Intricacies of the Molecular Machinery of Catecholamine Biosynthesis and Secretion by Chromaffin Cells of the Normal Adrenal Medulla and in Pheochromocytoma and Paraganglioma

 , and
, and
Abstract
:1. Introduction
2. Adrenomedullary Function
2.1. Biosynthesis of Catecholamines
2.1.1. Tyrosine Hydroxylase
2.1.2. Aromatic L-Amino Acid Decarboxylase
2.1.3. Dopamine β-hydroxylase
2.1.4. Phenylethanolamine-N-Methyltransferase
2.1.5. Co-Secreted Products
2.2. Storage and Secretion of Catecholamines
2.2.1. Storage and Vesicular Transmembrane Dynamics
2.2.2. Characteristics of Chromaffin Storage Vesicles
2.2.3. Secretion and Re-Uptake of Catecholamines
2.3. Regulation of Adrenomedullary Activity
2.3.1. Stimulus-Dependent Exocytosis in Adrenal Chromaffin Cells
2.3.2. Neuronal Regulation of the Calcium-Dependent Catecholamine Secretory Pathway
2.3.3. Non-Neuronal Regulation of Catecholamine Secretion
3. Pheochromocytoma and Paraganglioma
3.1. Increased Biosynthesis of Catecholamines
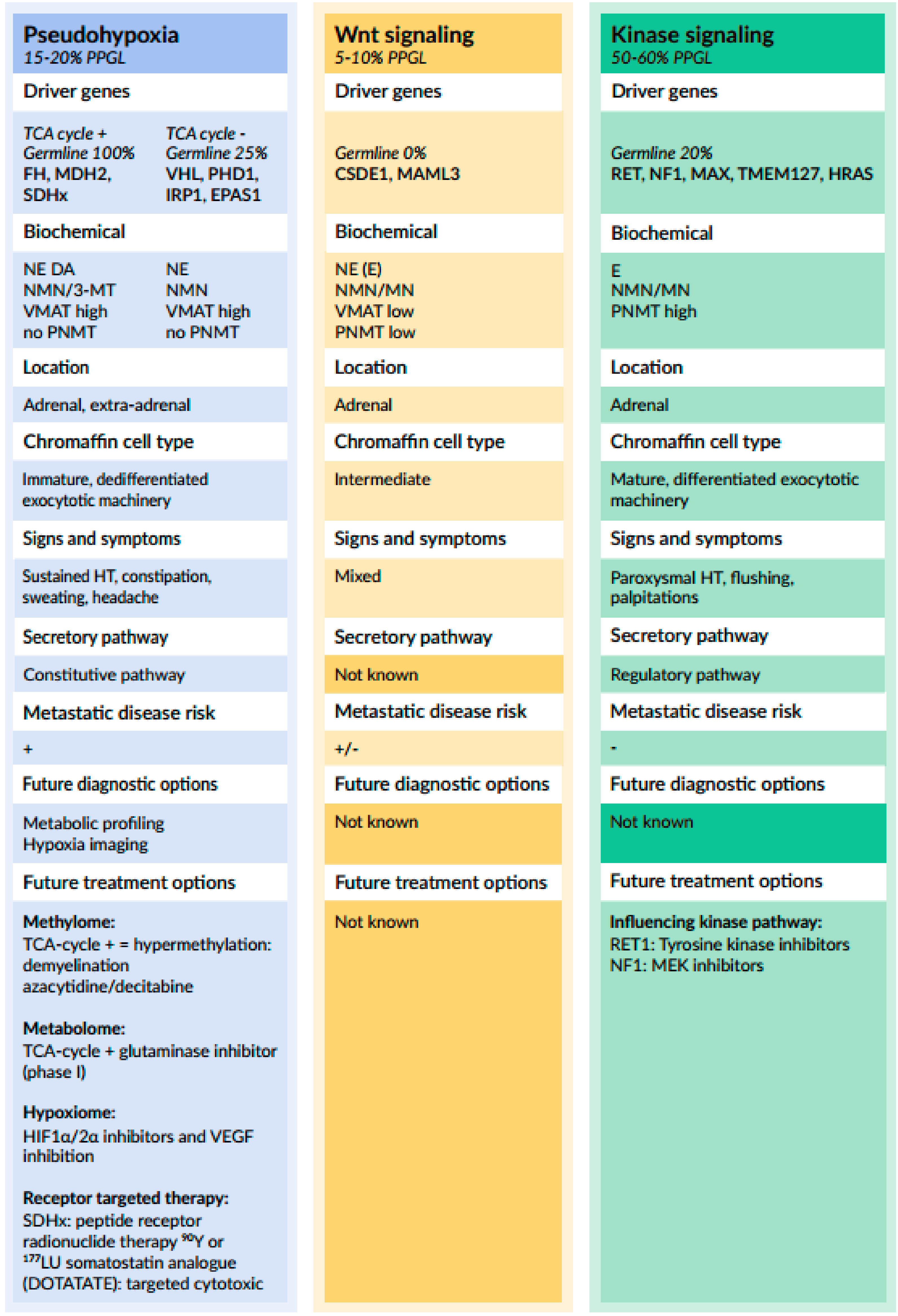
3.1.1. Relationship between Genotype and Catecholamine Biochemical Phenotype
3.1.2. Relationship between Genotype and Catecholamine Secretory Pathways
3.2. Alterations in Chromaffin Cell Pathways Associated with Metastatic Pheochromocytoma and Paraganglioma
3.2.1. Clinical Features and Risk Factors
3.2.2. Molecular Alterations in SDHx-Related PPGL and Their Effect on Catecholamine Biosynthesis
3.2.3. Relationship between Other Components of the Exocytotic Machinery and Metastatic Disease
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Melmed, S.; Polonsky, K.S.; Larsen, P.R.; Kronenberg, H.M. Williams Textbook Endocrinology, 13th ed.; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 978-0-323-29738-7. [Google Scholar]
- Eisenhofer, G.; Ehrhart-Bornstein, M.; Bornstein, S. The Adrenal Medulla. Physiology and Pathophysiology. In Handbook of the Autonomic Nervous System in Health and Disease; Bolis, C.L., Govoni, S., Eds.; Marcel Dekker Inc.: New York, NY, USA; Basel, Switzerland, 2003; pp. 185–224. [Google Scholar]
- Lenders, J.W.M.; Eisenhofer, G. Pathophysiology and diagnosis of disorder of the adrenal medulla: Focus on pheochromocytoma. Compr. Physiol. 2014, 4, 691–713. [Google Scholar] [PubMed]
- Pihlajoki, M.; Dörmer, J.; Cochran, R.S.; Heikinheimo, M.; Wilson, D.B. Adrenocortical zonation, renewal and remodeling. Front. Endocrinol. (Lausanne) 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Lumb, R.; Schwartz, Q. Sympathoadrenal neural crest cells: The known, unknown and forgotten? Dev. Growth Differ. 2015, 57, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.L. Why is the adrenal adrenergic (review). Endocr. Pathol. 2003, 14, 25–36. [Google Scholar] [CrossRef]
- Schinner, S.; Bornstein, S.R. Cortical-chromaffin cell interactions in the adrenal gland. Endocr. Pathol. 2005, 16, 91–98. [Google Scholar] [CrossRef]
- Merke, D.P.; Chrousos, G.P.; Eisenhofer, G.; Weise, M.; Keil, M.F.; Rogol, A.D.; van Wyk, J.J.; Bornstein, S.R. Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N. Engl. J. Med. 2000, 343, 1362–1368. [Google Scholar] [CrossRef]
- Haase, M.; Willenberg, H.S.; Bornstein, S.R. Update on the corticomedullary interaction in the adrenal gland. Endocr. Dev. 2011, 20, 28–37. [Google Scholar]
- Bornstein, S.R.; Gonzalez-Hernandez, J.A.; Ehrhart-Bornstein, M.; Adler, G.; Scherbaum, W.A. Intimate contact of chromaffin and cortical cells within the human adrenal gland forms the cellular basis for important intraadrenal interactions. J. Clin. Endocrinol. Metab. 1994, 78, 225–232. [Google Scholar]
- Eisenhofer, G.; Huynh, T.T.; Hiroi, M.; Pacak, K. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev. Endocr. Metab. Disord. 2001, 2, 297–311. [Google Scholar] [CrossRef]
- Tank, A.W.; Lee Wong, D. Peripheral and central effects of circulating catecholamines. Compr. Physiol. 2015. [Google Scholar] [CrossRef]
- McCarty, R. Learning about stress: Neuronal, endocrine and behavioural adaptations. Stress 2016, 19, 449–475. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Eisenhofer, G.; Mannelli, M.; Pacak, K. Phaeochromocytoma. Lancet 2005, 366, 665–675. [Google Scholar] [CrossRef]
- McNicol, A.M. Update on tumours of the adrenal cortex, pheochromocytoma and extra-adrenal paraganglioma. Histopathology 2011, 58, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.K. Update on adrenal tumours in 2017 World Health Organization (WHO) of endocrine tumours. Endocr. Pathol. 2017, 28, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.B.; Liu, C.F.; Chiang, C.W.; Kung, C.T.; Lee, C.W. Cardiovascular manifestations of pheochromocytoma. Am. J. Emerg. Med. 2000, 18, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, F.M.; Lenders, J.W.; Eisenhofer, G.; Pacak, K. Pheochromocytoma as an endocrine emergency. Rev. Endocr. Metab. Disord. 2003, 4, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Prejbisz, A.; Lenders, J.W.; Eisenhofer, G.; Januszewicz, A. Mortality associated with phaeochromocytoma. Horm. Metab. Res. 2013, 45, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Stolk, R.F.; Bakx, C.; Mulder, J.; Timmers, H.J.; Lenders, J.W. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J. Clin. Endocrinol. Metab. 2013, 98, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Berends, A.M.A.; Buitenwerf, E.; de Krijger, R.R.; Veeger, N.J.G.M.; van der Horst-Schrivers, A.N.A.; Links, T.P.; Kerstens, M.N. Incidence of pheochromocytoma and sympathetic paraganglioma in the Netherlands: A nationwide study and systematic review. Eur. J. Intern. Med. 2018, 51, 68–73. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Prejbisz, A.; Peitzsch, M.; Pamporaki, C.; Masjkur, J.; Rogowski-Lehmann, N.; Langton, K.; Tsourdi, E.; Peczkowska, M.; Fliedner, S.; et al. Biochemical diagnosis of chromaffin cell tumours in patients at high and low risk of disease: Plasma versus urinary free or deconjugated O-methylated catecholamine metabolites. Clin. Chem. 2018, 64, 1646–1656. [Google Scholar] [CrossRef]
- Bozkurt, M.F.; Virgolini, I.; Balogova, S.; Beheshti, M.; Rubello, D.; Decristoforo, C.; Ambrosini, V.; Kjaer, A.; Delgado-Bolton, R.; Kunikowska, J.; et al. Guideline for PET/CT imaging of neuroendocrine neoplasms with 68Ga-DOTA-conjugated somatostatin receptor targeting peptides and 18F-DOPA. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1588–1601. [Google Scholar] [CrossRef] [PubMed]
- Pacak, K. Pheochromocytoma: A catecholamine and oxidative stress disorder. Endocr. Regul. 2011, 45, 65–90. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Klink, B.; Richter, S.; Lenders, J.W.M.; Robledo, M. Metabologenomics of pheochromocytoma and paraganglioma: An integrated approach for personalised biochemical and genetic testing. Clin. Biochem. Rev. 2017, 38, 69–100. [Google Scholar] [PubMed]
- Gupta, G.; Pacak, K.; AACE Adrenal Scientific Committee. Precision medicine: An update on genotype/biochemical phenotype relationships in pheochromocytoma/paraganglioma patients. Endocr. Pract. 2017, 23, 690–704. [Google Scholar] [CrossRef]
- Lehnert, H. Regulation of catecholamine synthesizing enzyme gene expression in human pheochromocytoma. Eur. J. Endocrinol. 1998, 138, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garibotto, G.; Tessari, P.; Verzola, D.; Dertenois, L. The metabolic conversion of phenylalanine into tyrosine in the human kidney: Does it have nutritional implications in renal patients? J. Ren. Nutr. 2002, 12, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, K.P.; Neels, O.N.; Kema, I.P.; Elsinga, P.H.; Links, T.P.; de Vries, E.G.E.; Jager, P.L. Molecular imaging in neuroendocrine tumours: Molecular uptake mechanisms and clinical results. Crit. Rev. Oncol. Hematol. 2009, 71, 199–213. [Google Scholar] [CrossRef]
- Salisbury, T.B.; Arthur, S. The regulation and function of the L-type amino acid transporter 1 (LAT1) in cancer. Int. J. Mol. Sci. 2018, 19, 2373. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Rundquist, B.; Aneman, A.; Friberg, P.; Dakak, N.; Kopin, I.J.; Jacobs, M.C.; Lenders, J.W. Regional release and removal of catecholamines and extraneuronal metabolism to metanephrines. J. Clin. Endocrinol. Metab. 1995, 80, 3009–3017. [Google Scholar] [Green Version]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. Tyrosine hydroxylase: The initial step in norepinephrine biosynthesis. J. Biol. Chem. 1964, 239, 2910–2917. [Google Scholar]
- Wolf, M.E.; LeWitt, P.A.; Bannon, M.J.; Dragovic, L.J.; Kapatos, G. Effect of ageing on tyrosine hydroxylase protein content and the relative number of dopamine nerve terminals in human caudate. J. Neurochem. 1991, 56, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T. Tyrosine hydroxylase: Human isoforms, structure and regulation in physiology and pathology. Essays Biochem. 1995, 30, 15–35. [Google Scholar] [PubMed]
- Rao, F.; Zhang, K.; Zhang, L.; Rana, B.K.; Wessel, J.; Fung, M.M.; Rodriquez-Flores, J.L.; Taupenot, L.; Ziegler, M.G.; O’Connor, D.T. Human tyrosine hydroxylase natural allelic variation: Influcence on autonomic function and hypertension. Cell. Mol. Neurobiol. 2010, 30, 1391–1394. [Google Scholar] [CrossRef]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doskeland, A.P.; Flatmark, T. Ubiquitination of soluble and membrane bound tyrosine hydroxylase and degradation of the soluble form. Eur. J. Biochem. 2002, 269, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Best, J.A.; Nagamoto, K.; Tank, A.W. Regulation of tyrosine hydroxylase gene expression by the m1 muscarinic acetylcholine receptor in rat pheochromocytoma cells. Mol. Brain Res. 1996, 40, 42–54. [Google Scholar] [CrossRef]
- Sabban, E.L.; Kvetnansky, R. Stress-triggered activation of gene expression in catecholaminergic systems: Dynamics of transcriptional events. Trends Neurosci. 2001, 24, 91–98. [Google Scholar] [CrossRef]
- Eiden, L.E.; Jiang, S.Z. What’s new in endocrinology: The chromaffin cell. Front. Endocrinol. (Lausanne) 2018, 9, 711. [Google Scholar] [CrossRef]
- Lee, Y.H.; Gyun, Y.; Moon, J.Y.; Kim, J.S.; Jeong, K.H.; Lee, T.W.; Ihm, C.G.; Lee, S.H. Genetic variations of tyrosine hydroxylase in the pathogenesis of hypertension. Electrolyte Blood Press 2016, 14, 21–26. [Google Scholar] [CrossRef]
- Zhu, M.Y.; Juorio, A.V. Aromatic L-amino acid decarboxylase: Biological characterization and functional role. Gen. Pharmacol. 1995, 26, 681–696. [Google Scholar] [CrossRef]
- Barth, M.; Serre, V.; Hubert, L.; Chaabouni, Y.; Bahi-Buisson, N.; Cadoudal, M.; Rabier, D.; Tich, S.N.; Ribeiro, M.; Ricquier, D.; et al. Kinetic analyses guide the therapeutic decision in a novel form of moderate aromatic acid decarboxylase deficiency. JIMD Rep. 2012, 3, 25–32. [Google Scholar] [PubMed]
- Berends, A.M.A.; Kerstens, M.N.; Bolt, J.W.; Links, T.P.; Korpershoek, E.; de Krijger, R.R.; Walenkamp, A.M.E.; Noordzij, W.; van Etten, B.; Kats-Ugurlu, G.; et al. False-positive findings on 6-[18F]fluor-L-3,4-dihydroxyphenylalanine PET (18F-FDOPA-PET) performed for imaging of neuroendocrine tumors. Eur. J. Endocrinol. 2018, 179, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Waymire, J.C.; Haycock, J.W. Lack of regulation of aromatic L-amino acid decarboxylase in intact bovine chromaffin cells. J. Neurochem. 2002, 81, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Juorio, A.V.; Boulton, A.A. NSD-1015 alters the gene expression of aromatic L-amino acid decarboxylase in rat PC12 pheochromocytoma cells. Neurochem. Res. 1993, 18, 915–919. [Google Scholar] [CrossRef]
- Lee, J.J.; Jin, C.M.; Kim, Y.K.; Ryu, S.Y.; Lim, S.C.; Lee, M.K. Effects of anonaine on dopamine biosynthesis and L-dopa induced cytotoxicity in PC12 cells. Molecules 2008, 13, 475–487. [Google Scholar] [CrossRef]
- Friedman, S.; Kaufman, S. 3,4-Dihydroxyphenylethylamine β-hydroxylase. Physical properties, copper content, and role of copper in the catalytic activity. J. Biol. Chem. 1965, 240, 4763–4773. [Google Scholar]
- Weinshilboum, R.; Axelrod, J. Serum dopamine-beta-hydroxylase activity. Circ. Res. 1971, 28, 307–315. [Google Scholar] [CrossRef]
- Palatini, P. Fumarate is the cause of the apparent ping-pong kinetics of dopamine beta-hydroxylase. Biochem. Int. 1985, 11, 565–572. [Google Scholar]
- Wimalasena, K.; Dharmasena, S.; Wimalasena, D.S.; Hughbanks-Wheaton, D.K. Reduction of dopamine beta-monooxygenase. A unified model for apparent negative cooperativity and fumarate activation. J. Biol. Chem. 1996, 271, 26032–26043. [Google Scholar] [CrossRef]
- Patak, P.; Willenberg, H.S.; Bornstein, S.R. Vitamin C is an important cofactor for both adrenal cortex and adrenal medulla. Endocr. Res. 2004, 30, 871–875. [Google Scholar] [CrossRef]
- Trifaro, J. Molecular biology of the chromaffin cell. Ann. N. Y. Acad. Sci. 2002, 971, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.H. Phenylethanolamine N-methyltransferase from the brain and adrenal medulla of the rat: A comparison of their properties. Neurochem. Res. 1978, 3, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Livett, B.G. Adrenal medullary chromaffin cells in vitro. Physiol. Rev. 1984, 64, 1103–1161. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.L.; Lesage, A.; Siddall, B.; Funder, J.W. Glucocorticoid regulation of phenylethanolamine N-methyltransferase in vivo. FASEB J. 1992, 6, 3310–3315. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.L.; Siddall, B.J.; Ebert, S.N.; Bell, R.A.; Her, S. Phenylethanolamine N-methyltransferase gene expression: Synergistic activation by Egr-1, AP-2 and the glucocorticoid receptor. Brain Res. Mol. Brain Res. 1998, 61, 154–161. [Google Scholar] [CrossRef]
- Ceccatelli, S.; Dagerlind, A.; Schalling, M.; Wikstróm, A.C.; Okret, S.; Gustafsson, J.A.; Goldstein, M.; Hökfelt, T. The glucocorticoid receptor in the adrenal gland is localized in the cytoplasm of adrenaline cells. Acta Physiol. Scand. 1989, 137, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman-Levin, N.; Tiosano, D.; Eisenhofer, G.; Bornstein, S.; Hochberg, Z. The importance of adrenocortical glucocorticoids for adrenomedullary and physiological response to stress: A study in isolated glucocorticoid deficiency. J. Clin. Endocrinol. Metab. 2001, 86, 5920–5924. [Google Scholar] [CrossRef]
- Kennedy, B.; Bigby, T.D.; Ziegler, M.G. Nonadrenal epinephrine-forming enzymes in humans. Characteristics, distribution, regulation, and relationship to epinephrine levels. J. Clin. Investig. 1995, 95, 2896–2902. [Google Scholar] [CrossRef] [PubMed]
- Osuala, K.; Telusma, K.; Khan, S.M.; Wu, S.; Shah, M.; Baker, C.; Alam, S.; Abukenda, I.; Fuentes, A.; Seifein, H.B.; et al. Distinctive left-sided distribution of adrenergic derived cells in the adult mouse heart. PLoS ONE 2011, 6, e22811. [Google Scholar] [CrossRef]
- Winkler, H.; Apps, D.K.; Fischer-Colbrie, R. The molecular function of adrenal chromaffin granules: Established facts and unresolved topics. Neuroscience 1986, 18, 261–290. [Google Scholar] [CrossRef]
- Crivellato, E.; Nico, B.; Ribatti, D. The chromaffin vesicle: Advances in understanding the composition of a versatile, multifunctional secretory organelle. Anat. Rec. (Hoboken) 2008, 291, 1587–1602. [Google Scholar] [CrossRef] [PubMed]
- Estévez-Herrera, J.; González-Santana, A.; Baz-Dávila, R.; Machado, J.D.; Borges, R. The intravesicular cocktail and its role in the regulation of exocytosis. J. Neurochem. 2016, 137, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estevez-Herrera, J.; Pardo, M.R.; Dominguez, N.; Pereda, D.; Machado, J.D.; Borges, R. The role of chromogranins in the secretory pathway. Biomol. Concepts 2013, 4, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Tao-Cheng, J.H.; Eiden, L.E.; Loh, Y.P. Chromogranin A, an “on/off” switch controlling dense-core secretory granule biogenesis. Cell 2001, 106, 499–509. [Google Scholar] [CrossRef]
- Stenman, A.; Svahn, F.; Hoijat-Farsangi, M.; Zedenius, J.; Söderkvist, P.; Gimm, O.; Larsson, C.; Juhlin, C.C. Molecular profiling of pheochromocytoma and abdominal paraganglioma stratified by the PASS algorithm reveals chromogranin B as associated with histologic prediction of malignant behavior. Am. J. Surg. Pathol. 2019, 43, 409–421. [Google Scholar] [CrossRef]
- Zuber, S.; Wesley, R.; Prodanov, T.; Eisenhofer, G.; Pacak, K.; Kantorovich, V. Clinical utility of chromogranin A in SDHx-related paragangliomas. Eur. J. Clin. Investig. 2014, 44, 365–371. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.; Deftos, L. Secretion of chromogranin A by peptide producing endocrine neoplasms. N. Engl. J. Med. 1986, 314, 1145–1151. [Google Scholar] [CrossRef]
- Hsiao, R.J.; Parmer, R.J.; Takiyyuddin, M.A.; O’Connor, D.T. Chromogranin A storage and secretion: Sensitivity and specificity for the diagnosis of pheochromocytoma. Medicine (Baltimore) 1991, 70, 33–45. [Google Scholar] [CrossRef]
- Stridsberg, M.; Husebye, E.S. Chromogranin A and chromogranin B are sensitive circulating markers for pheochromocytoma. Eur. J. Endocrinol. 1997, 136, 67–73. [Google Scholar] [CrossRef]
- Brouwers, F.M.; Gläsker, S.; Nave, A.F.; Vortmeyer, A.O.; Lubensky, I.; Huang, S.; Abu-Asab, M.S.; Eisenhofer, G.; Weil, R.J.; Park, D.M.; et al. Proteomic profiling of von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 pheochromocytomas reveals different expression of chromogranin B. Endocr. Relat. Cancer 2007, 14, 463–471. [Google Scholar] [CrossRef]
- Guillemot, J.; Thouënnon, E.; Guérin, M.; Vallet-Erdtmann, V.; Ravni, A.; Montéro-Hadjadje, M.; Lefebvre, H.; Klein, M.; Muresan, M.; Seidah, N.G.; et al. Differential expression and processing of secretogranin II in relation to the status of pheochromocytoma: Implications for the production of the tumoral marker EM66. J. Mol. Endocrinol. 2012, 48, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Huh, Y.H.; Jeon, S.H.; Yoo, S.H. Chromogranin B-induced secretory granule biogenesis: Comparison with the similar role of chromogranin A. J. Biol. Chem. 2003, 278, 40581–40589. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, J.; Guerin, M.; Thouënnon, E.; Montero-Hadjadje, M.; Leprince, J.; Lefebvre, H.; Klein, M.; Muresan, M.; Anouar, Y.; Yon, L. Characterization and plasma measurement of the WE-14 peptide in patients with pheochromocytoma. PLoS ONE 2014, 9, e88698. [Google Scholar] [CrossRef] [PubMed]
- Cleary, S.; Phillips, J.K.; Huynh, T.T.; Pacak, K.; Elkahloun, A.G.; Barb, J.; Worrell, R.A.; Goldstein, D.S.; Eisenhofer, G. Neuropeptide Y expression in phaeochromocytomas: Relative absence in tumours from patients with von Hippel-Lindau syndrome. J. Endocrinol. 2007, 193, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, J.; Barbier, L.; Thouënnon, E.; Vallet-Erdtmann, V.; Montero-Hadjadje, M.; Lefebvre, H.; Klein, M.; Muresan, M.; Plouin, P.F.; Sei dah, N.; et al. Expression and processing of the neuroendocrine protein secretogranin II in benign and malignant pheochromocytomas. Ann. N. Y. Acad. Sci. 2006, 107, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Stridsberg, M.; Eriksson, B.; Janson, E.T. Measurement of secretogranins II, III, V and proconvertases 1/3 and 2 in plasma from patients with neuroendocrine tumours. Regul. Pept. 2008, 148, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Namimatsu, S.; Kitagawa, W.; Akasu, H.; Takatsu, K.; Sugisaki, Y.; Tanaka, S. Immunohistochemical, biochemical and immunoelectron microscopic analysis of antigenic proteins on neuroendocrine cell tumors using monoclonal antibody HISL-19. J. Nippon. Med. Sch. 2002, 69, 365–372. [Google Scholar] [CrossRef]
- Marcinkiewicz, M.; Benjannet, S.; Falgueyret, J.P.; Seidah, N.G.; Schurch, W.; Verdy, M.; Cantin, M.; Chretien, M. Identification and localization of 7B2 protein in human, porcine, and rat thyroid gland and in human medullary carcinoma. Endocrinology 1988, 123, 866–873. [Google Scholar] [CrossRef]
- Hacker, G.W.; Bishop, A.E.; Terenghi, G.; Varndell, I.M.; Aghahowa, J.; Pollard, K.; Thurner, J.; Polak, J.M. Multiple peptide production and presence of general markers detected in 12 cases of human pheochromocytoma and in mammalian adrenal glands. Virchows Arch. A Pathol. Anat. Histopathol. 1988, 412, 399–411. [Google Scholar] [CrossRef]
- Natori, S.; Iguchi, H.; Ohashi, M.; Nawata, H. Plasma 7B2 (a novel pituitary protein) immunoreactivity concentrations in patients with various endocrine disorders. Endocrinol. Jpn. 1988, 35, 651–654. [Google Scholar] [CrossRef]
- Srivastava, A.; Padilla, O.; Fischer-Colbrie, R.; Tischler, A.S.; Dayal, Y. Neuroendocrine secretory protein-55 (NESP-55) expression discriminates pancreatic endocrine tumors and pheochromocytomas from gastrointestinal and pulmonary carcinoids. Am. J. Surg. Pathol. 2004, 28, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, A.M.; Ahlman, H.; Kolby, L.; Abrahamsson, J.; Fischer-Colbrie, R.; Nilsson, O. NESP55, a novel chromogranin-like peptide, is expressed in endocrine tumours of the pancreas and adrenal medulla but not in ileal carcinoids. Br. J. Cancer 2003, 88, 1746–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindi, G.; Licini, L.; Necchi, V.; Bottarelli, L.; Campanini, N.; Azzoni, C.; Favret, M.; Giordano, G.; D’Amato, F.; Brancia, C.; et al. Peptide products of the neurotrophin-inducible gene vgf are produced in human neuroendocrine cells from early development and increase in hyperplasia and neoplasia. J. Clin. Endocrinol. Metab. 2007, 92, 2811–2815. [Google Scholar] [CrossRef] [PubMed]
- Salton, S.R. Neurotrophins, growth-factor-regulated genes and the control of energy balance. Mt. Sinai J. Med. 2003, 70, 93–100. [Google Scholar] [PubMed]
- Salton, S.R.; Ferri, G.L.; Hahm, S.; Snyder, S.E.; Wilson, A.J.; Possenti, R.; Levi, A. VGF: A novel role for this neuronal and neuroendocrine polypeptide in the regulation of energy balance. Front. Neuroendocr. 2000, 21, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Isobe, K.; Nakai, T.; Yukimasa, N.; Nanmoku, T.; Takekoshi, K.; Nomura, F. Expression of mRNA coding for four catecholaminesynthesizing enzymes in human adrenal pheochromocytomas. Eur. J. Endocrinol. 1998, 138, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Jarrot, B.; Louis, W.J. Abnormalities in enzymes involved in catecholamine synthesis and catabolism in phaeochromocytoma. Clin. Sci. Mol. Med. 1977, 53, 529–535. [Google Scholar] [CrossRef]
- Supek, F.; Supekova, L.; Mandiyan, S.; Pan, Y.C.E.; Nelson, H.; Nelson, N. A novel accessory subunit for vacuolar H1-ATPase from chromaffin granules. J. Biol. Chem. 1994, 269, 24102–24106. [Google Scholar]
- Balogh, A.; Cadel, S.; Foulon, T.; Picart, R.; der Garabedian, A.; Rousselet, A.; Tougard, C.; Cohen, P. Aminopeptidase B: A processing enzyme secreted and associated with the plasma membrane of rat pheochromocytoma (PC12) cells. J. Cell Sci. 1998, 111, 161–169. [Google Scholar]
- Azaryan, A.V.; Schiller, M.R.; Hook, V.Y. Chromaffin granule aspartic proteinase processes recombinant proopiomelanocortin (POMC). Biochem. Biophys. Res. Commun. 1995, 215, 937–944. [Google Scholar] [CrossRef]
- Murthy, S.R.; Pacak, K.; Loh, Y.P. Carboxypeptidase E: Elevated expression correlated with tumor growth and metastasis in pheochromocytomas and other cancers. Cell. Mol. Neurobiol. 2010, 30, 1377–1381. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.R.K.; Dupart, E.; Al-Sweel, N.; Chen, A.; Cawley, N.X.; Loh, Y.P. Carboxypeptidase E promotes cancer cell survival, but inhibits migration and invasion. Cancer Lett. 2013, 341, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasothornsrikul, S.; Greenbaum, D.; Medzihradszky, K.F.; Toneff, T.; Bundey, R.; Miller, R.; Schilling, B.; Petermann, I.; Dehnert, J.; Logvinova, A.; et al. Cathepsin L in secretory vesicles functions as a prohormone-processing enzyme for production of the enkephalin peptide neurotransmitter. Proc. Natl. Acad. Sci. USA 2003, 100, 9590–9595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funkelstein, L.; Beinfeld, M.; Minokadeh, A.; Zadina, J.; Hook, V. Unique biological function of cathepsin L in secretory vesicles for biosynthesis of neuropeptides. Neuropeptides 2010, 44, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laslop, A.; Weiss, C.; Savaria, D.; Eiter, C.; Tooze, S.A.; Seidah, N.G.; Winkler, H. Proteolytic processing of chromogranin B and secretogranin II by prohormone convertases. J. Neurochem. 1998, 70, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Parmer, R.J.; Mahata, M.; Mahata, S.; Sebald, M.T.; O’Connor, D.T.; Miles, L.A. Tissue plasminogen activator (t-PA) is targeted to the regulated secretory pathway. Catecholamine storage vesicles as a reservoir for the rapid release of t-PA. J. Biol. Chem. 1997, 272, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Parmer, R.J.; Mahata, M.; Gong, Y.; Mahata, S.K.; Jiang, Q.; O’Connor, D.T.; Xi, X.P.; Miles, L.A. Processing of chromogranin A by plasmin provides a novel mechanism for regulating catecholamine secretion. J. Clin. Investig. 2000, 106, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.R.; Bundey, R.; Toneff, T.; Hook, V. Endopin serpin protease inhibitors localize with neuropeptides in secretory vesicles and neuroendocrine tissues. Neuroendocrinology 2009, 89, 210–216. [Google Scholar] [CrossRef]
- Thouënnon, E.; Piere, A.; Yon, L.; Anouar, Y. Expression of trophic peptides and their receptors in chromaffin cells and pheochromocytoma. Cell. Mol. Neurobiol. 2010, 30, 1383–1389. [Google Scholar] [CrossRef]
- Hu, W.; Shi, L.; Zhou, P.H.; Zhang, X.B. Plasma concentrations of adrenomedullin and atrial and brain natriuretic peptides in patients with adrenal pheochromocytoma. Oncol. Lett. 2015, 10, 3163–3170. [Google Scholar] [CrossRef]
- Shimosawa, T.; Fujita, T. Adrenomedullin and its related peptide. Endocr. J. 2005, 52, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zudaire, E.; Martinez, A.; Cuttitta, F. Adrenomedullin and cancer. Regul. Pept. 2003, 112, 175–183. [Google Scholar] [CrossRef]
- Morimoto, R.; Satoh, F.; Murakami, O.; Hirose, T.; Totsune, K.; Imai, Y.; Arai, Y.; Suzuki, T.; Sasano, H.; Ito, S.; et al. Expression of adrenomedullin 2/intermedin in human adrenal tumors and attached non-neoplastic adrenal tissues. J. Endocrinol. 2008, 198, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chejfec, G.; Lee, I.; Warren, W.H.; Gould, V.E. Bombesin in human neuroendocrine (NE) neoplasms. Peptides 1985, 6, 107–112. [Google Scholar] [CrossRef]
- Bostwick, D.G.; Bensch, K.G. Gastrin releasing peptide in human neuroendocrine tumours. J. Pathol. 1985, 147, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Linnoila, R.I.; Lack, E.E.; Steinberg, S.M.; Keiser, H.R. Decreased expression of neuropeptides in malignant paragangliomas: An immunohistochemical study. Hum. Pathol. 1988, 19, 41–50. [Google Scholar] [CrossRef]
- Fedorak, I.; Prinz, R.A.; Fiscus, R.R.; Wang, X.; Chaumont, J.; Chejfec, G.; Glisson, S. Plasma calcitonin gene-related peptide and atrial natriuretic peptide levels during resection of pheochromocytoma. Surgery 1991, 110, 1094–1098. [Google Scholar] [PubMed]
- Mazzocchi, G.; Musajo, F.G.; Neri, G.; Gottardo, G.; Nussdorfer, G.G. Adrenomedullin stimulates steroid secretion by the isolated perfused rat adrenal gland in situ: Comparison with calcitonin gene-related peptide effects. Peptides 1996, 17, 853–857. [Google Scholar] [CrossRef]
- Mahata, S.K.; Mahata, M.; Fung, M.; O’Connor, D.T. Catestatin: A multifunctional peptide from chromogranin A. Regul. Pept. 2010, 162, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahata, S.K.; O’Connor, D.T.; Mahata, M.; Yoo, S.H.; Taupenot, L.; Wu, H.; Gill, B.M.; Parmer, R.J. Novel autocrine feedback control of catecholamine release. A discrete chromogranin a fragment is a noncompetitive nicotinic cholinergic antagonist. J. Clin. Investig. 1997, 100, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, J.; Ait-Ali, D.; Turquier, V.; Montero-Hadjadje, M.; Fournier, A.; Vaudry, H.; Anouar, Y.; Yon, L. Involvement of multiple signalling pathways in PACAP induced EM66 secretion from chromaffin cells. Regul. Pept. 2006, 137, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Yon, L.; Guillemot, J.; Montero-Hadjadje, M.; Grumolato, L.; Leprince, J.; Lefebvre, H.; Contesse, V.; Plouin, P.F.; Vaudry, H.; Anouar, Y. Identification of the secretogranin II-derived peptide EM66 in pheochromocytomas as a potential marker for discriminating benign versus malignant tumors. J. Clin. Endocrinol. Metab. 2003, 88, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Babinski, K.; Haddad, P.; Vallerand, D.; McNicoll, N.; De Lean, A.; Ong, H. Natriuretic peptides inhibit nicotine-induced whole-cell currents and catecholamine secretion in bovine chromaffin cells: Evidence for the involvement of the atrial natriuretic factor R2 receptors. J. Neurochem. 1995, 64, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Suga, S.; Nakao, K.; Mukoyama, M.; Arai, H.; Hosoda, K.; Ogawa, Y.; Imura, H. Characterization of natriuretic peptide receptors in cultured cells. Hypertension 1992, 19, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Brill, A.; McCrink, K.A. GPCRs of adrenal chromaffin cells and catecholamines: The plot thickens. Int. J. Biochem. Cell Biol. 2016, 77, 213–219. [Google Scholar] [CrossRef]
- Spinazzi, R.; Andreis, P.G.; Nussdorfer, G. Neuropeptide Y and Y receptors in the autocrine-paracrine regulation of adrenal gland under physiological and pathophysiological conditions (review). Int. J. Mol. Med. 2005, 15, 3–13. [Google Scholar] [CrossRef]
- Cavadas, C.; Cefai, D.; Rosmaninho-Salgado, J.; Vieira-Coelho, M.A.; Moura, E.; Busso, N.; Pedrazzini, T.; Grand, D.; Rotman, S.; Waeber, B.; et al. Deletion of the neuropeptide Y (NPY) Y1 receptor gene reveals a regulatory role of NPY on catecholamine synthesis and secretion. Proc. Natl. Acad. Sci. USA 2006, 103, 10497–10502. [Google Scholar] [CrossRef]
- Kitlinska, J.; Abe, K.; Kuo, L.; Pons, J.; Yu, M.; Li, L.; Tilan, J.; Everhart, L.; Lee, E.W.; Zukowska, Z.; et al. Differential effects of neuropeptide Y on the growth and vascularizaton of neural crest-derived tumors. Cancer Res. 2005, 65, 1719–1728. [Google Scholar] [CrossRef]
- Helman, L.J.; Cohen, P.S.; Averbuch, S.D.; Cooper, M.J.; Keiser, H.R.; Isreal, M.A. Neuropeptide Y expression distinguishes malignant from benign pheochromocytoma. J. Clin. Oncol. 1989, 7, 1720–1725. [Google Scholar] [CrossRef]
- Grouzmann, E.; Comoy, E.; Bohuon, C. Plasma neuropeptide Y concentration in patients with neuroendocrine tumors. J. Clin. Endocrinol. Metab. 1989, 68, 808–813. [Google Scholar] [CrossRef]
- Tischler, A.S.; Lee, Y.C.; Perlman, R.L.; Costopoulos, D.; Slayton, V.W.; Bloom, S.R. Production of “ectopic” vasoactive intestinal peptide-like and Neurotensin-like immunoreactivity in human pheochromocytoma cell cultures. J. Neurosci. 1984, 4, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S.; Lee, Y.C.; Slayton, V.W.; Bloom, S.R. Content and release of neurotensin in PC12 pheochromocytoma cell cultures: Modulation by dexamethasone and nerve growth factor. Regul. Pept. 1982, 3, 415–421. [Google Scholar] [CrossRef]
- Pellizzari, E.H.; Barontini, M.; Figuerola, M.; Cigorraga, S.B.; Levin, G. Possible autocrine enkephalin regulation of catecholamine release in human pheochromocytoma cells. Life Sci. 2008, 83, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Bark, S.J.; Lu, W.D.; Taupenot, L.; O’Connor, D.T.; Pevzner, P.; Hook, V. Mass spectrometry based neuropeptidomics of secretory vesicles form human adrenal medullary pheochromocytoma reveals novel peptide products of prohormone processing. J. Proteome Res. 2010, 9, 5065–5075. [Google Scholar] [CrossRef] [PubMed]
- Yoshimasa, T.; Nakao, K.; Li, S.; Ikeda, Y.; Suda, M.; Sakamoto, M.; Imura, H. Plasma methionine-enkephalin and leucine-enkephalin in normal subjects and patients with pheochromocytoma. J. Clin. Endocrinol. Metab. 1983, 57, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; Gandia, L.; Michelena, P.; Gilabert, J.A.; del Valle, M.; Carbone, E.; Garcia, A.G. The mechanism of calcium channel facilitation in bovine chromaffin cells. J. Physiol. 1996, 494, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Thouënnon, E.; Piere, A.; Tanguy, Y.; Guillemot, J.; Manecka, D.L.; Guérin, M.; Ouafik, L.; Muresan, M.; Klein, M.; Bertherat, J.; et al. Expression of trophic amidated peptides and their receptors in benign and malignant pheochromocytomas: High expression of adrenomedullin RDC1 receptor and implication in tumoral cell survival. Endocr. Relat. Cancer 2010, 17, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.B.; Eiden, L. Is PACAP the major neurotransmitter for stress transduction at the adrenomedullary synapse? J. Mol. Neurosci. 2012, 48, 403–412. [Google Scholar] [CrossRef]
- Turquier, V.; Yon, L.; Grumolato, L.; Alexandre, D.; Fournier, A.; Vaudry, H.; Anouar, Y. Pituitary adenylate cyclase-activating polypeptide stimulates secretoneurin release and secretogranin II gene transcription in bovine adrenochromaffin cells through multiple signalling pathways and increased binding of pre-existing activator protein-1-like transcription factors. Mol. Pharm. 2001, 60, 42–52. [Google Scholar]
- Fischer-Colbrie, R.; Kirchmair, R.; Kahler, C.M.; Wiedermann, C.J.; Saria, A. Secretoneurin: A new player in angiogenesis and chemotaxis linking nerves, blood vessels and the immune system. Curr. Protein Pept. Sci. 2005, 6, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Wiedermann, C.J. Secretoneurin: A functional neuropeptide in health and disease. Peptides 2000, 21, 1289–1298. [Google Scholar] [CrossRef]
- Unsicker, K.; Krieglstein, K. Growth factors in chromaffin cells. Prog. Neurobiol. 1996, 48, 307–324. [Google Scholar] [CrossRef]
- Combs, S.E.; Ernsberger, U.; Krieglstein, K.; Unsicker, K. Reduction of endogenous TGF-B does not affect phenotypic development of sympathoadrenal progenitors into adrenal chromaffin cells. Mech. Dev. 2001, 109, 295–302. [Google Scholar] [CrossRef]
- Flanders, K.C.; Lüdecke, G.; Engels, S.; Cissel, D.S.; Roberts, A.B.; Kondaiah, P.; Lafyatis, R.; Sporn, M.B.; Unsicker, K. Localization and actions of transforming growth factor-Bs in the embryonic nervous system. Development 1991, 113, 183–191. [Google Scholar] [PubMed]
- Lugardon, K.; Raffner, R.; Goumon, Y.; Corti, A.; Delmas, A.; Bulet, P.; Aunis, D.; Metz-Boutigue, M.H. Antibacterial and antifungal activities of vasostatin-1, the N-terminal fragment of chromogranin A. J. Biol. Chem. 2000, 275, 10745–10753. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.; Gee, P.; Liu, S.M.; Angeletti, R.H. Inhibition of parathyroid hormone secretion by amino-terminal chromogranin peptides. Endocrinology 1994, 135, 227–342. [Google Scholar] [CrossRef]
- Aardal, S.; Helle, K.B. The vasoinhibitory activity of bovine chromogranin A fragment (vasostatin) and its independence of extracellular calcium in isolated segments of human blood vessels. Regul. Pept. 1992, 41, 9–18. [Google Scholar] [CrossRef]
- Strub, J.M.; Goumon, Y.; Lugardon, K.; Capon, C.; Lopez, M.; Moniatte, M.; van Dorsselaer, A.; Aunis, D.; Metz-Boutigue, M.H. Antibacterial activity of glycosylated and phosphorylated chromogranin-A derived peptide 173–184 from bovine adrenal medullary chromaffin granules. J. Biol. Chem. 1996, 271, 28533–28540. [Google Scholar] [CrossRef]
- Strub, J.M.; Garcia-Sablone, P.; Lonning, K.; Taupenot, L.; Hubert, P.; Van Dorsselaer, A.; Aunis, D.; Metz-Boutigue, M.H. Processing of chromogranin B in bovine adrenal medulla. Identification of secretolytin, the endogenous C-terminal fragment of residues 614–626 with antibacterial activity. Eur. J. Biochem. 1995, 229, 356–368. [Google Scholar] [CrossRef]
- Metz-Boutigue, M.H.; Kieffer, A.E.; Goumon, Y.; Aunis, D. Innate immunity: Involvement of new neuropeptides. Trends Microbiol. 2003, 11, 585–592. [Google Scholar] [CrossRef]
- Levine, E.Y.; Levenberg, B.; Kaufman, S. The enzymatic conversion of 3,4-dihydroxyphenylethylamine to norepinephrine. J. Biol. Chem. 1960, 235, 2080–2086. [Google Scholar]
- Schlüter, H.; Meissner, M.; van der Giet, M.; Tepel, M.; Bachmann, J.; Gross, I.; Nordhoff, E.; Karas, M.; Spieker, C.; Witzel, H.; et al. Coenzyme A glutathione disulfide. A potent vasoconstrictor derived from the adrenal gland. Circ. Res. 1995, 76, 675–680. [Google Scholar] [CrossRef]
- Burgoyne, R.D.; Morgan, A. Secretory granule exocytosis. Physiol. Rev. 2003, 83, 581–632. [Google Scholar] [CrossRef] [PubMed]
- Tadros, T.S.; Strauss, R.M.; Cohen, C.; Gal, A.A. Galanin immunoreactivity in paragangliomas but not in carcinoid tumors. Appl. Immunohistochem. Mol. Morphol. 2003, 11, 250–252. [Google Scholar] [CrossRef]
- Bauer, F.E.; Hacker, G.W.; Terenghi, G.; Adrian, T.E.; Polak, J.M.; Bloom, S.R. Localization and molecular forms of galanin in human adrenals: Elevated levels in pheochromocytomas. J. Clin. Endocrinol. Metab. 1986, 63, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Painter, G.R.; Diliberto, E.J.; Knoth, J. 31P nuclear magnetic resonance study of the metabolic pools of adenosine triphosphate in cultured bovine adrenal medullary chromaffin cells. Proc. Natl. Acad. Sci. USA 1989, 86, 2239–2242. [Google Scholar] [CrossRef] [PubMed]
- Sala, F.; Nistri, A.; Criado, M. Nicotinic acetylcholine receptors of adrenal chromaffin cells. Acta Physiol. 2008, 192, 203–212. [Google Scholar] [CrossRef]
- Simasko, S.M.; Durkin, J.A.; Weiland, G.A. Effects of substance P on nicotinic acetylcholine receptor function in PC12 cells. J. Neurochem. 1987, 49, 253–260. [Google Scholar] [CrossRef]
- Zhou, X.F.; Livett, B.G. Substance P has biphasic effects on catecholamine secretion evoked by electrical stimulation of perfused rat adrenal glands in vitro. J. Auton. Nerv. Syst. 1990, 31, 31–39. [Google Scholar] [CrossRef]
- Oehme, P.; Hecht, H.D.; Faulhaber, K.; Nieber, I.; Roske, I.; Rathsack, R. Relationship of substance P to catecholamines, stress and hypertension. J. Cardiovasc. Pharmacol. 1987, 10, 109–111. [Google Scholar] [CrossRef]
- Vinik, A.I.; Shapiro, B.; Thompson, N.W. Plasma gut hormone levels in 37 patients with pheochromocytomas. World J. Surg. 1986, 10, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Cao, W.; Zhao, M. Octreotide reverses shock due to vasoactive intestinal peptide-secreting adrenal pheochromocytoma: A case report and review of literature. World J. Clin. Cases 2018, 6, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, L.; Wu, Z.; Ai, Z.; Hou, Y.; Lu, Z.; Gao, X. A rare case of watery diarrhea, hypokalemia and achlorhydria syndrome caused by pheochromocytoma. BMC Cancer 2014, 14, 533. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz-Amit, R.; Mete, O.; Asa, S.L.; Ezzat, S.; Joshua, A.M. Malignant pheochromocytoma secreting vasoactive intestinal peptide and response to Sunitinib: A case report and literature review. Endocr. Pr. 2014, 20, e145–e150. [Google Scholar] [CrossRef] [PubMed]
- Ghzili, H.; Grumolato, L.; Thouennon, E.; Tanguy, Y.; Turquier, V.; Vaudry, H.; Anouar, Y. Role of PACAP in the physiology and pathology of the sympathoadrenal system. Front. Neuroendocrinol. 2008, 29, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Henry, J.P.; Sagne, C.; Bedet, C.; Gasnier, B. The vesicular monoamine transporter: From chromaffin granule to brain. Neurochem. Int. 1998, 32, 227–246. [Google Scholar] [CrossRef]
- Wimalasena, K. Vesicular monoamine transporters: Structure-function, pharmacology, and medicinal chemistry. Med. Res. Rev. 2011, 31, 483–519. [Google Scholar] [CrossRef]
- Schuldiner, S.; Shirvan, A.; Linial, M. Vesicular neurotransmitter transporters: From bacteria to humans. Physiol. Rev. 1995, 75, 369–392. [Google Scholar] [CrossRef]
- Hillarp, N.A.; Hokfelt, B. Evidence of adrenaline and noradrenaline in separate adrenal medullary cells. Acta Physiol. Scand 1953, 30, 55–68. [Google Scholar] [CrossRef]
- Carmichael, S.W. The adrenal chromaffin vesicle: An historical perspective. J. Auton. Nerv. Syst. 1983, 7, 7–12. [Google Scholar] [CrossRef]
- Eisenhofer, G. The role of neuronal and extraneuronal plasma membrane transporters. Pharmacol. Ther. 2001, 91, 35–62. [Google Scholar] [CrossRef]
- Huynh, T.T.; Pacak, K.; Brouwers, F.M.; Abu-Asab, M.S.; Worrel, R.A.; Walther, M.M.; Elkahloun, A.G.; Goldstein, D.S.; Cleary, S.; Eisenhofer, G. Different expression of catecholamine transporters in phaeochromocytomas from patients with von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2. Eur. J. Endocrinol. 2005, 153, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H.J.; Insel, P.A. Adrenergic receptors in man: Direct identification, physiologic regulation, and clinical alterations. N. Engl. J. Med. 1982, 307, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, S.; Moura, D. Vascular Adrenoceptors: An update. Pharm. Rev. 2001, 53, 319–356. [Google Scholar] [PubMed]
- Aunis, D.; Langley, K. Physiological aspects of exocytosis in chromaffin cells of the adrenal medulla. Acta Physiol. Scand 1999, 167, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.L.; Anderson, L.J.; Tai, T.C. Cholinergic and peptidergic regulation of phenylethanolamine N-methyltransferase gene expression. Ann. N. Y. Acad. Sci. 2002, 971, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Huynh, T.T.; Elkahloun, A.; Morris, J.C.; Bratslavsky, G.; Linehan, W.M.; Zhuang, Z.; Balgley, B.M.; Lee, C.S.; Manelli, M.; et al. Differential expression of the regulated catecholamine secretory pathway in different hereditary forms of pheochromocytoma. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1223–E1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colomer, C.; Desarmenien, M.G.; Guerineau, N.C. Revisiting the stimulus-secretion coupling in the adrenal medulla: Role of gap junction-mediated intercellular communication. Mol. Neurobiol. 2009, 40, 87–100. [Google Scholar] [CrossRef] [PubMed]
- García, A.G.; Garciá-de Diego, A.M.; Gandía, L.; Borges, R.; García-Sancho, J. Calcium signalling and exocytosis in adrenal chromaffin cells. Physiol. Rev. 2006, 86, 1093–1131. [Google Scholar] [CrossRef] [PubMed]
- Marengo, F.D.; Cardenas, A.M. How does the stimulus define exocytosis in adrenal chromaffin cells (review). Pflug. Arch. 2018, 470, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Trifaro, J.M.; Gasman, S.; Gutierrez, L.M. Cytoskeletal control of vesicle transport and exocytosis in chromaffin cells. Acta Physiol. (Oxf.) 2008, 192, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.; Morgan, A. Analysis of regulated exocytosis in adrenal chromaffin cells: Insights into NSF/SNAP/SNARE function. BioEssays 1998, 20, 328–335. [Google Scholar] [CrossRef]
- Holz, R.W.; Brondyk, W.H.; Senter, R.A.; Kuizon, L.; Macara, I.G. Evidence for the involvement of Rab3A in Ca(2+)-dependent exocytosis from adrenal chromaffin cells. J. Biol. Chem. 1994, 269, 10229–10234. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D.; Morgan, A.; Robinson, I.; Pender, N.; Cheek, T.R. Exocytosis in adrenal chromaffin cells. J. Anat. 1993, 183, 309–314. [Google Scholar]
- Umbrecht-Jenck, E.; Demais, V.; Calco, V.; Bailly, Y.; Bader, M.F.; Chasserot-Golaz, S. S100A10 mediated translocation of annexin A2 to SNARE proteins in adrenergic chromaffin cells undergoing exocytosis. Traffic 2010, 11, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Gabel, A.G.; Chasserot-Golaz, S. Annexin A2, an essential partner of the exocytotic process in chromaffin cells. J. Neurochem. 2016, 137, 890–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavadas, C.; Silva, A.L.P.; Mosimann, F.; Cotrim, M.D.; Ribeiro, C.A.; Brunner, H.R.; Grouzmann, E. NPY regulates catecholamine secretion from human adrenal chromaffin cells. J. Clin. Endocrinol. Metab. 2001, 86, 5956–5963. [Google Scholar] [CrossRef]
- Fishbein, L.; Merrill, S.; Fraker, D.L.; Cohen, D.L.; Nathanson, K.L. Inherited mutations in pheochromocytoma and paraganglioma: Why all patients should be offered genetic testing. Ann. Surg. Oncol. 2013, 20, 1440–1450. [Google Scholar] [CrossRef]
- Dahia, P.L. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Crona, J.; Nordling, M.; Maharjan, R.; Granberg, D.; Stålberg, P.; Hellman, P.; Björklund, P. Integrative genetic characterization and phenotype correlations in pheochromocytoma and paraganglioma tumours. PLoS ONE 2014, 9, e86756. [Google Scholar] [CrossRef]
- Burnichon, N.; Buffet, A.; Gimenez-Roqueplo, A.P. Pheochromocytoma and paraganglioma: Molecular testing and personalized medicine. Curr. Opin. Oncol. 2016, 28, 5–10. [Google Scholar] [CrossRef] [PubMed]
- NGS in PPGL (NGSnPPGL) Study Group; Toledo, R.A.; Burnichon, N.; Cascon, N.; Cascon, A.; Benn, D.E.; Bayley, J.P.; Welander, J.; Tops, C.M.; Firth, H.; et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat. Rev. Endocrinol. 2017, 13, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L.; Ross, K.N.; Wright, M.E.; Hayashida, C.Y.; Santagata, S.; Barontini, M.; Kung, A.L.; Sanso, G.; Powers, J.F.; Tischler, A.S.; et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005, 1, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Huynh, T.T.; Pacak, K.; Brouwers, F.M.; Walther, M.M.; Linehan, W.M.; Munson, P.J.; Mannelli, M.; Goldstein, D.S.; Elkahloun, A.G. Distinct gene expression profiles in norepinephrine- and epinephrine- producing hereditary and sporadic pheochromocytomas: Activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr. Relat. Cancer 2004, 11, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Crona, J.; Taïeb, D.; Pacak, K. New perspectives on pheochromocytoma and paraganglioma: Toward a molecular classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Wilkerson, M.D. Chromaffin cell biology: Interferences from the Cancer Genome Atlas. Cell Tissue Res. 2018, 372, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Calsina, B.; Curras-Freixes, M.; Buffet, A.; Pons, T.; Contreras, L.; Leton, R.; Comino-Mendez, I.; Remacha, L.; Calatayud, M.; Obispo, B.; et al. Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients. Genet. Med. 2018, 20, 1652–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, S.; Gieldon, L.; Pang, Y.; Peitzsch, M.; Huynh, T.; Leton, R.; Viana, B.; Ercolino, T.; Mangelis, A.; Rapizzi, E.; et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet. Med. 2019, 21, 705–717. [Google Scholar] [CrossRef]
- Yang, C.; Zhuang, Z.; Fliedner, S.M.; Shankavaram, U.; Sun, M.G.; Bullova, P.; Zhu, R.; Elkahloun, A.G.; Kourlas, P.J.; Merino, M. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J. Mol. Med. 2015, 93, 93–104. [Google Scholar] [CrossRef]
- Alrezk, R.; Suarez, A.; Tena, I.; Pacak, K. Update of pheochromocytoma syndromes: Genetics, biochemical evaluation and imaging. Front. Endocrinol. (Lausanne) 2018, 9, 515. [Google Scholar] [CrossRef] [PubMed]
- Crona, J.; Delgado Verdugo, A.; Maharjan, R.; Stålberg, P.; Granberg, D.; Hellman, P.; Björklund, P. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J. Clin. Endocrinol. Metab. 2013, 98, E1266–E1271. [Google Scholar] [CrossRef] [PubMed]
- Nakada, T.; Furuta, H.; Katayama, T. Catecholamine metabolism in pheochromocytoma and normal adrenal medullae. J. Urol. 1988, 140, 1348–1351. [Google Scholar] [CrossRef]
- Kimura, N.; Miura, Y.; Nagatsu, I.; Nagura, H. Catecholamine synthesizing enzymes in 70 cases of functioning and non-functioning phaeochromocytoma and extra-adrenal paraganglioma. Virchows Arch. A Pathol. Anat. Histopathol. 1992, 421, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, H.; Imai, T.; Tanaka, Y.; Tobinaga, J.; Wada, M.; Matsuyama, T.; Tsukamura, K.; Yamada, F.; Takagi, H.; Narita, T.; et al. Discrepancy between PNMT presence and relative lack of adrenaline production in extra-adrenal pheochromocytoma. J. Surg. Oncol. 1994, 57, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Meijer, W.G.; Copray, S.C.; Hollema, H.; Kema, I.P.; Zwart, N.; Mantingh-Otter, I.; Links, T.P.; Willemse, P.H.; de Vries, E.G. Catecholamine-synthesizing enzymes in carcinoid tumors and pheochromocytomas. Clin. Chem. 2003, 49, 586–593. [Google Scholar] [CrossRef]
- Grouzmann, E.; Tschopp, O.; Triponez, F.; Matter, M.; Bilz, S.; Brändle, M.; Drechser, T.; Sigrist, S.; Zulewski, H.; Henzen, C.; et al. Catecholamine metabolism in paraganglioma and pheochromocytoma: Similar tumors in different sites? PLoS ONE 2015, 10, e0125426. [Google Scholar] [CrossRef] [PubMed]
- Konosu-Fukaya, S.; Omata, K.; Tezuka, Y.; Ono, Y.; Aoyama, Y.; Satoh, F.; Fujishima, F.; Sasano, H.; Nakamura, Y. Catecholamine-synthesizing enzymes in pheochromocytoma and extra adrenal paraganglioma. Endocr. Pathol. 2018, 29, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Iwase, K.; Nagasaka, A.; Nagatsu, I.; Kikuchi, K.; Nagatsu, T.; Funahashi, H.; Tsujimura, T.; Inagaki, A.; Nakai, A.; Kishikawa, T.; et al. Tyrosine hydroxylase induces cell differentiation of catecholamine biosynthesis in neuroendocrine tumors. J. Endocrinol. Investig. 1994, 17, 235–239. [Google Scholar] [CrossRef]
- Feldman, J.M. Phenylethanolamine-N-methyltransferase activity determines the epinephrine concentration of pheochromocytomas. Res. Commun. Chem. Pathol. Pharm. 1981, 34, 389–398. [Google Scholar] [CrossRef]
- Fliedner, S.M.; Breza, J.; Kvetnansky, R.; Powers, J.F.; Tischler, A.S.; Wesley, R.; Merino, M.; Lehnert, H.; Pacak, K. Tyrosine hydroxylase, chromogranin A, and steroidogenic acute regulator as markers for successful separation of human adrenal medulla. Cell Tissue Res. 2010, 340, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Feldman, J.M.; Blalock, J.A.; Zern, R.T.; Wells, S.A., Jr. The relationship between enzyme activity and the catecholamine content and secretion of pheochromocytomas. J. Clin. Endocrinol. Metab. 1979, 49, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.A.; Gray, D.K. Dopamine-secreting pheochromocytomas: In search of a syndrome. World J. Surg. 2005, 29, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Lenders, J.W.; Timmers, H.; Mannelli, M.; Grebe, S.K.; Hofbauer, L.C.; Bornstein, S.R.; Tiebel, O.; Adams, K.; Bratslavsky, G.; et al. Measurements of plasma Methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin. Chem. 2011, 57, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Pacak, K.; Huynh, T.T.; Qin, N.; Bratslavsky, G.; Linehan, W.M.; Mannelli, M.; Friberg, P.; Timmers, H.J.; Bornstein, S.R.; et al. Catecholamine metabolomics and secretory phenotypes in phaeochromocytoma. Endocr. Relat. Cancer 2011, 18, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Timmers, H.J.; Pacak, K.; Huynh, T.T.; Abu-Asab, M.; Tsokos, M.; Merino, M.J.; Baysal, B.E.; Adams, K.T.; Eisenhofer, G. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J. Clin. Endocrinol. Metab. 2008, 93, 4826–4832. [Google Scholar] [CrossRef]
- Dreijerink, K.M.A.; Rijken, J.A.; Compaijen, C.J.A.C.; Timmers, H.J.L.M.; van der Horst-Schrivers, A.N.A.; van Leeuwaarde, R.S.; van Dam, S.; Leemans, C.R.; van Dam, E.W.C.M.; Dickhoff, C.; et al. Biochemically silent sympathetic paraganglioma, pheochromocytoma or metastatic disease in SDHD mutation carriers. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Walther, M.; Huynh, T.T.; Li, S.T.; Bornstein, S.R.; Vortmeyer, A.; Mannelli, M.; Goldstein, D.S.; Linehan, W.M.; Lenders, J.W.; et al. Pheochromocytomas in von Hippel-Lindau Syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J. Clin. Endocrinol. Metab. 2001, 86, 1999–2008. [Google Scholar] [CrossRef]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef]
- Favier, J.; Briere, J.J.; Burnichon, N.; Riviere, J.; Vescovo, L.; Benit, P.; Giscos-Douriez, I.; De Reynies, A.; Bertherat, J.; Badoual, C.; et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS ONE 2009, 4, e7094. [Google Scholar] [CrossRef]
- Lopez-Jimenez, E.; Gomez-Lopez, G.; Leandro-Garcia, L.J.; Munoz, I.; Schiavi, F.; Montero-Conde, C.; de Cubas, A.A.; Ramires, R.; Landa, I.; Leskelä, S.; et al. Research resource: Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol. Endocrinol. 2010, 24, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Vescovo, L.; Amar, L.; Libé, R.; de Reynies, A.; Venisse, A.; Jouanno, E.; Laurendeau, I.; Parfait, B.; Bertherat, J.; et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum. Mol. Genet. 2011, 20, 3974–3985. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; de Cubas, A.A.; Garcia-Martin, R.; Richter, S.; Peitzsch, M.; Menschikowski, M.; Lenders, J.W.; Timmers, H.J.; Mannelli, M.; Opocher, G.; et al. Opposing effects of HIF1a and HIF2a on chromaffin cell phenotypic features and tumor cell proliferation: Insights from MYC associated factor X. Int. J. Cancer 2014, 135, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Hammer, R.E.; Matsumoto, A.M.; Russell, D.W.; McKnight, S.L. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998, 12, 3320–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favier, J.; Kempf, H.; Corvol, P.; Casc, J.M. Cloning and expression pattern of EPAS1 in the chicken embryo. Colocalization with tyrosine hydroxylase. FEBS Lett. 1999, 462, 19–24. [Google Scholar] [CrossRef]
- Nilsson, H.; Jögi, A.; Beckman, S.; Harris, A.L.; Poellinger, L.; Pahlman, S. HIF-2alpha expression in human fetal paraganglia and neuroblastoma: Relation to sympathetic differentiation, glucose deficiency, and hypoxia. Exp. Cell Res. 2005, 303, 447–456. [Google Scholar] [CrossRef]
- Wang, H.; Cui, J.; Yang, C.; Rosenblum, R.S.; Zhang, Q.; Song, Q.; Pang, Y.; Fang, F.; Sun, M.; Dmitriev, P. A transgenic mouse model of Pacak-Zhuang syndrome with an EPAS1 gain of function mutation. Cancers 2019, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Cascon, A.; Schiavi, F.; Morales, N.P.; Comino-Mendez, I.; Abermil, N.; Inglada-Perez, L.; de Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin. Cancer Res. 2012, 18, 2828–2837. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.V.; Osamura, R.Y.; Kloppel, G.; Rosai, J. WHO Classification of Tumours: Pathology and Genetics of Tumours of Endocrine Organs, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Tischler, A.S.; de Krijger, R.R. Pathology of pheochromocytoma and paraganglioma. Endocr. Relat. Cancer 2015, 22, 123–133. [Google Scholar] [CrossRef]
- Thompson, L.D. Pheochromocytoma of the adrenal gland scaled score (PASS) to separate benign from malignant neoplasms: A clinicopathological and immunophenotypic study of 100 cases. Am. J. Surg. Pathol. 2002, 26, 551–566. [Google Scholar] [CrossRef]
- Wu, D.; Tischler, A.S.; Lloyd, R.V.; DeLellis, R.A.; de Krijger, R.; van Nederveen, F.; Nosé, V. Obserer variation in the application of the pheochromocytoma of the adrenal gland scaled score. Am. J. Surg. Pathol. 2009, 33, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Takayanagi, R.; Takizawa, N.; Itagaki, E.; Katabami, T.; Kakoi, N.; Rakugi, H.; Ikeda, Y.; Tanabe, A.; Nigawara, T.; et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr. Relat. Cancer 2014, 21, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalat, A.; Fraenkel, M.; Doviner, V.; Salmon, A.; Gross, D.J. Malignant pheochromocytoma: Predictive factors of malignancy and clinical course in 16 patients at a single tertiary medical center. Endocrine 2010, 39, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Feng, L.; Johnson, M.M.; Ejaz, S.; Habra, M.A.; Rich, T.; Busaidy, N.; Cote, G.J.; Perrier, N.; Phan, A.; et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: Primary tumor size and primary tumor location as prognostic indicators. J. Clin. Endocrinol. Metab. 2011, 96, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Goffredo, P.; Sosa, J.A.; Roman, S.A. Malignant pheochromocytoma and paraganglioma; a population level analysis of long-term survival over two decades. J. Surg. Oncol. 2013, 107, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, O.; Young, W.F., Jr.; Iniguez-Ariza, N.M.; Kittah, N.E.; Gruber, L.; Bancos, C.; Tamhane, S.; Bancos, I. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J. Clin. Endocrinol. Metab. 2017, 102, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Hescot, S.; Curras-Freixes, M.; Deutschbein, T.; van Berkel, A.; Vezzosi, D.; Amar, L.; de la Fouchardiere, C.; Valdes, N.; Riccardi, F.; Do Cao, C.; et al. Prognosis of malignant pheochromocytoma and paraganglioma (MAPP Prono Study): A European Network for the Study of Adrenal Tumors Retrospective Study. J. Clin. Endocrinol. Metab. 2019, 104, 2367–2374. [Google Scholar] [CrossRef]
- Amar, L.; Bertherat, J.; Baudin, E.; Ajzenberg, C.; Bressac-de Paillerets, B.; Chabre, O.; Chamontin, B.; Delemer, B.; Giraud, S.; Murat, A. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 2005, 23, 8812–8818. [Google Scholar] [CrossRef]
- Amar, L.; Baudin, E.; Burnichon, N.; Peyrard, S.; Silvera, S.; Bertherat, J.; Bertagna, X.; Schlumberger, M.; Jeunemaitre, X.; Gimenez-Roqueplo, A.P.; et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 3822–3828. [Google Scholar] [CrossRef]
- Burnichon, N.; Rohmer, V.; Amar, L.; Herman, P.; Leboulleux, S.; Darrouzet, V.; Niccoli, P.; Gaillard, D.; Chabrier, G.; Chabolle, F.; et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 2817–2827. [Google Scholar] [CrossRef]
- Castro-Vega, L.J.; Buffet, A.; De Cubas, A.A.; Cascon, A.; Menara, M.; Khalifa, E.; Amar, L.; Azriel, S.; Bourdeau, I.; Chabre, O.; et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 2014, 23, 2440–2446. [Google Scholar] [CrossRef] [PubMed]
- Buffet, A.; Morin, A.; Castro-Vega, L.J.; Habarou, F.; Lussey-Lepoutre, C.; Letouzé, E.; Lefebvre, H.; Guilhem, I.; Haissaguerre, M.; Raingeard, I.; et al. Germline mutations in the mitochrondrial 2-oxoglutarate/malate carrier SLC25A11 gene confer a predisposition to metastatic paragangliomas. Cancer Res. 2018, 78, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Bausch, B.; Schiavi, F.; Ni, Y.; Welander, J.; Patocs, A.; Ngeow, J.; Wellner, U.; Malinoc, A.; Taschin, E.; Barbon, G.; et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 2017, 3, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Lenders, J.W.; Siegert, G.; Bornstein, S.R.; Friberg, P.; Milosevic, D.; Mannelli, M.; Linehan, W.M.; Adams, K.; Timmers, H.J.; et al. Plasma methoxytyramine; a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumor size, location and SDHB mutation status. Eur. J. Cancer 2012, 48, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, M.; Prejbisz, A.; Kroiβ, M.; Beuschlein, F.; Arlt, W.; Januszewicz, A.; Siegert, G.; Eisenhofer, G. Analysis of plasma 3-methoxytyramine, normetanephrine and metanephrine by ultraperformance liquid chromatography tandem mass spectrometry: Utility for diagnosis of dopamineproducing metastatic phaeochromocytoma. Ann. Clin. Biochem. 2013, 50, 147–155. [Google Scholar] [CrossRef]
- Van der Harst, E.; de Herder, W.W.; de Krijger, R.R.; Bruining, H.A.; Bonjer, H.J.; Lamberts, S.W.; van den Meiracker, A.H.; Stijnen, T.H.; Boomsma, F. The value of plasma markers for the clinical behaviour of phaeochromocytomas. Eur. J. Endocrinol. 2002, 147, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sue, M.; Martucci, V.; Frey, F.; Lenders, J.M.; Timmers, H.J.; Peczkowska, M.; Prejbisz, A.; Swantje, B.; Bornstein, S.R.; Arlt, W.; et al. Lack of utility of SDHB mutation testing in adrenergic metastatic phaeochromocytoma. Eur. J. Endocrinol. 2015, 172, 89–95. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Richter, S.; Peitzsch, M.; Rapizzi, E.; Lenders, J.W.; Qin, N.; de Cubas, A.A.; Schiavi, F.; Rao, J.U.; Beuschlein, F.; Quinkler, M.; et al. Krebs cycle metabolite profiling for identification and stratification of pheochromocytoma/paragangliomas due to succinate dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 2014, 99, 3903–3911. [Google Scholar] [CrossRef]
- Ploumakis, A.; Coleman, M.L. OH, the places you’ll go! Hydroxylation, gene expression and cancer. Mol. Cell 2015, 58, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Barollo, S.; Bertazza, L.; Watutantrige-Fernando, S.; Censi, S.; Cavedon, E.; Galuppini, F.; Pennelli, G.; Fassina, A.; Citton, M.; Rubin, B.; et al. Overexpression of L-type amino acid transporter 1 (LAT1) and 2 (LAT2): Novel markers of neuroendocrine tumors. PLoS ONE 2016, 11, e0156044. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, O.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Segawa, H.; Nii, T.; Cha, S.H.; Matsuo, H.; Fukushima, J.; Fukasawa, Y.; et al. Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumour cell lines. Biochem. Biophys. Acta 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, L.; Pan, J. The role of L-type amino acid transporter 1 in human tumours. Intractable Rare Dis. Res. 2015, 4, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Kongpracha, P.; Nagamori, S.; Wiriyasermkul, P.; Tanaka, Y.; Kaneda, K.; Okuda, S.; Ohgaki, R.; Kanai, Y. Structure-activity relationship of novel series of inhibitors for cancer type transporter L-type amino acid transporter 1 (LAT1). J. Pharmacol. Sci. 2017, 133, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, G.; Brighi, N.; Maggio, I.; Manuzzi, L.; Peterle, C.; Ambrosini, V.; Ricci, C.; Casadei, R.; Campana, D. The role of the mTOR in neuroendocrine tumors: Future cornerstone of a winning strategy? Int. J. Mol. Sci. 2018, 19, 747. [Google Scholar] [CrossRef]
- Willenberg, H.S.; Schott, M.; Saeger, W.; Tries, A.; Scherbaum, W.A.; Bornstein, S.R. Expression of connexins in chromaffin cells of normal human adrenals and in benign and malignant pheochromocytomas. Ann. N. Y. Acad. Sci. 2006, 1073, 578–583. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Component | Function in Human Adrenal Medulla | Reported Alterations in PPGL |
|---|---|---|
| Granins | ||
| Chromogranin A–C [65,66,67,68,69,70,71,72,73,74,75,76,77] | Role in vesiculogenesis, vesicle protein stability, hormone storage within vesicles. Sorting proteins in the regulated secretory pathway. Precursor protein for several peptides; chromogranin A (vasostatin I–II, catestatin, cateslytin, chromacin, chromofungin, pancreastatin, parastatin, WE-14, EL35), chromogranin B (secretolytin), chromogranin C (secretoneurin, EM66, manserin). | Higher plasma levels of chromogranin A and B are reported in PPGL compared to healthy volunteers. Chromogranin C mRNA was overexpressed in PPGL compared to non-tumoral chromaffin tissue. Higher expression of chromogranin A and B in RET associated PPGL compared to VHL associated PPGL at both the mRNA and protein levels. Downregulation of chromogranin B and chromogranin C observed to be associated with malignant behavior. |
| Secretogranins III–VII [78,79,80,81,82,83,84,85,86,87] | Secretogranin III (syn. 1B1075) not found in adrenal medulla. Presence of secretogranin IV (syn. HISL-19), V (syn. 7B2), VI (syn. NESP55) and VII (VGF) reported in human adrenal medulla, exact function still mainly unknown. Proposed role of secretogranin VII (syn. VGF) in the regulation of energy homeostasis. | More pronounced immunoreactivity of secretogranin IV in malignant PPGL compared to benign PPGL. Significantly higher plasma levels of secretogranin V in PPGL compared to age-matched normal subjects. Secretogranin VI immunoreactivity found in PPGL with no differences between benign and malignant tumors. Variable proVGF-immunoreactive fragments are observed in human PPGL. |
| Glycoproteins | ||
| Glycoprotein I–V [88,89,90] | Glycoprotein I, i.e., DBH, catalyzes the conversion of dopamine into norepinephrine. Glycoprotein IV, i.e., the H+-ATP-ase subunit M45, provides the driving force for vesicular uptake of catecholamines by VMAT. | High expression levels of glycoprotein I (DBH) are reported in PPGL. |
| Prohormone processing enzymes | ||
| Aminopeptidase B (Ap-B) [91] | Exopeptidase involved in final conversion of proenkephalin to enkephalin. | Association of Ap-B with the secretory machinery is suggested in rat pheochromocytoma (PC12) cells. |
| Aspartic Proteinase [92] | Contributes to enkephalin precursor cleaving activity. | Unknown |
| Carboxypeptidase E (CPE) [93,94] | Role in peptide processing and sorting of prohormones. | High expression of CPE mRNA are reported. Elevated expression correlated with tumor growth and metastasis in pheochromocytomas. CPE promotes survival of pheochromocytoma (PC12) cells under nutrient starvation and hypoxic conditions by upregulation of pro-survival genes possibly by activation of the ERK1/2 pathway. |
| Cathepsin L [95,96] | Endopeptidase involved in proteolysis of proenkephalin into (Met)enkephalin. Proprotein convertase for biosynthesis of NPY and catestatin. | Unknown |
| Prohormone convertase 1/3 and 2 [73,78,97] | Conversion of chromogranin C into secretoneurin and EM66. | PC1 and PC2 mRNA expression levels are significantly higher in benign and malignant PPGL compared to normal adrenal medulla. mRNA expression and protein levels of PC1 and PC2 is 3–4 times higher in benign tumors compared to malignant tumors. |
| Tissue-Type Plasminogen Activator (t-PA) [98,99] | Participation in plasmin-dependent processing of bioactive peptides including chromogranin A and indirectly modulate chromogranin A release (negative-feedback loop). | Marked expressions of t-PA mRNA are reported in human pheochromocytomas. |
| Inhibitors of endogenous proteases | ||
| Endopin 1–2 [100] | Endopin 1 inhibits trypsin-like serine proteases. Endopin 2 inhibits papain-like cysteine proteases, including cathepsin L, as well as the serine protease elastase. | Unknown |
| Transmitter peptides | ||
| Adrenomedullin [65,101,102,103,104,105] | Increases blood flow in the adrenal gland. Increases catecholamine release. Induces systemic vasodilation. Increases natriuresis. | High plasma levels are reported, especially in PPGL patients with high blood pressure. |
| Bombesin [106,107,108] | Modulation of stress response. Paracrine regulatory effects on growth, structure and function of the adrenal cortex. | Highly variable immunoreactivity in pheochromocytomas and paragangliomas. An association between clinically malignant PPGL and lower expression of bombesin is postulated. |
| Calcitonin Gene-Related Peptide [109,110] | Vasodilatation, enhances aldosterone and corticosterone release by adrenocortical cells. | Slightly elevated levels are reported. |
| Catestatin, Cateslytin [111,112] | Inhibits release of catecholamines, chromogranin A, NPY, and ATP by acting as noncompetitive antagonist of the nicotinic receptor. | Unknown |
| EM66 (via PACAP) [73,113,114] | Derivate from chromogranin C (syn. secretogranin II). Synthesis and secretion regulated by PACAP. Paracrine regulation of steroidogenic cells in the adrenal gland. | Elevated in PPGL. Higher levels of EM66 reported in benign vs. malignant PPGL. |
| Natriuretic peptides (ANP, BNP, CNP) [102,115,116] | Autocrine/paracrine inhibition of catecholamine secretion via the ANF-R2 receptor subtype. Inhibition of aldosterone production by a direct action on the adrenal cortex both in vivo and in vitro. | Elevated in PPGL patients with high blood pressure. |
| Neuropeptide Y (NPY) [65,76,101,117,118,119,120,121,122] | Increases catecholamine biosynthesis (stimulates TH gene expression) and secretion. Potentiates catecholamine induced vasoconstriction. | Influence on tumorigenesis and stimulation of neoangiogenesis is described. High levels of NPY mRNA have been found in benign tumors, whereas its plasma levels are elevated in patients with malignant PPGL. Significantly lower expression level of NPY in VHL associated PPGL compared to other hereditary and sporadic PPGL is reported. |
| Neurotensin [123,124] | Not detected in human adrenal medulla. Various central and peripheral effects have been postulated in bovine, cat and rat, e.g., hypotension, hypothermia, analgesia. | Neurotensin has rarely been demonstrated in human PPGL. |
| Opioid peptides (enkephalins, endorphins) [96,117,125,126,127,128] | Decrease catecholamine release via binding Gi protein coupled receptor resulting in inhibition of Ca2+ channels Analgesia, enhancement of immune reaction. | Enkephalin decreases norepinephrine release in human pheochromocytomas. Different enkephalins (i.e., (Met)enkephalin, (Leu)enkephalin) are observed in human pheochromocytomas. Expression of (Met) enkephalin and (Leu)enkephalin is highly variable compared to normal adrenal medulla. Possible association between malignant PPGL and lower expression of enkephalins. |
| PACAP [40,63,101,117,129,130] | Induces transcription and stimulates activity of TH, DBH, and PNMT Stimulates expression and secretion of several other peptides e.g., brain natriuretic peptide, enkephalins, EM66, and secretoneurin. | High mRNA expression of PACAP and PAC1-R in PPGL reported. Comparable expression levels in benign and metastatic PPGL found in a relatively small series. |
| Secretoneurin (via PACAP) [131,132,133] | Derivate from chromogranin C (syn. secretogranin II). Synthesis and secretion regulated by PACAP. Role in angiogenesis and modulation of inflammatory response by chemoattractant effects on monocytes, eosinophils, fibroblasts, vascular smooth muscle cells, and endothelial cells. | Unknown |
| Transforming Growth Factor β [134,135,136] | Role in regulation of chromaffin cell proliferation and differentiation. Reduction of TGF β has been shown to increase proliferation of chromaffin cell in vivo. | Unknown |
| Vasostatins [137,138,139] | The N-terminal fragment of chromogranin A Inhibition of endothelin-induced vasoconstriction Antibacterial and antifungal activity. | Unknown |
| Anti-bacterial/anti-fungal peptides | ||
| Chromacin P, G and PG [140] | Antibacterial activity against Gram positive bacteria. | Unknown |
| Secretolytin [141] | Antibacterial activity against Gram positive bacteria. | Unknown |
| Ubifungin [142] | Antifungal activity. | Unknown |
| Other minor components | ||
| Ascorbic acid [143] | Regulation of DBH activity. | Unknown |
| Coenzyme A glutathione disulfide [144] | Vasoconstriction, modulation of AngII effects. | Unknown |
| Ions (Ca2+, Na+, K+, Mg2+, Cl−) [145] | Regulation of exocytosis. | Unknown |
| Galanin [81,146,147] | Stimulation of norepinephrine and glucocorticoid secretion. | Immunoreactivity in human PPGL. Higher levels reported in PPGL compared to normal adrenal medulla. Variable expression; galanin was predominantly found in noradrenergic pheochromocytoma cells. Induction of apoptosis in PC12 cells. Inhibition of dopamine secretion in pheochromocytoma cells. |
| Nucleotides (ATP, ADP, GTP) [148] | Formation of intravesicular complex with catecholamines, buffer function, decreases intravesicular osmotic pressure, neuromodulation. | Unknown |
| Substance P [149,150,151,152,153] | Inhibition of nicotinic acetylcholine receptor mediated catecholamine release. Vasodilation. | Variable immunoreactivity demonstrated in human pheochromocytomas. Elevated plasma levels in minority of patients. |
| Vasoactive intestinal polypeptide [154,155,156] | Stimulation of catecholamine release, stimulation of steroid secretion. | Few cases described of human PPGL with concomitant excessive VIP secretion. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berends, A.M.A.; Eisenhofer, G.; Fishbein, L.; van der Horst-Schrivers, A.N.A.; Kema, I.P.; Links, T.P.; Lenders, J.W.M.; Kerstens, M.N. Intricacies of the Molecular Machinery of Catecholamine Biosynthesis and Secretion by Chromaffin Cells of the Normal Adrenal Medulla and in Pheochromocytoma and Paraganglioma. Cancers 2019, 11, 1121. https://doi.org/10.3390/cancers11081121
Berends AMA, Eisenhofer G, Fishbein L, van der Horst-Schrivers ANA, Kema IP, Links TP, Lenders JWM, Kerstens MN. Intricacies of the Molecular Machinery of Catecholamine Biosynthesis and Secretion by Chromaffin Cells of the Normal Adrenal Medulla and in Pheochromocytoma and Paraganglioma. Cancers. 2019; 11(8):1121. https://doi.org/10.3390/cancers11081121
Chicago/Turabian StyleBerends, Annika M.A., Graeme Eisenhofer, Lauren Fishbein, Anouk N.A. van der Horst-Schrivers, Ido P. Kema, Thera P. Links, Jacques W.M. Lenders, and Michiel N. Kerstens. 2019. "Intricacies of the Molecular Machinery of Catecholamine Biosynthesis and Secretion by Chromaffin Cells of the Normal Adrenal Medulla and in Pheochromocytoma and Paraganglioma" Cancers 11, no. 8: 1121. https://doi.org/10.3390/cancers11081121
APA StyleBerends, A. M. A., Eisenhofer, G., Fishbein, L., van der Horst-Schrivers, A. N. A., Kema, I. P., Links, T. P., Lenders, J. W. M., & Kerstens, M. N. (2019). Intricacies of the Molecular Machinery of Catecholamine Biosynthesis and Secretion by Chromaffin Cells of the Normal Adrenal Medulla and in Pheochromocytoma and Paraganglioma. Cancers, 11(8), 1121. https://doi.org/10.3390/cancers11081121