The Benefit of Reactivating p53 under MAPK Inhibition on the Efficacy of Radiotherapy in Melanoma

 ,
,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
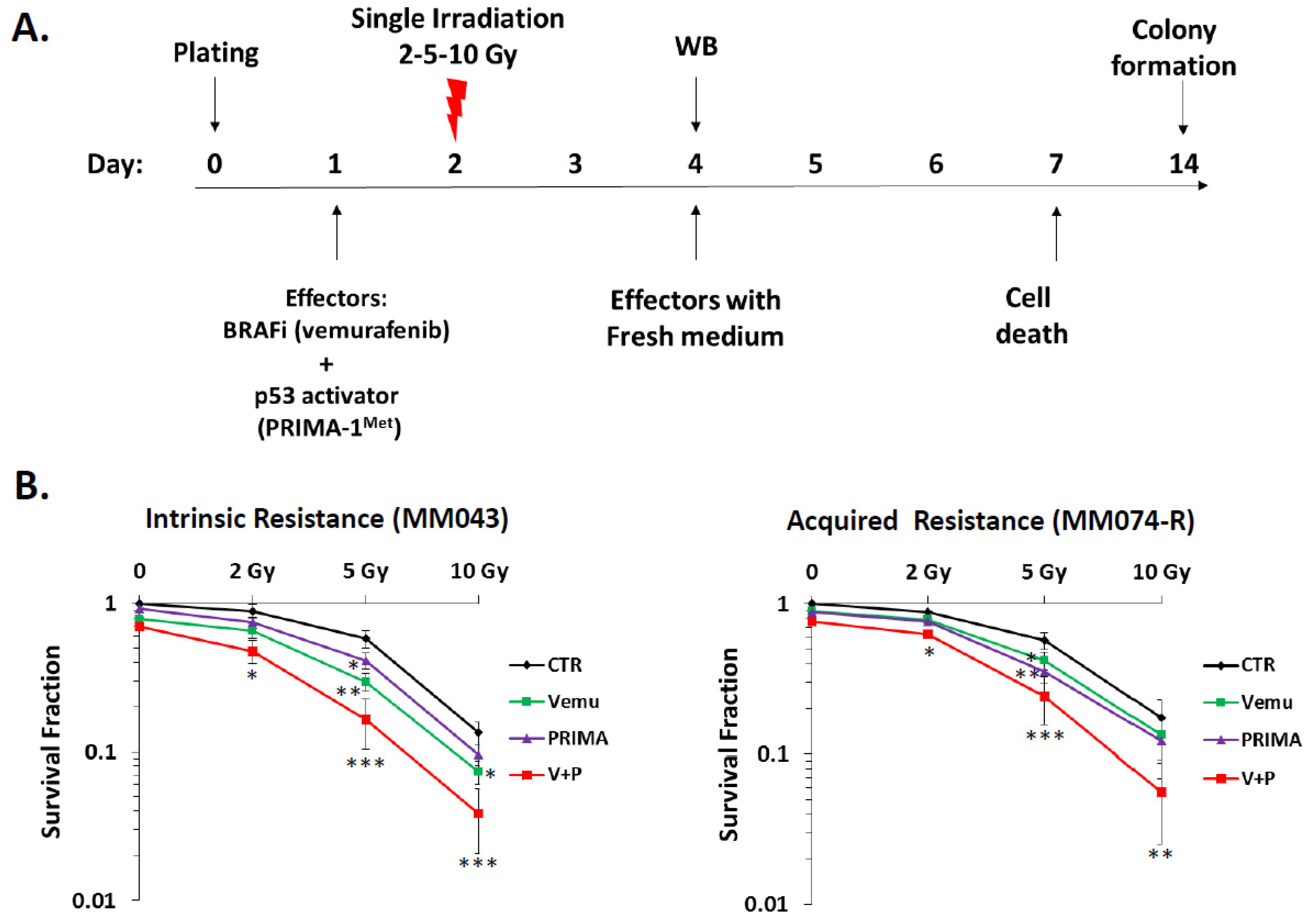
2.1. p53 Activator (PRIMA-1Met) Improves the Radiosensitizing Effect of BRAF Inhibitor (Vemurafenib) in V600EBRAF Mutant Melanoma Cells
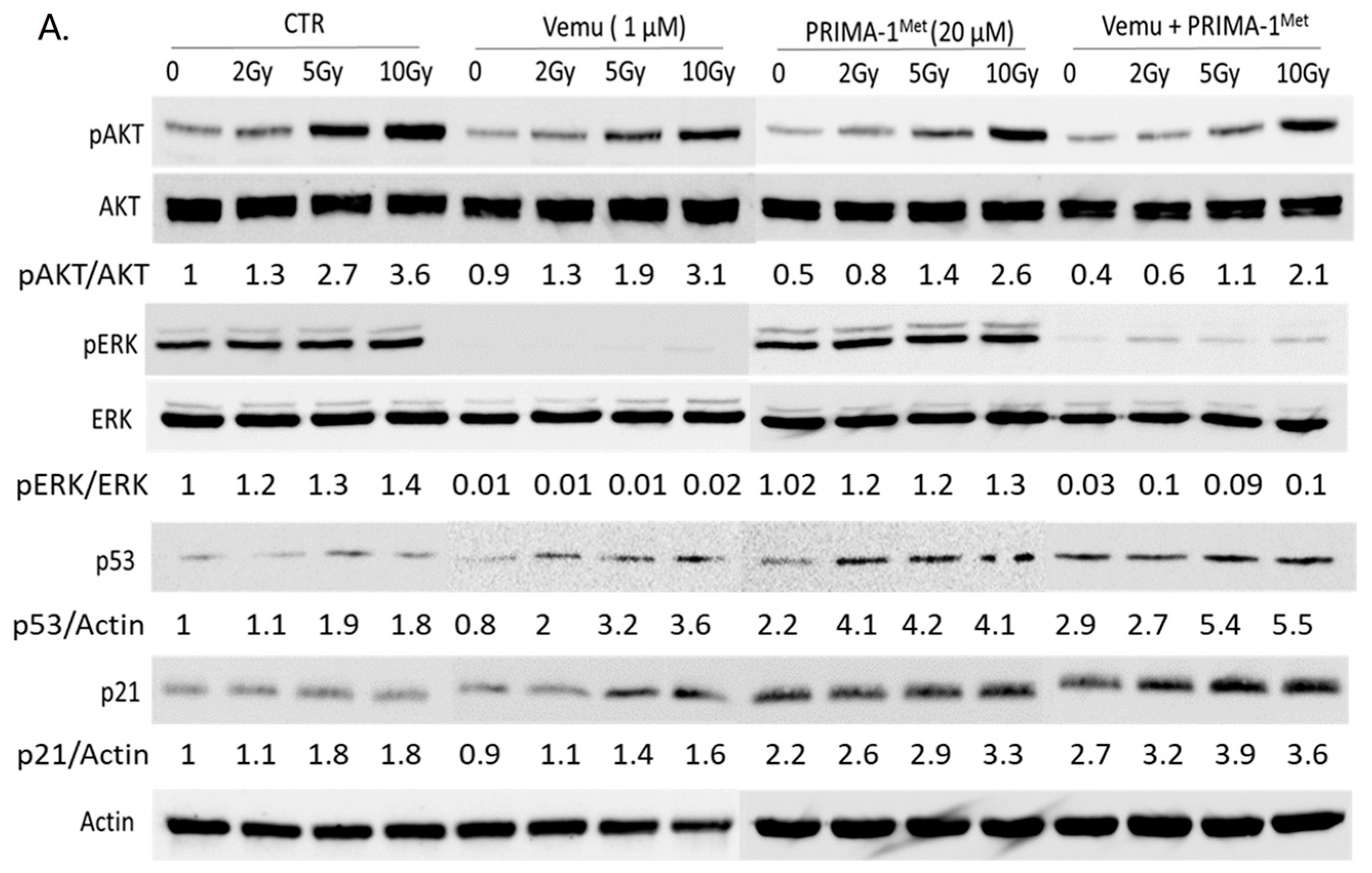
2.2. Activation of Both MAPK and PI3K/AKT Pathways Are a Frequent Event in Melanoma Radioresistance
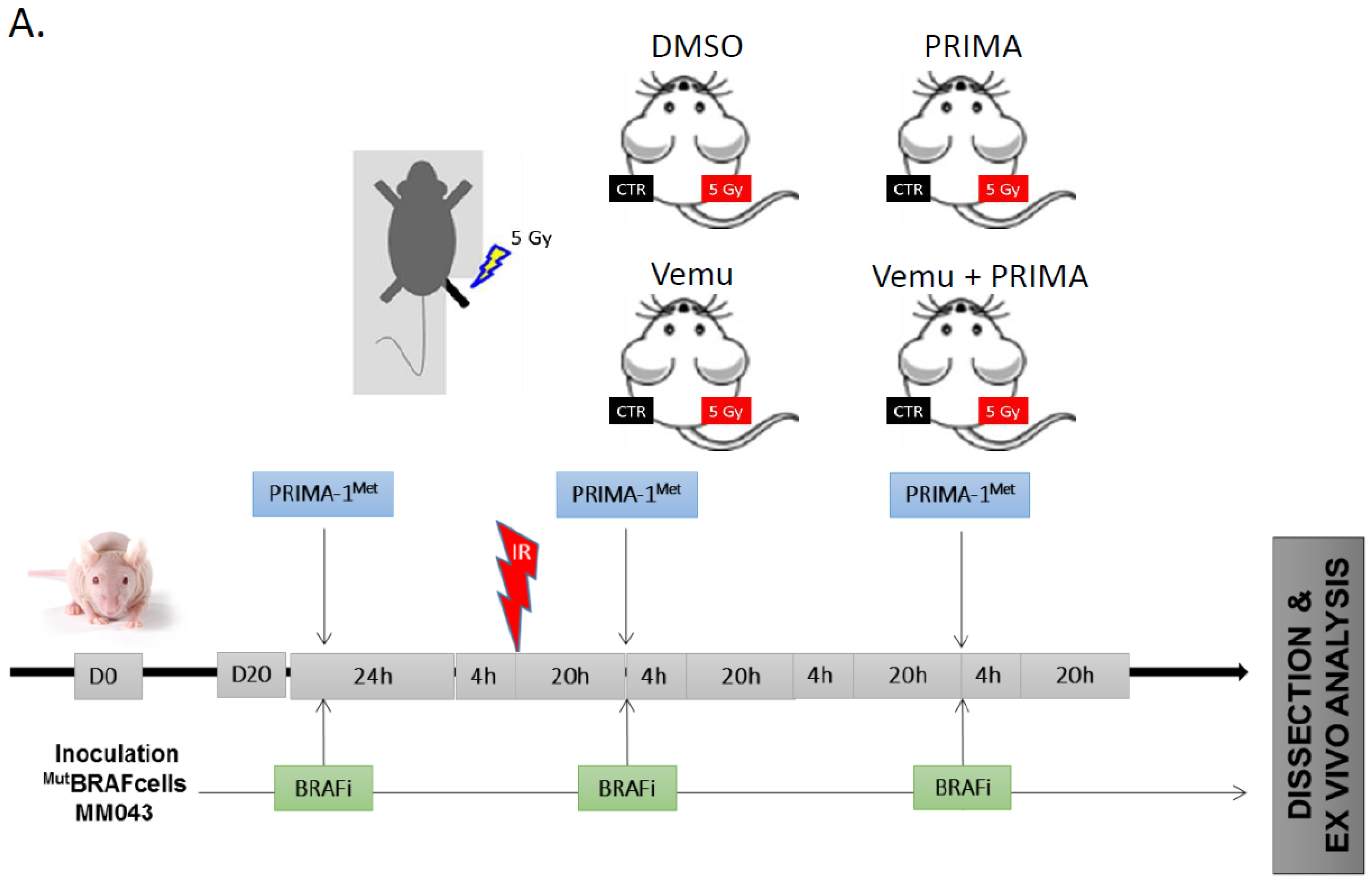
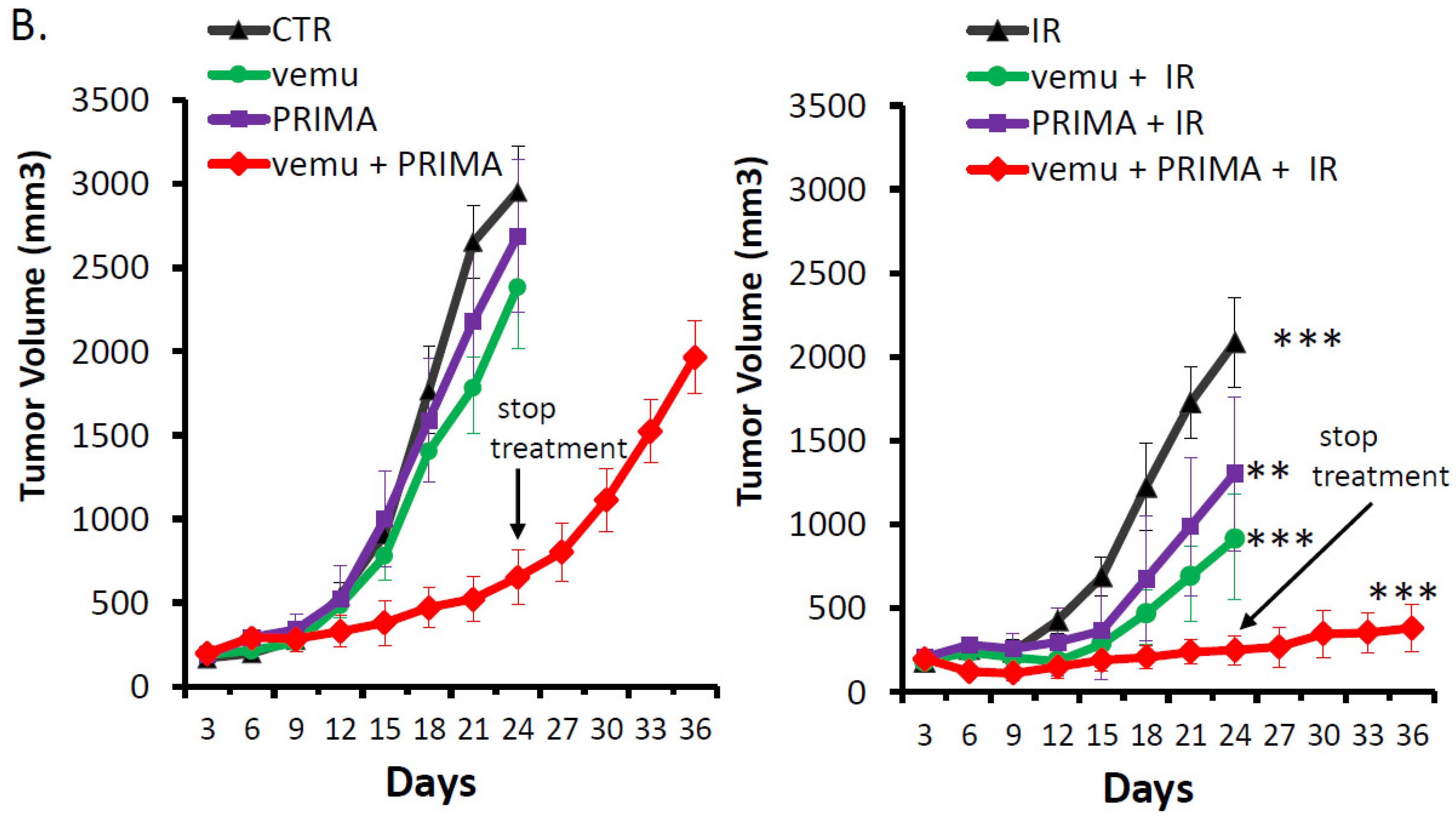
2.3. Evaluation of the Radiosensitizing Effect of Reactivating p53 under BRAF Inhibition In Vivo
3. Discussion
4. Materials and Methods
4.1. Inhibitors
4.2. Melanoma Cell Lines
4.3. Cell Culture Conditions
4.4. Clonogenic Assay
4.5. Western Blot Analysis
4.6. Cell Death Determination
4.7. Human Melanoma Xenografts
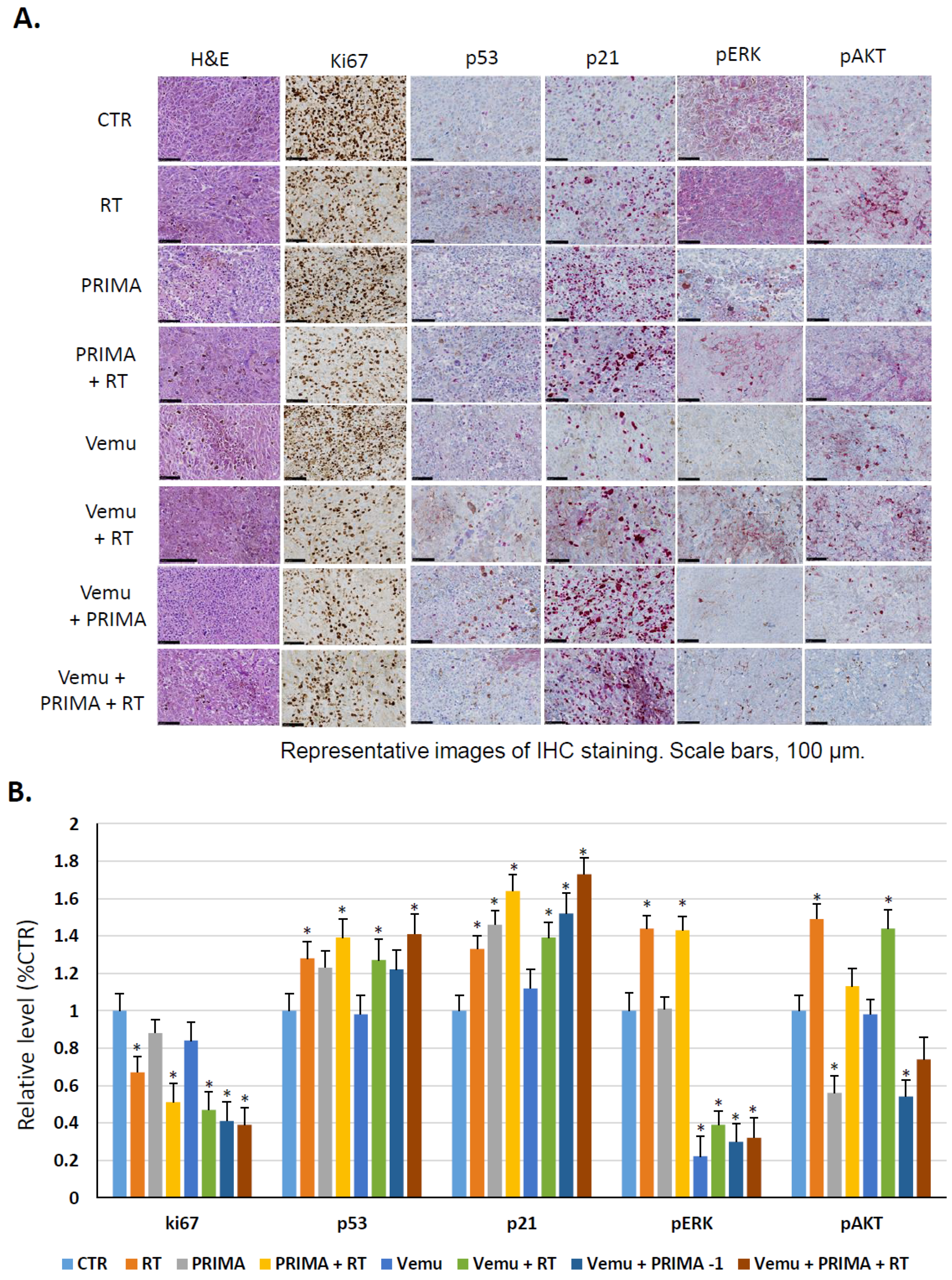
4.8. Immunohistochemistry Staining
4.9. Statistical Analysis
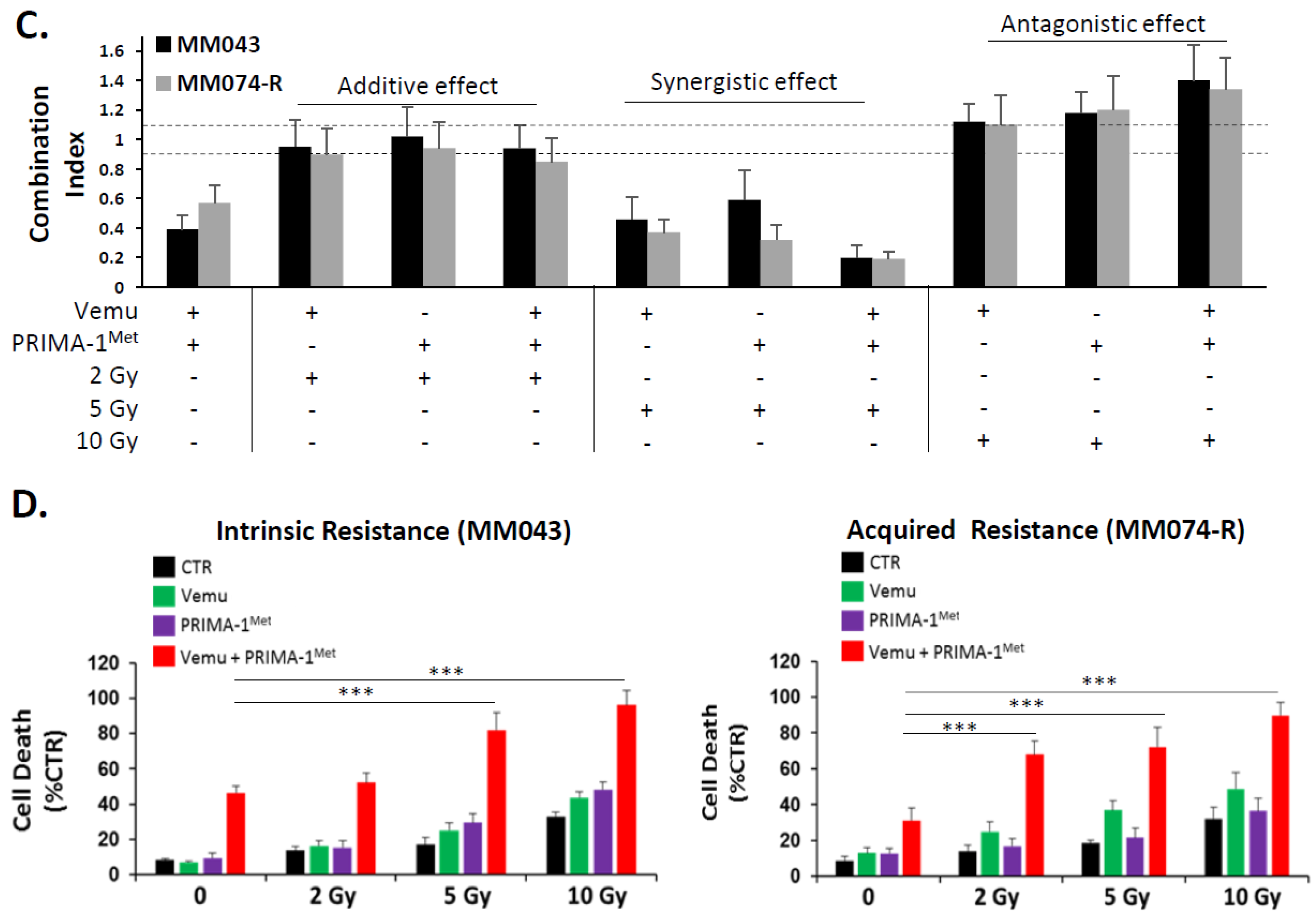
4.10. Combination Index Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Capasso, C.; Hirvinen, M.; Garofalo, M.; Romaniuk, D.; Kuryk, L.; Sarvela, T.; Vitale, A.; Antopolsky, M.; Magarkar, A.; Viitala, T.; et al. Oncolytic adenoviruses coated with MHC-I tumor epitopes increase the antitumor immunity and efficacy against melanoma. Oncoimmunology 2016, 5, e1105429. [Google Scholar] [CrossRef] [PubMed]
- Ressler, J.; Silmbrod, R.; Stepan, A.; Tuchmann, F.; Cicha, A.; Uyanik-Ünal, K.; Hoeller, C. Talimogene laherparepvec (T-VEC) in advanced melanoma: Complete response in a heart and kidney transplant patient. A case report. Br. J. Derm. 2019, 181, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, A.; Patel, V.L.; Dagoglu, N. Radiation Therapy in the Management of Malignant Melanoma. Oncology (Williston Park) 2015, 29, 743–751. [Google Scholar] [PubMed]
- Burmeister, B.H.; Henderson, M.A.; Ainslie, J.; Fisher, R.; Di Iulio, J.; Smithers, B.M.; Hong, A.; Shannon, K.; Scolyer, R.A.; Carruthers, S.; et al. Adjuvant radiotherapy versus observation alone for patients at risk of lymph-node field relapse after therapeutic lymphadenectomy for melanoma: A randomised trial. Lancet Oncol. 2012, 13, 589–597. [Google Scholar] [CrossRef]
- Bibault, J.-E.; Dewas, S.; Mirabel, X.; Mortier, L.; Penel, N.; Vanseymortier, L.; Lartigau, E. Adjuvant radiation therapy in metastatic lymph nodes from melanoma. Radiat. Oncol. Lond. Engl. 2011, 6, 12. [Google Scholar] [CrossRef]
- Khan, N.; Khan, M.K.; Almasan, A.; Singh, A.D.; Macklis, R. The Evolving Role of Radiation Therapy in the Management of Malignant Melanoma. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 645–654. [Google Scholar] [CrossRef]
- Hecht, M.; Meier, F.; Zimmer, L.; Polat, B.; Loquai, C.; Weishaupt, C.; Forschner, A.; Gutzmer, R.; Utikal, J.S.; Goldinger, S.M.; et al. Clinical outcome of concomitant vs. interrupted BRAF inhibitor therapy during radiotherapy in melanoma patients. Br. J. Cancer 2018, 118, 785–792. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, K.; Kruse, V.; Sundahl, N.; van Gele, M.; Chevolet, I.; Speeckaert, R.; Brochez, L.; Ost, P. A phase II trial of stereotactic body radiotherapy with concurrent anti-PD1 treatment in metastatic melanoma: Evaluation of clinical and immunologic response. J. Transl. Med. 2017, 15, 21. [Google Scholar] [CrossRef]
- Wang, H.; Mu, X.; He, H.; Zhang, X.-D. Cancer Radiosensitizers. Trends Pharm. Sci. 2018, 39, 24–48. [Google Scholar] [CrossRef]
- Tao, Z.; Le Blanc, J.M.; Wang, C.; Zhan, T.; Zhuang, H.; Wang, P.; Yuan, Z.; Lu, B. Coadministration of Trametinib and Palbociclib Radiosensitizes KRAS-Mutant Non-Small Cell Lung Cancers In Vitro and In Vivo. Clin. Cancer Res. 2016, 22, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Aroca, D.M.; Roche, O.; Sabater, S.; Pascual-Serra, R.; Ortega-Muelas, M.; Sánchez Pérez, I.; Belandia, B.; Ruiz-Hidalgo, M.J.; Sánchez-Prieto, R. P53 pathway is a major determinant in the radiosensitizing effect of Palbociclib: Implication in cancer therapy. Cancer Lett. 2019, 451, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Bouchy, S.; Penninckx, S.; Marega, R.; Fichera, O.; Gallez, B.; Feron, O.; Martinive, P.; Heuskin, A.-C.; Michiels, C.; et al. Antibody-functionalized gold nanoparticles as tumor-targeting radiosensitizers for proton therapy. Nanomed. 2019, 14, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Solit, D.B.; Rosen, N. Resistance to BRAF inhibition in melanomas. N. Engl. J. Med. 2011, 364, 772–774. [Google Scholar] [CrossRef]
- Alcalá, A.M.; Flaherty, K.T. BRAF inhibitors for the treatment of metastatic melanoma: Clinical trials and mechanisms of resistance. Clin. Cancer Res. 2012, 18, 33–39. [Google Scholar] [CrossRef]
- Krayem, M.; Najem, A.; Journe, F.; Morandini, R.; Sales, F.; Awada, A.; Ghanem, G.E. Acquired resistance to BRAFi reverses senescence-like phenotype in mutant BRAF melanoma. Oncotarget 2018, 9, 31888–31903. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Narayana, A.; Mathew, M.; Tam, M.; Kannan, R.; Madden, K.M.; Golfinos, J.G.; Parker, E.C.; Ott, P.A.; Pavlick, A.C. Vemurafenib and radiation therapy in melanoma brain metastases. J. Neurooncol. 2013, 113, 411–416. [Google Scholar] [CrossRef]
- Chowdhary, M.; Patel, K.R.; Danish, H.H.; Lawson, D.H.; Khan, M.K. BRAF inhibitors and radiotherapy for melanoma brain metastases: Potential advantages and disadvantages of combination therapy. OncoTargets Ther. 2016, 9, 7149–7159. [Google Scholar] [CrossRef]
- Leszczynska, K.B.; Foskolou, I.P.; Abraham, A.G.; Anbalagan, S.; Tellier, C.; Haider, S.; Span, P.N.; O’Neill, E.E.; Buffa, F.M.; Hammond, E.M. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via AKT. J. Clin. Investig. 2015, 125, 2385–2398. [Google Scholar] [CrossRef]
- Kumar, A.; Chandna, S. Evidence for a radiation-responsive ‘p53 gateway’ contributing significantly to the radioresistance of lepidopteran insect cells. Sci. Rep. 2018, 8, 2. [Google Scholar] [CrossRef]
- Wang, X.; Wei, L.; Cramer, J.M.; Leibowitz, B.J.; Judge, C.; Epperly, M.; Greenberger, J.; Wang, F.; Li, L.; Stelzner, M.G.; et al. Pharmacologically blocking p53-dependent apoptosis protects intestinal stem cells and mice from radiation. Sci. Rep. 2015, 5, 8566. [Google Scholar] [CrossRef]
- Arya, A.K.; El-Fert, A.; Devling, T.; Eccles, R.M.; Aslam, M.A.; Rubbi, C.P.; Vlatković, N.; Fenwick, J.; Lloyd, B.H.; Sibson, D.R.; et al. Nutlin-3, the small-molecule inhibitor of MDM2, promotes senescence and radiosensitises laryngeal carcinoma cells harbouring wild-type p53. Br. J. Cancer 2010, 103, 186–195. [Google Scholar] [CrossRef]
- Fei, P.; El-Deiry, W.S. P53 and radiation responses. Oncogene 2003, 22, 5774–5783. [Google Scholar] [CrossRef]
- Supiot, S.; Zhao, H.; Wiman, K.; Hill, R.P.; Bristow, R.G. PRIMA-1(met) radiosensitizes prostate cancer cells independent of their MTp53-status. Radiother. Oncol. J. Eur. Soc. Radiol. Oncol. 2008, 86, 407–411. [Google Scholar] [CrossRef]
- Houben, R.; Hesbacher, S.; Schmid, C.P.; Kauczok, C.S.; Flohr, U.; Haferkamp, S.; Müller, C.S.L.; Schrama, D.; Wischhusen, J.; Becker, J.C. High-level expression of wild-type p53 in melanoma cells is frequently associated with inactivity in p53 reporter gene assays. PLoS ONE 2011, 6, e22096. [Google Scholar] [CrossRef]
- Box, N.F.; Vukmer, T.O.; Terzian, T. Targeting p53 in melanoma. Pigment Cell Melanoma Res. 2014, 27, 8–10. [Google Scholar] [CrossRef]
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef]
- Hocker, T.; Tsao, H. Ultraviolet radiation and melanoma: A systematic review and analysis of reported sequence variants. Hum. Mutat. 2007, 28, 578–588. [Google Scholar] [CrossRef]
- De Lange, J.; Ly, L.V.; Lodder, K.; Verlaan-de Vries, M.; Teunisse, A.F.A.S.; Jager, M.J.; Jochemsen, A.G. Synergistic growth inhibition based on small-molecule p53 activation as treatment for intraocular melanoma. Oncogene 2012, 31, 1105–1116. [Google Scholar] [CrossRef]
- Ji, Z.; Njauw, C.N.; Taylor, M.; Neel, V.; Flaherty, K.T.; Tsao, H. p53 Rescue through HDM2 Antagonism Suppresses Melanoma Growth and Potentiates MEK Inhibition. J. Investig. Derm. 2012, 132, 356–364. [Google Scholar] [CrossRef]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.-C.; et al. p53 Reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K)BRAF melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar]
- Lu, M.; Breyssens, H.; Salter, V.; Zhong, S.; Hu, Y.; Baer, C.; Ratnayaka, I.; Sullivan, A.; Brown, N.R.; Endicott, J.; et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell 2013, 23, 618–633. [Google Scholar] [CrossRef]
- Yu, X.; Narayanan, S.; Vazquez, A.; Carpizo, D.R. Small molecule compounds targeting the p53 pathway: Are we finally making progress? Apoptosis 2014, 19, 1055–1068. [Google Scholar] [CrossRef]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef]
- Bando, S.-I.; Hatano, O.; Takemori, H.; Kubota, N.; Ohnishi, K. Potentiality of syringetin for preferential radiosensitization to cancer cells. Int. J. Radiat. Biol. 2017, 93, 286–294. [Google Scholar] [CrossRef]
- Yi, H.; Yan, X.; Luo, Q.; Yuan, L.; Li, B.; Pan, W.; Zhang, L.; Chen, H.; Wang, J.; Zhang, Y.; et al. A novel small molecule inhibitor of MDM2-p53 (APG-115) enhances radiosensitivity of gastric adenocarcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 97. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.N.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Najem, A.; Krayem, M.; Salès, F.; Hussein, N.; Badran, B.; Robert, C.; Awada, A.; Journe, F.; Ghanem, G.E. P53 and MITF/Bcl-2 identified as key pathways in the acquired resistance of NRAS-mutant melanoma to MEK inhibition. Eur. J. Cancer 2017, 83, 154–165. [Google Scholar] [CrossRef]
- Rubner, Y.; Muth, C.; Strnad, A.; Derer, A.; Sieber, R.; Buslei, R.; Frey, B.; Fietkau, R.; Gaipl, U.S. Fractionated radiotherapy is the main stimulus for the induction of cell death and of Hsp70 release of p53 mutated glioblastoma cell lines. Radiat. Oncol. Lond. Engl. 2014, 9, 89. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Fertil, B.; Malaise, E.P. Inherent cellular radiosensitivity as a basic concept for human tumor radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1981, 7, 621–629. [Google Scholar] [CrossRef]
- Shahbazian, D.; Bindra, R.S.; Kluger, H.M.; Glazer, P.M. Radiation sensitivity and sensitization in melanoma. Pigment Cell Melanoma Res. 2013, 26, 928–930. [Google Scholar] [CrossRef]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef]
- Ahmed, K.A.; Abuodeh, Y.A.; Echevarria, M.I.; Arrington, J.A.; Stallworth, D.G.; Hogue, C.; Naghavi, A.O.; Kim, S.; Kim, Y.; Patel, B.G.; et al. Clinical outcomes of melanoma brain metastases treated with stereotactic radiosurgery and anti-PD-1 therapy, anti-CTLA-4 therapy, BRAF/MEK inhibitors, BRAF inhibitor, or conventional chemotherapy. Ann. Oncol. 2016, 27, 2288–2294. [Google Scholar] [CrossRef]
- Harding, J.J.; Barker, C.A.; Carvajal, R.D.; Wolchok, J.D.; Chapman, P.B.; Lacouture, M.E. Cutis Verticis Gyrata in Association With Vemurafenib and Whole-Brain Radiotherapy. J. Clin. Oncol. 2014, 32, e54–e56. [Google Scholar] [CrossRef]
- Schulze, B.; Meissner, M.; Wolter, M.; Rödel, C.; Weiss, C. Unusual acute and delayed skin reactions during and after whole-brain radiotherapy in combination with the BRAF inhibitor vemurafenib. Two case reports. Strahlentherapie und Onkologie 2014, 190, 229–232. [Google Scholar] [CrossRef]
- Merten, R.; Hecht, M.; Haderlein, M.; Distel, L.; Fietkau, R.; Heinzerling, L.; Semrau, S. Increased skin and mucosal toxicity in the combination of vemurafenib with radiation therapy. Strahlentherapie und Onkologie 2014, 190, 1169–1172. [Google Scholar] [CrossRef]
- Anker, C.J.; Ribas, A.; Grossmann, A.H.; Chen, X.; Narra, K.K.; Akerley, W.; Andtbacka, R.H.I.; Noyes, R.D.; Shrieve, D.C.; Grossmann, K.F. Severe liver and skin toxicity after radiation and vemurafenib in metastatic melanoma. J. Clin. Oncol. 2013, 31, e283–e287. [Google Scholar] [CrossRef]
- Sambade, M.J.; Peters, E.C.; Thomas, N.E.; Kaufmann, W.K.; Kimple, R.J.; Shields, J.M. Melanoma cells show a heterogeneous range of sensitivity to ionizing radiation and are radiosensitized by inhibition of B-RAF with PLX-4032. Radiother. Oncol. J. Eur. Soc. Radiol. Oncol. 2011, 98, 394–399. [Google Scholar] [CrossRef]
- Genotype-Dependent Cooperation of Ionizing Radiation with BRAF Inhibition in BRAF V600E-mutated…—Abstract—Europe PMC. Available online: http://europepmc.org/abstract/MED/23354848 (accessed on 3 December 2018).
- Walter, L.; Heinzerling, L. BRAF Inhibitors and Radiation Do Not Act Synergistically to Inhibit WT and V600E BRAF Human Melanoma. Anticancer Res. 2018, 38, 1335–1341. [Google Scholar]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Knoops, L.; Haas, R.; de Kemp, S.; Majoor, D.; Broeks, A.; Eldering, E.; de Boer, J.P.; Verheij, M.; van Ostrom, C.; de Vries, A.; et al. In vivo p53 response and immune reaction underlie highly effective low-dose radiotherapy in follicular lymphoma. Blood 2007, 110, 1116–1122. [Google Scholar] [CrossRef]
- Barberi-Heyob, M.; Védrine, P.-O.; Merlin, J.-L.; Millon, R.; Abecassis, J.; Poupon, M.-F.; Guillemin, F. Wild-type p53 gene transfer into mutated p53 HT29 cells improves sensitivity to photodynamic therapy via induction of apoptosis. Int. J. Oncol. 2004, 24, 951–958. [Google Scholar] [CrossRef]
- Camp, E.R.; Wang, C.; Little, E.C.; Watson, P.M.; Pirollo, K.F.; Rait, A.; Cole, D.J.; Chang, E.H.; Watson, D.K. Transferrin receptor targeting nanomedicine delivering wild-type p53 gene sensitizes pancreatic cancer to gemcitabine therapy. Cancer Gene Ther. 2013, 20, 222–228. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Gudkov, A.V.; Komarova, E.A. The role of p53 in determining sensitivity to radiotherapy. Nat. Rev. Cancer 2003, 3, 117. [Google Scholar] [CrossRef]
- El-Deiry, W.S. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003, 22, 7486–7495. [Google Scholar] [CrossRef]
- Siroy, A.E.; Boland, G.M.; Milton, D.R.; Roszik, J.; Frankian, S.; Malke, J.; Haydu, L.; Prieto, V.G.; Tetzlaff, M.; Ivan, D.; et al. Beyond BRAF(V600): Clinical mutation panel testing by next-generation sequencing in advanced melanoma. J. Investig. Derm. 2015, 135, 508–515. [Google Scholar] [CrossRef]
- Burrows, N.; Williams, J.; Telfer, B.A.; Resch, J.; Valentine, H.R.; Fitzmaurice, R.J.; Eustace, A.; Irlam, J.; Rowling, E.J.; Hoang-Vu, C.; et al. Phosphatidylinositide 3-kinase (PI3K) and PI3K-related kinase (PIKK) activity contributes to radioresistance in thyroid carcinomas. Oncotarget 2016, 7, 63106–63123. [Google Scholar] [CrossRef]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013, 4, e875. [Google Scholar] [CrossRef]
- Xu, S.; Li, Y.; Lu, Y.; Huang, J.; Ren, J.; Zhang, S.; Yin, Z.; Huang, K.; Wu, G.; Yang, K. LZTS2 inhibits PI3K/AKT activation and radioresistance in nasopharyngeal carcinoma by interacting with p85. Cancer Lett. 2018, 420, 38–48. [Google Scholar] [CrossRef]
- Che, Y.; Li, Y.; Zheng, F.; Zou, K.; Li, Z.; Chen, M.; Hu, S.; Tian, C.; Yu, W.; Guo, W.; et al. TRIP4 promotes tumor growth and metastasis and regulates radiosensitivity of cervical cancer by activating MAPK, PI3K/AKT, and hTERT signaling. Cancer Lett. 2019, 452, 1–13. [Google Scholar] [CrossRef]
- Ponten, F.; Lindman, H.; Bostrom, A.; Berne, B.; Bergh, J. Induction of p53 Expression in Skin by Radiotherapy and UV Radiation: A Randomized Study. JNCI J. Natl. Cancer Inst. 2001, 93, 128–133. [Google Scholar] [CrossRef]
- Astanehe, A.; Arenillas, D.; Wasserman, W.W.; Leung, P.C.K.; Dunn, S.E.; Davies, B.R.; Mills, G.B.; Auersperg, N. Mechanisms underlying p53 regulation of PIK3CA transcription in ovarian surface epithelium and in ovarian cancer. J. Cell Sci. 2008, 121, 664–674. [Google Scholar] [CrossRef]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN Transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Hecht, M.; Zimmer, L.; Loquai, C.; Weishaupt, C.; Gutzmer, R.; Schuster, B.; Gleisner, S.; Schulze, B.; Goldinger, S.M.; Berking, C.; et al. Radiosensitization by BRAF inhibitor therapy-mechanism and frequency of toxicity in melanoma patients. Ann. Oncol. 2015, 26, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Paik, A.; Li, J.J. p53 activation in chronic radiation-treated breast cancer cells: Regulation of MDM2/p14ARF. Cancer Res. 2004, 64, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Djehal, A.; Krayem, M.; Najem, A.; Hammoud, H.; Cresteil, T.; Nebigil, C.G.; Wang, D.; Yu, P.; Bentouhami, E.; Ghanem, G.E.; et al. Targeting prohibitin with small molecules to promote melanogenesis and apoptosis in melanoma cells. Eur. J. Med. Chem. 2018, 155, 880–888. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krayem, M.; Sabbah, M.; Najem, A.; Wouters, A.; Lardon, F.; Simon, S.; Sales, F.; Journe, F.; Awada, A.; Ghanem, G.E.; et al. The Benefit of Reactivating p53 under MAPK Inhibition on the Efficacy of Radiotherapy in Melanoma. Cancers 2019, 11, 1093. https://doi.org/10.3390/cancers11081093
Krayem M, Sabbah M, Najem A, Wouters A, Lardon F, Simon S, Sales F, Journe F, Awada A, Ghanem GE, et al. The Benefit of Reactivating p53 under MAPK Inhibition on the Efficacy of Radiotherapy in Melanoma. Cancers. 2019; 11(8):1093. https://doi.org/10.3390/cancers11081093
Chicago/Turabian StyleKrayem, Mohammad, Malak Sabbah, Ahmad Najem, An Wouters, Filip Lardon, Stephane Simon, François Sales, Fabrice Journe, Ahmad Awada, Ghanem E. Ghanem, and et al. 2019. "The Benefit of Reactivating p53 under MAPK Inhibition on the Efficacy of Radiotherapy in Melanoma" Cancers 11, no. 8: 1093. https://doi.org/10.3390/cancers11081093
APA StyleKrayem, M., Sabbah, M., Najem, A., Wouters, A., Lardon, F., Simon, S., Sales, F., Journe, F., Awada, A., Ghanem, G. E., & Van Gestel, D. (2019). The Benefit of Reactivating p53 under MAPK Inhibition on the Efficacy of Radiotherapy in Melanoma. Cancers, 11(8), 1093. https://doi.org/10.3390/cancers11081093