SLMP53-2 Restores Wild-Type-Like Function to Mutant p53 through Hsp70: Promising Activity in Hepatocellular Carcinoma

 , , , , , ,
, , , , , ,  ,
, 
 , ,
, ,  and
and  add
Show full author list
add
Show full author list
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
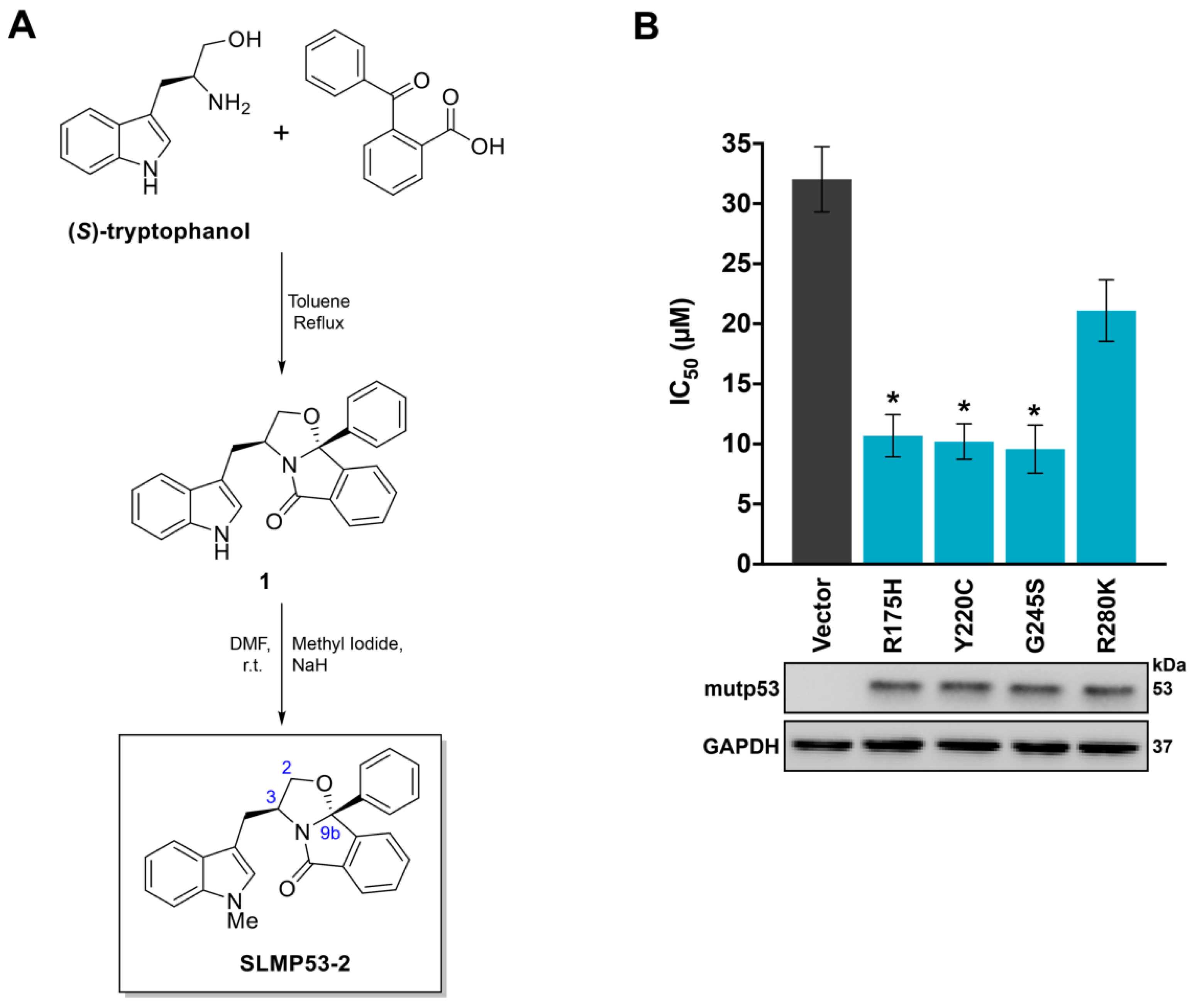
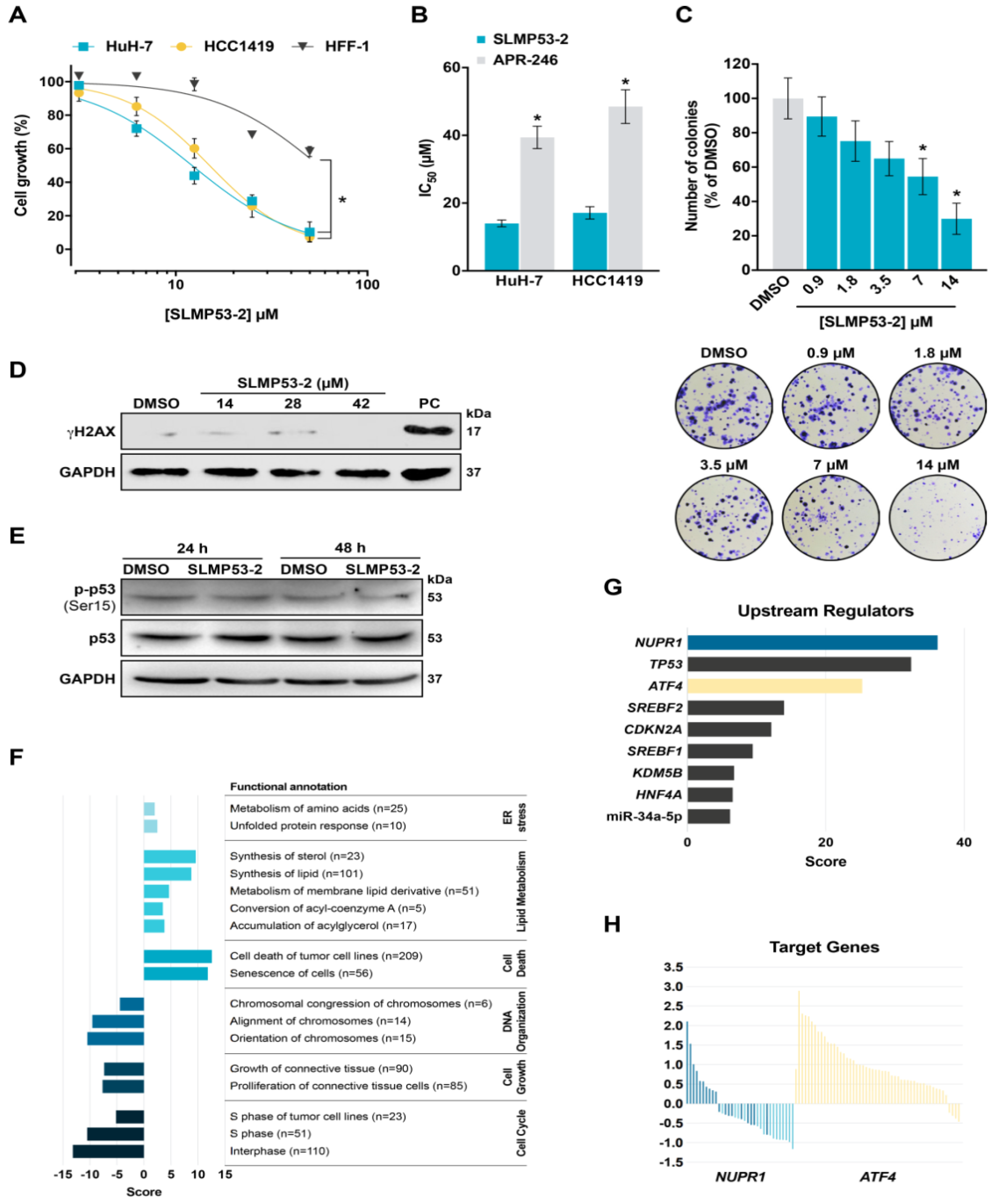
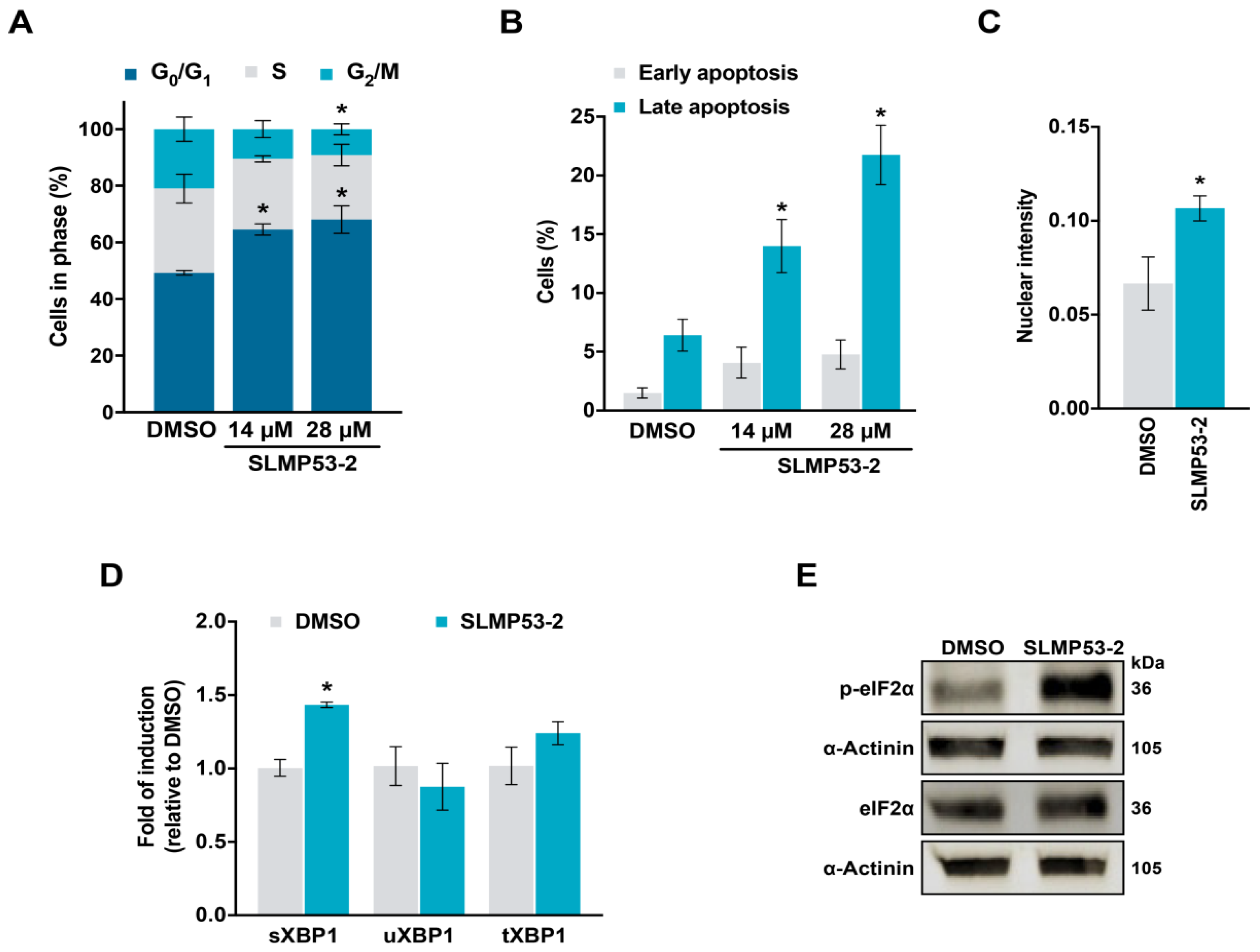
2.1. SLMP53-2 Displays mutp53-Dependent Growth Inhibitory Effect in Human Tumor Cells, Leading to Cell Cycle Arrest, Apoptosis and Endoplasmic Reticulum (ER) Stress
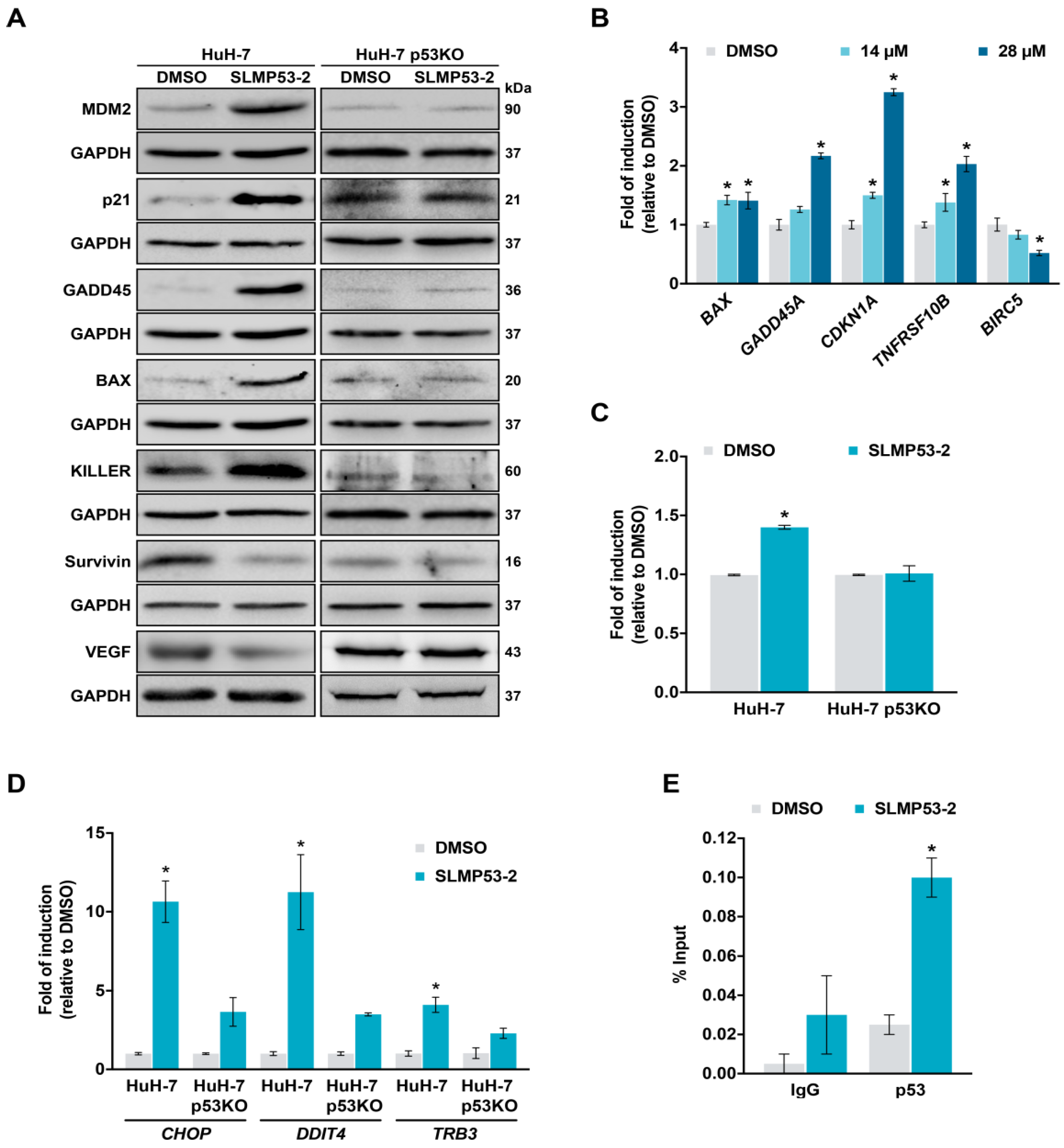
2.2. SLMP53-2 Restores WT-Like Conformation and Transcriptional Activity to mutp53-Y220C in HCC Cells
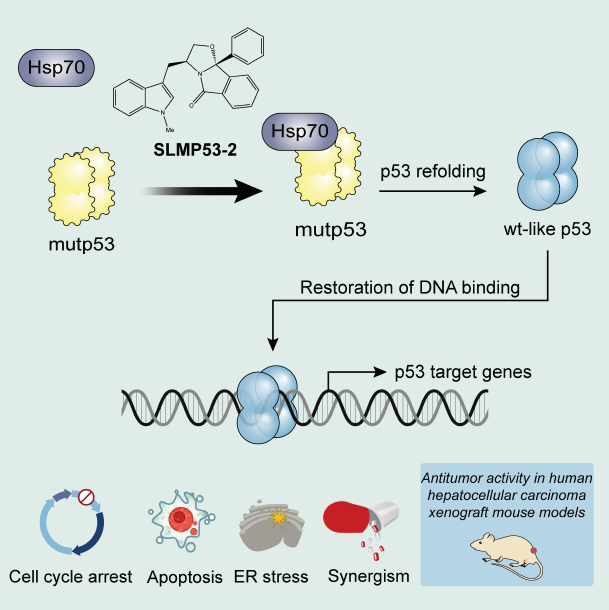
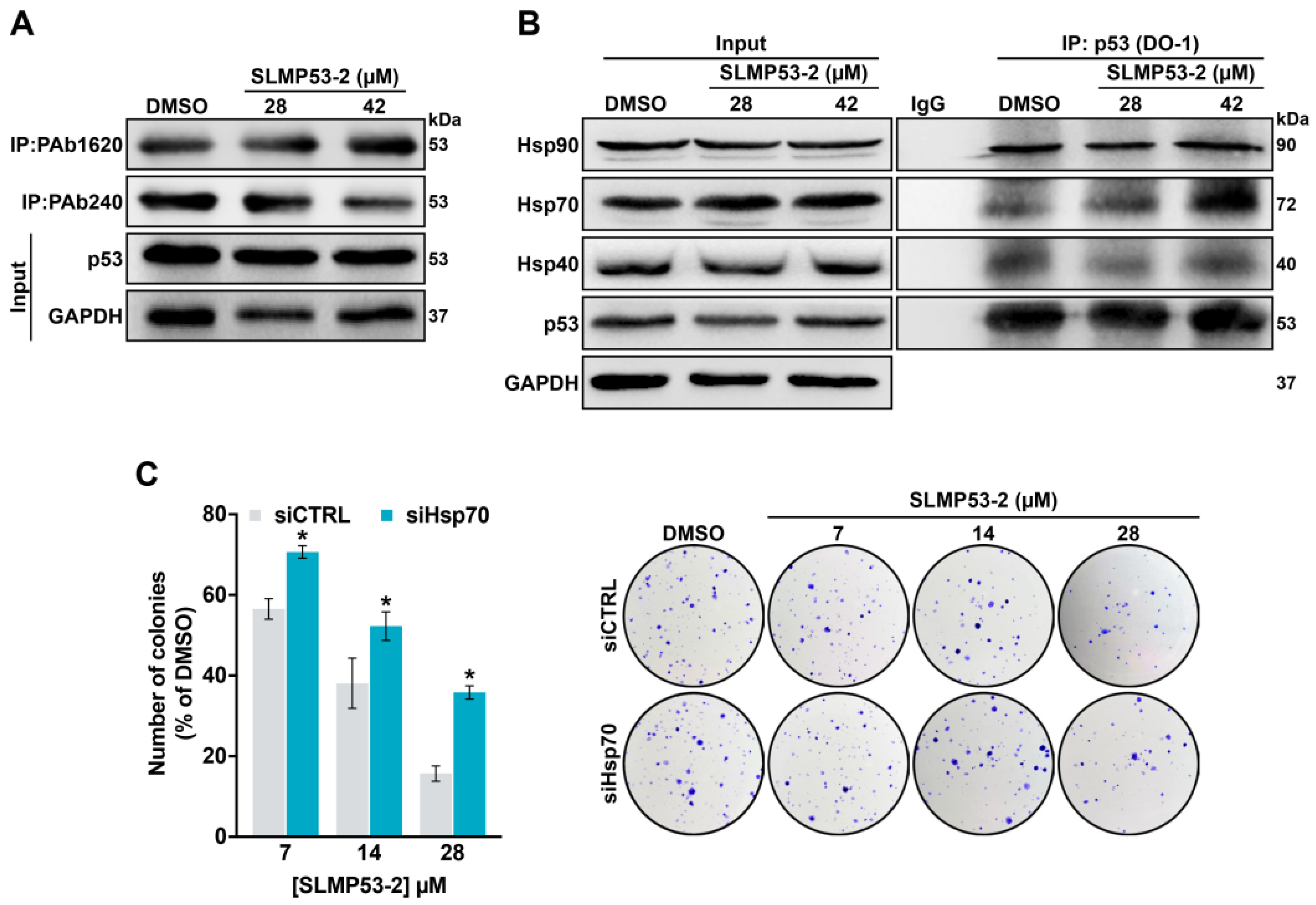
2.3. Hsp70 is a Potential Mediator of mutp53-Y220C Reactivation by SLMP53-2
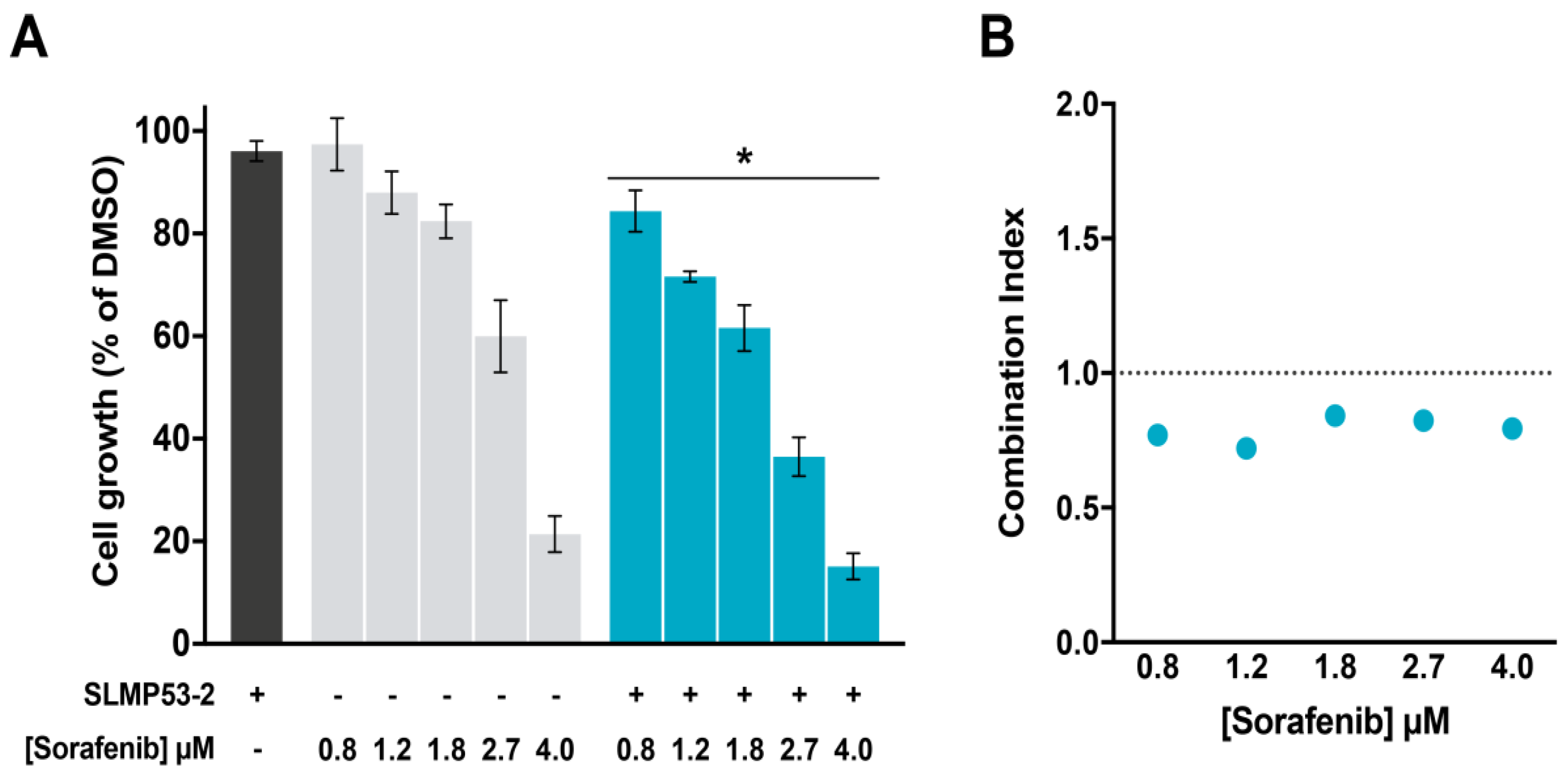
2.4. SLMP53-2 Sensitizes HCC Cells to Sorafenib
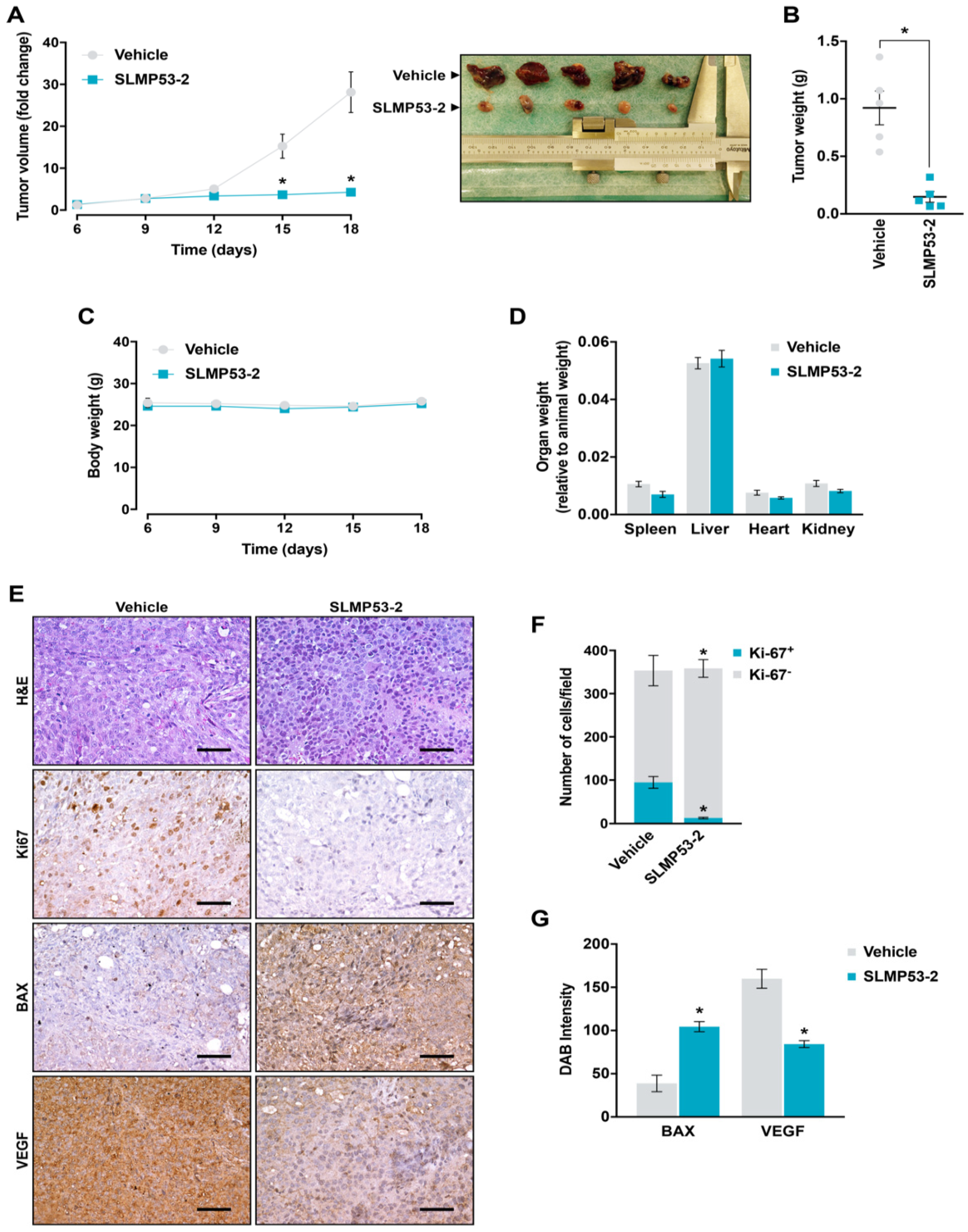
2.5. SLMP53-2 Displays In Vivo Anti-Tumor Activity in HCC Xenograft Mouse Models, with No Apparent Toxic Side Effects
3. Discussion
4. Materials and Methods
4.1. Compounds and Antibodies
4.2. Chemical Synthesis of SLMP53-2
4.2.1. General Methods
4.2.2. Chemical Synthesis of SLMP53-2
4.3. Human Cell Culture Conditions
4.4. Transfection, Cell Proliferation, and Combination Therapy Assays
4.4.1. Construction of Mammalian Expression Vectors
4.4.2. Transfection
4.4.3. SRB Assay
4.4.4. Colony Formation Assay
4.5. Cell Cycle and Apoptosis Analysis
4.6. Western Blot
4.7. IP and co-IP
4.8. RNA Extraction and RT-qPCR
4.8.1. Gene Expression Analysis
4.8.2. miRNA Analysis
4.9. ChIP
4.10. Microarray Experiments
4.11. Immunofluorescence
4.11.1. XBP1 Staining
4.11.2. PAb240/PAb1620 Staining
4.12. HSQC-NMR
4.13. In Vivo Anti-Tumor and Toxicity Assays
4.14. Immunohistochemical (IHC) Analysis
4.15. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [PubMed]
- Lopes, E.A.; Gomes, S.; Saraiva, L.; Santos, M. Small Molecules Targeting Mutant P53: A Promising Approach for Cancer Treatment. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2018; Available online: https://gco.iarc.fr/today (accessed on 4 February 2019).
- Noureddin, M.; Rinella, M.E. Nonalcoholic Fatty liver disease, diabetes, obesity, and hepatocellular carcinoma. Clin. Liver Dis. 2015, 19, 361–379. [Google Scholar] [CrossRef]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.; Meyer, T.; Nault, J.C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv238–iv255. [Google Scholar] [CrossRef]
- Lin, S.; Hoffmann, K.; Schemmer, P. Treatment of hepatocellular carcinoma: A systematic review. Liver Cancer 2012, 1, 144–158. [Google Scholar] [CrossRef]
- Bevant, K.; Coulouarn, C. Landscape of genomic alterations in hepatocellular carcinoma: Current knowledge and perspectives for targeted therapies. Hepatobiliary Surg. Nutr. 2017, 6, 404–407. [Google Scholar] [CrossRef]
- Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341. [CrossRef]
- Soares, J.; Raimundo, L.; Pereira, N.A.; Monteiro, A.; Gomes, S.; Bessa, C.; Pereira, C.; Queiroz, G.; Bisio, A.; Fernandes, J.; et al. Reactivation of wild-type and mutant p53 by tryptophanolderived oxazoloisoindolinone SLMP53-1, a novel anticancer small-molecule. Oncotarget 2016, 7, 4326–4343. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Yogosawa, S.; Yoshida, K. Tumor suppressive role for kinases phosphorylating p53 in DNA damage-induced apoptosis. Cancer Sci. 2018, 109, 3376–3382. [Google Scholar] [CrossRef] [PubMed]
- Niessner, H.; Sinnberg, T.; Kosnopfel, C.; Smalley, K.S.M.; Beck, D.; Praetorius, C.; Mai, M.; Beissert, S.; Kulms, D.; Schaller, M.; et al. BRAF Inhibitors Amplify the Proapoptotic Activity of MEK Inhibitors by Inducing ER Stress in NRAS-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 6203–6214. [Google Scholar] [CrossRef] [PubMed]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Laurell, C.; Selivanova, G.; Lundeberg, J.; Nilsson, P.; Wiman, K.G. Hypoxia induces p53-dependent transactivation and Fas/CD95-dependent apoptosis. Cell Death Differ. 2007, 14, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Di Como, C.J.; Prives, C. Human tumor-derived p53 proteins exhibit binding site selectivity and temperature sensitivity for transactivation in a yeast-based assay. Oncogene 1998, 16, 2527–2539. [Google Scholar] [CrossRef] [Green Version]
- Hainaut, P.; Milner, J. Interaction of heat-shock protein 70 with p53 translated in vitro: Evidence for interaction with dimeric p53 and for a role in the regulation of p53 conformation. EMBO J. 1992, 11, 3513–3520. [Google Scholar] [CrossRef]
- Walerych, D.; Olszewski, M.B.; Gutkowska, M.; Helwak, A.; Zylicz, M.; Zylicz, A. Hsp70 molecular chaperones are required to support p53 tumor suppressor activity under stress conditions. Oncogene 2009, 28, 4284–4294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wawrzynow, B.; Zylicz, A.; Zylicz, M. Chaperoning the guardian of the genome. The two-faced role of molecular chaperones in p53 tumor suppressor action. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Giknis, M.; Clifford, C.B. Clinical Laboratory Parameters for CRL: WI (Han); Charles River Laboratories: Wilmington, MA, USA, 2008. [Google Scholar]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef]
- Yang, P.M.; Lin, Y.T.; Shun, C.T.; Lin, S.H.; Wei, T.T.; Chuang, S.H.; Wu, M.S.; Chen, C.C. Zebularine inhibits tumorigenesis and stemness of colorectal cancer via p53-dependent endoplasmic reticulum stress. Sci. Rep. 2013, 3, 3219. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.R.; Joerger, A.C.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 2010, 29, 1329–1338. [Google Scholar] [CrossRef]
- Liu, J.L.; Zhang, X.J.; Zhang, Z.; Zhang, A.H.; Wang, W.; Dong, J.H. Meta-analysis: Prognostic value of survivin in patients with hepatocellular carcinoma. PLoS ONE 2013, 8, e83350. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, J.; Feng, Z.; Fan, W.; Wang, Y.; Li, J.; Tong, D. The expression of Survivin and NF-kappaB associated with prognostically worse clinicopathologic variables in hepatocellular carcinoma. Tumour Biol. 2014, 35, 9905–9910. [Google Scholar] [CrossRef]
- Su, C. Survivin in survival of hepatocellular carcinoma. Cancer Lett. 2016, 379, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Slabakova, E.; Culig, Z.; Remsik, J.; Soucek, K. Alternative mechanisms of miR-34a regulation in cancer. Cell Death Dis. 2017, 8, e3100. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fu, H.; Tie, Y.; Hu, Z.; Kong, W.; Wu, Y.; Zheng, X. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009, 275, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Daige, C.L.; Wiggins, J.F.; Priddy, L.; Nelligan-Davis, T.; Zhao, J.; Brown, D. Systemic delivery of a miR34a mimic as a potential therapeutic for liver cancer. Mol. Cancer Ther. 2014, 13, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Hiraki, M.; Hwang, S.Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polireddy, K.; Singh, K.; Pruski, M.; Jones, N.C.; Manisundaram, N.V.; Ponnela, P.; Ouellette, M.; Van Buren, G.; Younes, M.; Bynon, J.S.; et al. Mutant p53(R175H) promotes cancer initiation in the pancreas by stabilizing HSP70. Cancer Lett. 2019, 453, 122–130. [Google Scholar] [CrossRef]
- Raoul, J.L.; Gilabert, M.; Adhoute, X.; Edeline, J. An in-depth review of chemical angiogenesis inhibitors for treating hepatocellular carcinoma. Expert Opin. Pharm. 2017, 18, 1467–1476. [Google Scholar] [CrossRef]
- Kudo, M. Systemic Therapy for Hepatocellular Carcinoma: 2017 Update. Oncology 2017, 93 (Suppl. S1), 135–146. [Google Scholar] [CrossRef]
- Pereira, N.A.; Monteiro, A.; Machado, M.; Gut, J.; Molins, E.; Perry, M.J.; Dourado, J.; Moreira, R.; Rosenthal, P.J.; Prudencio, M.; et al. Enantiopure Indolizinoindolones with in vitro Activity against Blood- and Liver-Stage Malaria Parasites. Chem. Med. Chem. 2015, 10, 2080–2089. [Google Scholar] [CrossRef]
- Baud, M.G.J.; Bauer, M.R.; Verduci, L.; Dingler, F.A.; Patel, K.J.; Horil Roy, D.; Joerger, A.C.; Fersht, A.R. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 2018, 152, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leao, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New inhibitor of the TAp73 interaction with MDM2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019, 446, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Raimundo, L.; Pereira, N.A.; dos Santos, D.J.; Perez, M.; Queiroz, G.; Leao, M.; Santos, M.M.; Saraiva, L. A tryptophanol-derived oxazolopiperidone lactam is cytotoxic against tumors via inhibition of p53 interaction with murine double minute proteins. Pharmacol. Res. 2015, 95–96, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Raimundo, L.; Espadinha, M.; Soares, J.; Loureiro, J.B.; Alves, M.G.; Santos, M.M.M.; Saraiva, L. Improving anticancer activity towards colon cancer cells with a new p53-activating agent. Br. J. Pharm. 2018, 175, 3947–3962. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Pereira, N.A.; Monteiro, A.; Leao, M.; Bessa, C.; Dos Santos, D.J.; Raimundo, L.; Queiroz, G.; Bisio, A.; Inga, A.; et al. Oxazoloisoindolinones with in vitro antitumor activity selectively activate a p53-pathway through potential inhibition of the p53-MDM2 interaction. Eur. J. Pharm. Sci. 2015, 66, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Espadinha, M.; Raimundo, L.; Ramos, H.; Gomes, A.S.; Gomes, S.; Loureiro, J.B.; Inga, A.; Reis, F.; Gomes, C.; et al. DIMP53-1: A novel small-molecule dual inhibitor of p53-MDM2/X interactions with multifunctional p53-dependent anticancer properties. Mol. Oncol. 2017, 11, 612–627. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Alessandrini, F.; Pezze, L.; Menendez, D.; Resnick, M.A.; Ciribilli, Y. ETV7-Mediated DNAJC15 Repression Leads to Doxorubicin Resistance in Breast Cancer Cells. Neoplasia 2018, 20, 857–870. [Google Scholar] [CrossRef]
- Vazquez, C.; Beachboard, D.C.; Horner, S.M. Methods to Visualize MAVS Subcellular Localization. Methods Mol. Biol. 2017, 1656, 131–142. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, S.; Bosco, B.; Loureiro, J.B.; Ramos, H.; Raimundo, L.; Soares, J.; Nazareth, N.; Barcherini, V.; Domingues, L.; Oliveira, C.; et al. SLMP53-2 Restores Wild-Type-Like Function to Mutant p53 through Hsp70: Promising Activity in Hepatocellular Carcinoma. Cancers 2019, 11, 1151. https://doi.org/10.3390/cancers11081151
Gomes S, Bosco B, Loureiro JB, Ramos H, Raimundo L, Soares J, Nazareth N, Barcherini V, Domingues L, Oliveira C, et al. SLMP53-2 Restores Wild-Type-Like Function to Mutant p53 through Hsp70: Promising Activity in Hepatocellular Carcinoma. Cancers. 2019; 11(8):1151. https://doi.org/10.3390/cancers11081151
Chicago/Turabian StyleGomes, Sara, Bartolomeo Bosco, Joana B. Loureiro, Helena Ramos, Liliana Raimundo, Joana Soares, Nair Nazareth, Valentina Barcherini, Lucília Domingues, Carla Oliveira, and et al. 2019. "SLMP53-2 Restores Wild-Type-Like Function to Mutant p53 through Hsp70: Promising Activity in Hepatocellular Carcinoma" Cancers 11, no. 8: 1151. https://doi.org/10.3390/cancers11081151
APA StyleGomes, S., Bosco, B., Loureiro, J. B., Ramos, H., Raimundo, L., Soares, J., Nazareth, N., Barcherini, V., Domingues, L., Oliveira, C., Bisio, A., Piazza, S., Bauer, M. R., Brás, J. P., Almeida, M. I., Gomes, C., Reis, F., Fersht, A. R., Inga, A., ... Saraiva, L. (2019). SLMP53-2 Restores Wild-Type-Like Function to Mutant p53 through Hsp70: Promising Activity in Hepatocellular Carcinoma. Cancers, 11(8), 1151. https://doi.org/10.3390/cancers11081151