The Tumor Microenvironment in Colorectal Cancer Therapy


Abstract
:1. Cancer: Immune Surveillance, Escape, and Immunotherapy
2. Metabolic Reprogramming in Tumor Cells and Associated Immune Cells
3. Metastatic Colorectal Cancer
4. Reprogramming of CRC in Response to Chemo- or Radio-Therapy: Angiogenesis, Immune Response, and Metabolism
5. Immune Microenvironments in CRC
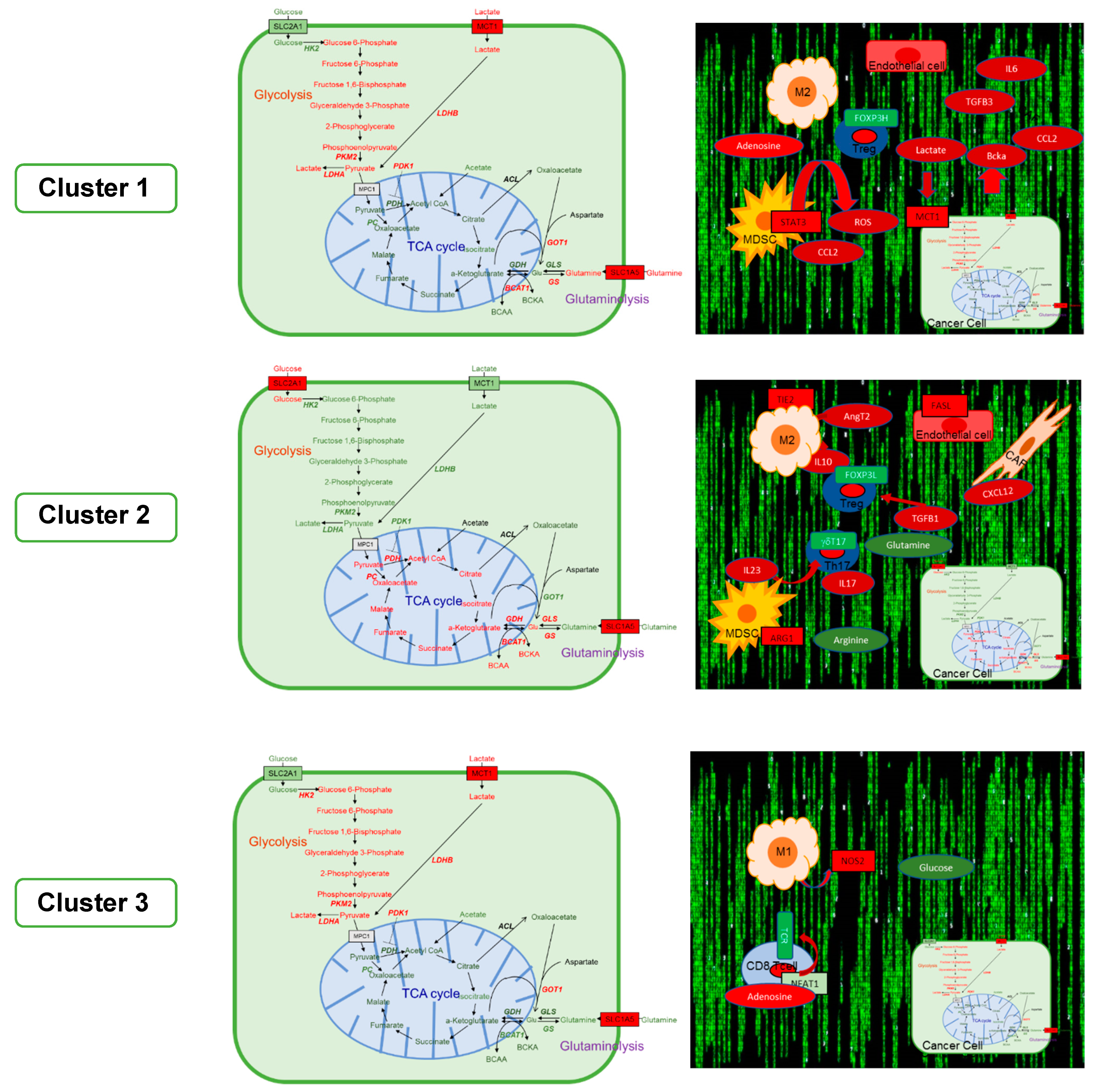
6. The GAIS-42 Immune-Metabolic Classification of mCRC
6.1. Cluster 1: Inflamed-Stromal Dependent
6.1.1. Metabolism and Microenvironment
6.1.2. Rationale for the Design of Clinical Trials
6.2. Cluster 2: Inflamed Non-Stromal Dependent
6.2.1. Metabolism and Microenvironment
6.2.2. Rationale for the Design of Clinical Trials
6.3. Cluster 3: Non-Inflamed/Cold
6.3.1. Metabolism and Microenvironment
6.3.2. Rationale for the Design of Clinical Trials
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rhoads, C.P. Paul Ehrlich and the cancer problem. Ann. N. Y. Acad. Sci. 1954, 59, 190–197. Available online: https://www.ncbi.nlm.nih.gov/pubmed/13229207 (accessed on 15 July 2019). [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. Available online: https://www.ncbi.nlm.nih.gov/pubmed/15032581 (accessed on 15 July 2019). [CrossRef] [PubMed]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765.e16. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30318143 (accessed on 15 July 2019). [CrossRef] [PubMed] [Green Version]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21832238 (accessed on 15 July 2019). [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1746. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8596936 (accessed on 15 July 2019). [CrossRef] [PubMed]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24240160 (accessed on 15 July 2019). [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29544515 (accessed on 15 July 2019). [CrossRef]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 33, 581–598. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29634946 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Kim, T.K.; Herbst, R.S.; Chen, L. Defining and Understanding Adaptive Resistance in Cancer Immunotherapy. Trends Immunol. 2018, 39, 624–631. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29802087 (accessed on 15 July 2019). [CrossRef]
- Paulson, K.G.; Voillet, V.; McAfee, M.S.; Hunter, D.S.; Wagener, F.D.; Perdicchio, M.; Valente, W.J.; Koelle, S.J.; Church, C.D.; Vandeven, N.; et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun. 2018, 9, 3868. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30250229 (accessed on 15 July 2019). [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12091876 (accessed on 15 July 2019). [CrossRef] [PubMed]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27789795 (accessed on 15 July 2019). [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26858199 (accessed on 15 July 2019). [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26321679 (accessed on 15 July 2019). [CrossRef] [PubMed] [Green Version]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28416194 (accessed on 15 July 2019). [CrossRef] [PubMed] [Green Version]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29798856 (accessed on 15 July 2019). [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22439925 (accessed on 15 July 2019). [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. Available online: https://www.ncbi.nlm.nih.gov/pubmed/13351639 (accessed on 15 July 2019).
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28741521 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Baba, Y.; Nosho, K.; Shima, K.; Irahara, N.; Chan, A.T.; Meyerhardt, J.A.; Chung, D.C.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am. J. Pathol. 2010, 176, 2292–2301. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20363910 (accessed on 15 July 2019). [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. Available online: https://www.ncbi.nlm.nih.gov/pubmed/16517405 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. Available online: https://www.ncbi.nlm.nih.gov/pubmed/16452478 (accessed on 15 July 2019). [CrossRef]
- Maciolek, J.A.; Pasternak, J.A.; Wilson, H.L. Metabolism of activated T lymphocytes. Curr. Opin. Immunol. 2014, 27, 60–74. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24556090 (accessed on 15 July 2019). [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21317389 (accessed on 15 July 2019). [CrossRef]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21358638 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26420908 (accessed on 15 July 2019). [CrossRef]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30392958 (accessed on 15 July 2019). [CrossRef]
- Loftus, R.M.; Assmann, N.; Kedia-Mehta, N.; O’Brien, K.L.; Garcia, A.; Gillespie, C.; Hukelmann, J.L.; Oefner, P.J.; Lamond, A.I.; Gardiner, C.M.; et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Saltz, L.B.; Clarke, S.; Díaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Lichinitser, M.; Yang, T.S.; Rivera, F.; et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: A randomized phase III study. J. Clin. Oncol. 2008, 26, 2013–2019. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18421054 (accessed on 15 July 2019). [CrossRef]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19339720 (accessed on 15 July 2019). [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25358689 (accessed on 15 July 2019). [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26028255 (accessed on 15 July 2019). [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28596308 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28734759 (accessed on 15 July 2019). [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29355075 (accessed on 15 July 2019). [CrossRef]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31038663 (accessed on 15 July 2019). [CrossRef]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12750523 (accessed on 15 July 2019). [CrossRef]
- Truman, J.P.; García-Barros, M.; Kaag, M.; Hambardzumyan, D.; Stancevic, B.; Chan, M.; Fuks, Z.; Kolesnick, R.; Haimovitz-Friedman, A. Endothelial membrane remodeling is obligate for anti-angiogenic radiosensitization during tumor radiosurgery. PLoS ONE 2010, 5, e12310. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20808818 (accessed on 15 July 2019). [CrossRef]
- Goedegebuure, R.S.A.; Klerk, L.K.; Bass, A.J.; Derks, S.; Thijssen, V.L.J.L. Combining Radiotherapy with Anti-angiogenic Therapy and Immunotherapy; A Therapeutic Triad for Cancer? Front. Immunol. 2018, 9, 3107. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30692993 (accessed on 15 July 2019). [CrossRef]
- Khoo, L.T.; Chen, L.Y. Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep. 2018, 19, e46935. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30446584 (accessed on 15 July 2019). [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, CD-19. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31197017 (accessed on 15 July 2019). [CrossRef]
- Aleynick, M.; Svensson-Arvelund, J.; Flowers, C.R.; Marabelle, A.; Brody, J.D. Pathogen molecular pattern receptor agonists: Treating cancer by mimicking infection. Clin. Cancer Res. 2019. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31123052 (accessed on 15 July 2019).
- Giuliani, A.L.; Sarti, A.C.; Falzoni, S.; Di Virgilio, F. The P2X7 Receptor-Interleukin-1 Liaison. Front. Pharmacol. 2017, 8, 123. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28360855 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Sturtzel, C. Endothelial Cells. Adv. Exp. Med. Biol. 2017, 1003, 71–91. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28667554 (accessed on 15 July 2019).
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24332029 (accessed on 15 July 2019). [CrossRef]
- Mandai, M.; Hamanishi, J.; Abiko, K.; Matsumura, N.; Baba, T.; Konishi, I. Dual Faces of IFNgamma in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin. Cancer Res. 2016, 22, 2329–2334. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27016309 (accessed on 15 July 2019). [CrossRef]
- Bosco, M.C.; Pierobon, D.; Blengio, F.; Raggi, F.; Vanni, C.; Gattorno, M.; Eva, A.; Novelli, F.; Cappello, P.; Giovarelli, M.; et al. Hypoxia modulates the gene expression profile of immunoregulatory receptors in human mature dendritic cells: Identification of TREM-1 as a novel hypoxic marker in vitro and in vivo. Blood 2011, 117, 2625–2639. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21148811 (accessed on 15 July 2019). [CrossRef]
- Tseng, C.W.; Hung, C.F.; Alvarez, R.D.; Trimble, C.; Huh, W.K.; Kim, D.; Chuang, C.M.; Lin, C.T.; Tsai, Y.C.; He, L.; et al. Pretreatment with cisplatin enhances E7-specific CD8+ T-Cell-mediated antitumor immunity induced by DNA vaccination. Clin. Cancer Res. 2008, 14, 3185–3192. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18483387 (accessed on 15 July 2019). [CrossRef]
- Chakraborty, M.; Abrams, S.I.; Camphausen, K.; Liu, K.; Scott, T.; Coleman, C.N.; Hodge, J.W. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J. Immunol. 2003, 170, 6338–6347. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12794167 (accessed on 15 July 2019). [CrossRef]
- Droeser, R.A.; Hirt, C.; Viehl, C.T.; Frey, D.M.; Nebiker, C.; Huber, X.; Zlobec, I.; Eppenberger-Castori, S.; Tzankov, A.; Rosso, R.; et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur. J. Cancer 2013, 49, 2233–2242. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23478000 (accessed on 15 July 2019). [CrossRef]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Prev. Biomark. 2014, 23, 2965–2970. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25392179 (accessed on 15 July 2019). [CrossRef]
- Li, Y.; Liang, L.; Dai, W.; Cai, G.; Xu, Y.; Li, X.; Li, Q.; Cai, S. Prognostic impact of programed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor infiltrating lymphocytes in colorectal cancer. Mol. Cancer. 2016, 15, 55. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27552968 (accessed on 15 July 2019). [CrossRef]
- De Smedt, L.; Lemahieu, J.; Palmans, S.; Govaere, O.; Tousseyn, T.; Van Cutsem, E.; Prenen, H.; Tejpar, S.; Spaepen, M.; Matthijs, G.; et al. Microsatellite instable vs stable colon carcinomas: Analysis of tumour heterogeneity, inflammation and angiogenesis. Br. J. Cancer 2015, 113, 500–509. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26068398 (accessed on 15 July 2019). [CrossRef]
- Maby, P.; Tougeron, D.; Hamieh, M.; Mlecnik, B.; Kora, H.; Bindea, G.; Angell, H.K.; Fredriksen, T.; Elie, N.; Fauquembergue, E.; et al. Correlation between Density of CD8+ T-cell Infiltrate in Microsatellite Unstable Colorectal Cancers and Frameshift Mutations: A Rationale for Personalized Immunotherapy. Cancer Res. 2015, 75, 3446–3455. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26060019 (accessed on 15 July 2019). [CrossRef]
- Liu, G.C.; Liu, R.Y.; Yan, J.P.; An, X.; Jiang, W.; Ling, Y.H.; Chen, J.W.; Bei, J.X.; Zuo, X.Y.; Cai, M.Y.; et al. The Heterogeneity Between Lynch-Associated and Sporadic MMR Deficiency in Colorectal Cancers. J. Natl. Cancer Inst. 2018, 110, 975–984. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29471527 (accessed on 15 July 2019). [CrossRef]
- Mlecnik, B.; Bindea, G.; Kirilovsky, A.; Angell, H.K.; Obenauf, A.C.; Tosolini, M.; Church, S.E.; Maby, P.; Vasaturo, A.; Angelova, M.; et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci. Transl. Med. 2016, 8, 327ra26. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26912905 (accessed on 15 July 2019). [CrossRef]
- Mlecnik, B.; Van den Eynde, M.; Bindea, G.; Church, S.E.; Vasaturo, A.; Fredriksen, T.; Lafontaine, L.; Haicheur, N.; Marliot, F.; Debetancourt, D.; et al. Comprehensive Intrametastatic Immune Quantification and Major Impact of Immunoscore on Survival. J. Natl. Cancer Inst. 2018, 110, 97–108. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28922789 (accessed on 15 July 2019). [CrossRef]
- Girardin, A.; McCall, J.; Black, M.A.; Edwards, F.; Phillips, V.; Taylor, E.S.; Reeve, A.E.; Kemp, R.A. Inflammatory and regulatory T cells contribute to a unique immune microenvironment in tumor tissue of colorectal cancer patients. Int. J. Cancer 2013, 132, 1842–1850. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23002055 (accessed on 15 July 2019). [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26771115 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Poznanski, S.M.; Ashkar, A.A. What Defines NK Cell Functional Fate: Phenotype or Metabolism? Front. Immunol. 2019, 10, 1414. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31275330 (accessed on 15 July 2019). [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30309915 (accessed on 15 July 2019). [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26997480 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Pare, L.; Pascual, T.; Seguí, E.; Teixidó, C.; Gonzalez-Cao, M.; Galván, P.; Rodríguez, A.; González, B.; Cuatrecasas, M.; Pineda, E.; et al. Association between PD1 mRNA and response to anti-PD1 monotherapy across multiple cancer types. Ann. Oncol. 2018, 29, 2121–2128. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30165419 (accessed on 15 July 2019). [CrossRef]
- Prat, A.; Navarro, A.; Paré, L.; Reguart, N.; Galván, P.; Pascual, T.; Martínez, A.; Nuciforo, P.; Comerma, L.; Alos, L.; et al. Immune-Related Gene Expression Profiling After PD-1 Blockade in Non-Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res. 2017, 77, 3540–3550. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28487385 (accessed on 15 July 2019). [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28650338 (accessed on 15 July 2019). [CrossRef]
- Becht, E.; de Reynies, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26994146 (accessed on 15 July 2019). [CrossRef]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29510987 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Pitroda, S.P.; Khodarev, N.N.; Huang, L.; Uppal, A.; Wightman, S.C.; Ganai, S.; Joseph, N.; Pitt, J.; Brown, M.; Forde, M.; et al. Integrated molecular subtyping defines a curable oligometastatic state in colorectal liver metastasis. Nat. Commun. 2018, 9, 1793. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29728604 (accessed on 15 July 2019). [CrossRef]
- Pedrosa, L.; Moreno, R.; Esposito, F.; Jares, P.; Cuatrecasas, M.; Pineda, E.; Español, M.; Paré, L.; de la Iglesia, N.; Benitez, D.; et al. Immune signatures identify three immune clusters in mCRC, with potential clinical implications. Ann. Oncol. 2019, 30 (Suppl. 4). [Google Scholar] [CrossRef]
- Chun, E.; Lavoie, S.; Michaud, M.; Gallini, C.A.; Kim, J.; Soucy, G.; Odze, R.; Glickman, J.N.; Garrett, W.S. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015, 12, 244–257. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26146082 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Gao, Y.; Sun, W.; Shang, W.; Li, Y.; Zhang, D.; Wang, T.; Zhang, X.; Zhang, S.; Zhang, Y.; Yang, R. Lnc-C/EBPbeta Negatively Regulates the Suppressive Function of Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2018, 6, 1352–1363. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30171135 (accessed on 15 July 2019). [CrossRef]
- Hutton, J.E.; Wang, X.; Zimmerman, L.J.; Slebos, R.J.; Trenary, I.A.; Young, J.D.; Li, M.; Liebler, D.C. Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer. Mol. Cell. Proteom. 2016, 15, 2924–2938. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27340238 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22541435 (accessed on 15 July 2019). [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23535601 (accessed on 15 July 2019). [CrossRef]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24825347 (accessed on 15 July 2019). [CrossRef]
- Sprowl-Tanio, S.; Habowski, A.N.; Pate, K.T.; McQuade, M.M.; Wang, K.; Edwards, R.A.; Grun, F.; Lyou, Y.; Waterman, M.L. Lactate/pyruvate transporter MCT-1 is a direct Wnt target that confers sensitivity to 3-bromopyruvate in colon cancer. Cancer Metab. 2016, 4, 20. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27729975 (accessed on 15 July 2019). [CrossRef]
- Dawson, H.; Galvan, J.A.; Helbling, M.; Muller, D.E.; Karamitopoulou, E.; Koelzer, V.H.; Economou, M.; Hammer, C.; Lugli, A.; Zlobec, I. Possible role of Cdx2 in the serrated pathway of colorectal cancer characterized by BRAF mutation, high-level CpG Island methylator phenotype and mismatch repair-deficiency. Int. J. Cancer 2014, 134, 2342–2351. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24166180 (accessed on 15 July 2019). [CrossRef]
- Guo, R.J.; Funakoshi, S.; Lee, H.H.; Kong, J.; Lynch, J.P. The intestine-specific transcription factor Cdx2 inhibits beta-catenin/TCF transcriptional activity by disrupting the beta-catenin-TCF protein complex. Carcinogenesis 2010, 31, 159–166. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19734199 (accessed on 15 July 2019). [CrossRef]
- Codony-Servat, J.; Cuatrecasas, M.; Asensio, E.; Montironi, C.; Martínez-Cardús, A.; Marín-Aguilera, M.; Horndler, C.; Martínez-Balibrea, E.; Rubini, M.; Jares, P.; et al. Nuclear IGF-1R predicts chemotherapy and targeted therapy resistance in metastatic colorectal cancer. Br. J. Cancer 2017, 117, 1777–1786. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29123263 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013, 73, 3007–3018. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23514705 (accessed on 15 July 2019). [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R.; et al. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27216177 (accessed on 15 July 2019). [CrossRef]
- Cortes, M.; Sanchez-Moral, L.; Barrios, O.; Fernández-Aceñero, M.J.; Martínez-Campanario, M.C.; Esteve-Codina, A.; Darling, D.S.; Győrffy, B.; Lawrence, T.; Dean, D.C.; et al. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. EMBO J. 2017, 36, 3336–3355. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29038174 (accessed on 15 July 2019). [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25348003 (accessed on 15 July 2019). [CrossRef]
- Seth, P.; Csizmadia, E.; Hedblom, A.; Vuerich, M.; Xie, H.; Li, M.; Longhi, M.S.; Wegiel, B. Deletion of Lactate Dehydrogenase-A in Myeloid Cells Triggers Antitumor Immunity. Cancer Res. 2017, 77, 3632–3643. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28446465 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. Available online: https://www.ncbi.nlm.nih.gov/pubmed/16278308 (accessed on 15 July 2019). [CrossRef]
- Xiao, W.; Klement, J.D.; Lu, C.; Ibrahim, M.L.; Liu, K. IFNAR1 Controls Autocrine Type I IFN Regulation of PD-L1 Expression in Myeloid-Derived Suppressor Cells. J. Immunol. 2018, 201, 264–277. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29752314 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1247135. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28123883 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25043024 (accessed on 15 July 2019). [CrossRef]
- Haverkamp, J.M.; Smith, A.M.; Weinlich, R.; Dillon, C.P.; Qualls, J.E.; Neale, G.; Koss, B.; Kim, Y.; Bronte, V.; Herold, M.J.; et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity 2014, 41, 947–959. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25500368 (accessed on 15 July 2019). [CrossRef]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20814239 (accessed on 15 July 2019). [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. Available online: https://www.ncbi.nlm.nih.gov/pubmed/17255361 (accessed on 15 July 2019). [CrossRef]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30006565 (accessed on 15 July 2019). [CrossRef]
- Ito, T.K.; Ishii, G.; Chiba, H.; Ochiai, A. The VEGF angiogenic switch of fibroblasts is regulated by MMP-7 from cancer cells. Oncogene 2007, 26, 7194–7203. Available online: https://www.ncbi.nlm.nih.gov/pubmed/17525740 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Suarez-Lopez, L.; Sriram, G.; Kong, Y.W.; Morandell, S.; Merrick, K.A.; Hernandez, Y.; Haigis, K.M.; Yaffe, M.B. MK2 contributes to tumor progression by promoting M2 macrophage polarization and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E4236–E4244. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29666270 (accessed on 15 July 2019). [CrossRef]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21300765 (accessed on 15 July 2019). [CrossRef]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27354473 (accessed on 15 July 2019). [CrossRef]
- Takeuchi, S.; Baghdadi, M.; Tsuchikawa, T.; Wada, H.; Nakamura, T.; Abe, H.; Nakanishi, S.; Usui, Y.; Higuchi, K.; Takahashi, M.; et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015, 75, 2629–2640. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25952647 (accessed on 15 July 2019). [CrossRef]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rébé, C.; Derangère, V.; Chevriaux, A.; Boidot, R.; Végran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 Cells in Patients with Metastatic Colorectal Cancer Predicts the Efficacy of a FOLFOX-Bevacizumab Drug Treatment Regimen. Cancer Res. 2016, 76, 5241–5252. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27496709 (accessed on 15 July 2019). [CrossRef]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23108136 (accessed on 15 July 2019). [CrossRef]
- Xu, B.; Yu, Z.; Xiang, S.; Li, Y.; Zhang, S.L.; He, Y. Rational design of mitochondria-targeted pyruvate dehydrogenase kinase 1 inhibitors with improved selectivity and antiproliferative activity. Eur. J. Med. Chem. 2018, 155, 275–284. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29890389 (accessed on 15 July 2019). [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20133848 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Beloueche-Babari, M.; Wantuch, S.; Casals Galobart, T.; Koniordou, M.; Parkes, H.G.; Arunan, V.; Chung, Y.L.; Eykyn, T.R.; Smith, P.D.; Leach, M.O. MCT1 Inhibitor AZD3965 Increases Mitochondrial Metabolism, Facilitating Combination Therapy and Noninvasive Magnetic Resonance Spectroscopy. Cancer Res. 2017, 77, 5913–5924. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28923861 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23221383 (accessed on 15 July 2019). [CrossRef]
- Grossman, J.G.; Nywening, T.M.; Belt, B.A.; Panni, R.Z.; Krasnick, B.A.; DeNardo, D.G.; Hawkins, W.G.; Goedegebuure, S.P.; Linehan, D.C.; Fields, R.C. Recruitment of CCR2(+) tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology 2018, 7, e1470729. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30228938 (accessed on 15 July 2019). [CrossRef]
- Sagiv-Barfi, I.; Kohrt, H.E.; Czerwinski, D.K.; Ng, P.P.; Chang, B.Y.; Levy, R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA 2015, 112, E966–E972. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25730880 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Stiff, A.; Trikha, P.; Wesolowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 2016, 76, 2125–2136. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26880800 (accessed on 15 July 2019). [CrossRef]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci. Transl. Med. 2018, 10, eaan5488. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29343622 (accessed on 15 July 2019). [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29443964 (accessed on 15 July 2019). [CrossRef]
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30674873 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19033189 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941–946. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29025995 (accessed on 15 July 2019). [CrossRef] [Green Version]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30220459 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Grim, J.E.; Knoblaugh, S.E.; Guthrie, K.A.; Hagar, A.; Swanger, J.; Hespelt, J.; Delrow, J.J.; Small, T.; Grady, W.M.; Nakayama, K.I.; et al. Fbw7 and p53 cooperatively suppress advanced and chromosomally unstable intestinal cancer. Mol. Cell. Biol. 2012, 32, 2160–2167. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22473991 (accessed on 15 July 2019). [CrossRef]
- Tong, J.; Tan, S.; Zou, F.; Yu, J.; Zhang, L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene 2017, 36, 787–796. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27399335 (accessed on 15 July 2019). [CrossRef]
- Davis, R.J.; Gonen, M.; Margineantu, D.H.; Handeli, S.; Swanger, J.; Hoellerbauer, P.; Paddison, P.J.; Gu, H.; Raftery, D.; Grim, J.E.; et al. Pan-cancer transcriptional signatures predictive of oncogenic mutations reveal that Fbw7 regulates cancer cell oxidative metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 5462–5467. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29735700 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22195744 (accessed on 15 July 2019). [CrossRef]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24792914 (accessed on 15 July 2019). [CrossRef]
- Lee, K.W.; Yeo, S.Y.; Sung, C.O.; Kim, S.H. Twist1 is a key regulator of cancer-associated fibroblasts. Cancer Res. 2015, 75, 73–85. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25368021 (accessed on 15 July 2019). [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24277834 (accessed on 15 July 2019). [CrossRef]
- Timperi, E.; Pacella, I.; Schinzari, V.; Focaccetti, C.; Sacco, L.; Farelli, F.; Caronna, R.; Del Bene, G.; Longo, F.; Ciardi, A.; et al. Regulatory T cells with multiple suppressive and potentially pro-tumor activities accumulate in human colorectal cancer. Oncoimmunology 2016, 5, e1175800. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27622025 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. gammadeltaT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 2014, 40, 785–800. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24816404 (accessed on 15 July 2019). [CrossRef]
- Hu, X.; Bardhan, K.; Paschall, A.V.; Yang, D.; Waller, J.L.; Park, M.A.; Nayak-Kapoor, A.; Samuel, T.A.; Abrams, S.I.; Liu, K. Deregulation of apoptotic factors Bcl-xL and Bax confers apoptotic resistance to myeloid-derived suppressor cells and contributes to their persistence in cancer. J. Biol. Chem. 2013, 288, 19103–19115. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23677993 (accessed on 15 July 2019). [CrossRef]
- Coffelt, S.B.; Tal, A.O.; Scholz, A.; De Palma, M.; Patel, S.; Urbich, C.; Biswas, S.K.; Murdoch, C.; Plate, K.H.; Reiss, Y.; et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010, 70, 5270–5280. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20530679 (accessed on 15 July 2019). [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28514441 (accessed on 15 July 2019). [CrossRef]
- Hu, G.; Wu, P.; Cheng, P.; Zhang, Z.; Wang, Z.; Yu, X.; Shao, X.; Wu, D.; Ye, J.; Zhang, T.; et al. Tumor-infiltrating CD39(+)gammadeltaTregs are novel immunosuppressive T cells in human colorectal cancer. Oncoimmunology 2017, 6, e1277305. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28344891 (accessed on 15 July 2019). [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29769722 (accessed on 15 July 2019). [CrossRef]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016, 17, 1322–1333. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27595233 (accessed on 15 July 2019). [CrossRef]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26880715 (accessed on 15 July 2019). [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27111280 (accessed on 15 July 2019). [CrossRef]
- Norton, S.E.; Ward-Hartstonge, K.A.; McCall, J.L.; Leman, J.K.H.; Taylor, E.S.; Munro, F.; Black, M.A.; Fazekas de St Groth, B.; McGuire, H.M.; Kemp, R.A. High-Dimensional Mass Cytometric Analysis Reveals an Increase in Effector Regulatory T Cells as a Distinguishing Feature of Colorectal Tumors. J. Immunol. 2019, 202, 1871–1884. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30728210 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Numasaki, M.; Fukushi, J.; Ono, M.; Narula, S.K.; Zavodny, P.J.; Kudo, T.; Robbins, P.D.; Tahara, H.; Lotze, M.T. Interleukin-17 promotes angiogenesis and tumor growth. Blood 2003, 101, 2620–2627. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12411307 (accessed on 15 July 2019). [CrossRef]
- Wu, W.K.; Llewellyn, O.P.; Bates, D.O.; Nicholson, L.B.; Dick, A.D. IL-10 regulation of macrophage VEGF production is dependent on macrophage polarisation and hypoxia. Immunobiology 2010, 215, 796–803. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20692534 (accessed on 15 July 2019). [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24793239 (accessed on 15 July 2019). [CrossRef]
- Pander, J.; Heusinkveld, M.; van der Straaten, T.; Jordanova, E.S.; Baak-Pablo, R.; Gelderblom, H.; Morreau, H.; van der Burg, S.H.; Guchelaar, H.J.; van Hall, T. Activation of tumor-promoting type 2 macrophages by EGFR-targeting antibody cetuximab. Clin. Cancer Res. 2011, 17, 5668–5673. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21788356 (accessed on 15 July 2019). [CrossRef]
- Xu, L.; Duda, D.G.; Tomaso, E.; Ancukiewicz, M.; Chung, D.C.; Lauwers, G.Y.; Samuel, R.; Shellito, P.; Czito, B.G.; Lin, P.C.; et al. Direct evidence that bevacizumab, an anti-VEGF antibody, up-regulates SDF1alpha, CXCR4, CXCL6, and neuropilin 1 in tumors from patients with rectal cancer. Cancer Res. 2009, 69, 7905–7910. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19826039 (accessed on 15 July 2019). [CrossRef]
- Jung, K.; Heishi, T.; Incio, J.; Huang, Y.; Beech, E.Y.; Pinter, M.; Ho, W.W.; Kawaguchi, K.; Rahbari, N.N.; Chung, E.; et al. Targeting CXCR4-dependent immunosuppressive Ly6C(low) monocytes improves antiangiogenic therapy in colorectal cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 10455–10460. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28900008 (accessed on 15 July 2019). [CrossRef]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29254508 (accessed on 15 July 2019). [CrossRef]
- Shi, T.; Ma, Y.; Cao, L.; Zhan, S.; Xu, Y.; Fu, F.; Liu, C.; Zhang, G.; Wang, Z.; Wang, R.; et al. B7-H3 promotes aerobic glycolysis and chemoresistance in colorectal cancer cells by regulating HK2. Cell Death Dis. 2019, 10, 308. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30952834 (accessed on 15 July 2019). [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26321681 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Henriques, F.S.; Sertie, R.A.L.; Franco, F.O.; Knobl, P.; Neves, R.X.; Andreotti, S.; Lima, F.B.; Guilherme, A.; Seelaender, M.; Batista, M.L., Jr. Early suppression of adipocyte lipid turnover induces immunometabolic modulation in cancer cachexia syndrome. FASEB J. 2017, 31, 1976–1986. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28138038 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Lawless, S.J.; Kedia-Mehta, N.; Walls, J.F.; McGarrigle, R.; Convery, O.; Sinclair, L.V.; Navarro, M.N.; Murray, J.; Finlay, D.K. Glucose represses dendritic cell-induced T cell responses. Nat. Commun. 2017, 8, 15620. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28555668 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Mennonna, D.; Maccalli, C.; Romano, M.C.; Garavaglia, C.; Capocefalo, F.; Bordoni, R.; Severgnini, M.; De Bellis, G.; Sidney, J.; Sette, A.; et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut 2017, 66, 454–463. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26681737 (accessed on 15 July 2019). [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28886381 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Seaman, S.; Zhu, Z.; Saha, S.; Zhang, X.M.; Yang, M.Y.; Hilton, M.B.; Morris, K.; Szot, C.; Morris, H.; Swing, D.A.; et al. Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer Cell 2017, 31, 501–515.e8. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28399408 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29101163 (accessed on 15 July 2019). [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29539425 (accessed on 15 July 2019). [CrossRef] [Green Version]
- Tarrado-Castellarnau, M.; Atauri, P.; Tarragó-Celada, J.; Perarnau, J.; Yuneva, M.; Thomson, T.M.; Cascante, M. De novo MYC addiction as an adaptive response of cancer cells to CDK4/6 inhibition. Mol. Syst. Biol. 2017, 13, 940. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28978620 (accessed on 15 July 2019). [CrossRef]
- Bendell, J.; Ciardiello, F.; Tabernero, J.; Tebbutt, N.; Eng, C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.; et al. Efficacy and safety results from IMblaze370, a randomised Phase III study comparing atezolizumab+cobimetinib and atezolizumab monotherapy vs regorafenib in chemotherapy-refractory metastatic colorectal cancer. Ann. Oncol. 2018, 29, mdy208.003. [Google Scholar] [CrossRef]


{kind=link}
| Features | Cluster 1 | Cluster 2 | Cluster 3 |
|---|---|---|---|
| Inflamed-Stromal Dependent | Inflamed-Non Stromal Dependent | Non-Inflamed Cold | |
| BRAF, RAS Mutations | BRAF | RAS | none |
| EMT | +++ | + | - |
| Angiogenesis | +++ | +++ | + |
| CAF | +++ | - | - |
| MDSC/M | M2 (pSTAT3) PMF-MDSC (ROS) | M2 (ARG1) PMF-MDSC (ARG1) | M1 (iNOS2) |
| TILs | Treg+++ (CD39+CD25−) | Treg+++ (CD39− CD25+). TH2-like Treg, γδT17 | TH17, TH1 |
| Metabolism in Tumoral Cells | Glycolytic (LDHA, PDK1, PKM2, MCT1) | Glutaminolytic and OXPHOS (GLS1, PC, GLUD1) | Glycolytic (LDHA, PDK1, PKM2, MCT1, HK2) |
| Extracellular Metabolites | High lactate High adenosine High BCKAs | Low glutamine Low arginine | Low glucose High adenosine |
| Potential Therapeutic Targets | PD-1, PI3Kδ, STAT3, BTK LDH, PDK1, MCT1 | PD-1, ARG1, BTK, CCR5 GLS1, PC | PD-1, CDK4/6 PDK1, MCT1 HLDCV, TVEC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedrosa, L.; Esposito, F.; Thomson, T.M.; Maurel, J. The Tumor Microenvironment in Colorectal Cancer Therapy. Cancers 2019, 11, 1172. https://doi.org/10.3390/cancers11081172
Pedrosa L, Esposito F, Thomson TM, Maurel J. The Tumor Microenvironment in Colorectal Cancer Therapy. Cancers. 2019; 11(8):1172. https://doi.org/10.3390/cancers11081172
Chicago/Turabian StylePedrosa, Leire, Francis Esposito, Timothy M. Thomson, and Joan Maurel. 2019. "The Tumor Microenvironment in Colorectal Cancer Therapy" Cancers 11, no. 8: 1172. https://doi.org/10.3390/cancers11081172
APA StylePedrosa, L., Esposito, F., Thomson, T. M., & Maurel, J. (2019). The Tumor Microenvironment in Colorectal Cancer Therapy. Cancers, 11(8), 1172. https://doi.org/10.3390/cancers11081172