47, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.C.; Galle, P.R.; Marquardt, J.U. The role of molecular enrichment on future therapies in hepatocellular carcinoma. J. Hepatol. 2018, 69, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Saborowski, A.; Czauderna, C.; Vogel, A. The Changing Landscape of Systemic Treatment of Advanced Hepatocellular Carcinoma: New Targeted Agents and Immunotherapies. Target. Oncol. 2019, 14, 115–123. [Google Scholar] [CrossRef]
- Matter, M.S.; Marquardt, J.U.; Andersen, J.B.; Quintavalle, C.; Korokhov, N.; Stauffer, J.K.; Kaji, K.; Decaens, T.; Quagliata, L.; Elloumi, F.; et al. Oncogenic driver genes and the inflammatory microenvironment dictate liver tumor phenotype. Hepatology 2016, 63, 1888–1899. [Google Scholar] [CrossRef]
- Wu, J.; Ding, J.; Yang, J.; Guo, X.; Zheng, Y. MicroRNA Roles in the Nuclear Factor Kappa B Signaling Pathway in Cancer. Front. Immunol. 2018, 9, 546. [Google Scholar] [CrossRef] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czauderna, C.; Castven, D.; Mahn, F.L.; Marquardt, J.U. Context-Dependent Role of NF-κB Signaling in Primary Liver Cancer—from Tumor Development to Therapeutic Implications. Cancers 2019, 11, 1053. https://doi.org/10.3390/cancers11081053
Czauderna C, Castven D, Mahn FL, Marquardt JU. Context-Dependent Role of NF-κB Signaling in Primary Liver Cancer—from Tumor Development to Therapeutic Implications. Cancers. 2019; 11(8):1053. https://doi.org/10.3390/cancers11081053
Chicago/Turabian StyleCzauderna, Carolin, Darko Castven, Friederike L. Mahn, and Jens U. Marquardt. 2019. "Context-Dependent Role of NF-κB Signaling in Primary Liver Cancer—from Tumor Development to Therapeutic Implications" Cancers 11, no. 8: 1053. https://doi.org/10.3390/cancers11081053
APA StyleCzauderna, C., Castven, D., Mahn, F. L., & Marquardt, J. U. (2019). Context-Dependent Role of NF-κB Signaling in Primary Liver Cancer—from Tumor Development to Therapeutic Implications. Cancers, 11(8), 1053. https://doi.org/10.3390/cancers11081053