BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors


Abstract
:1. Introduction
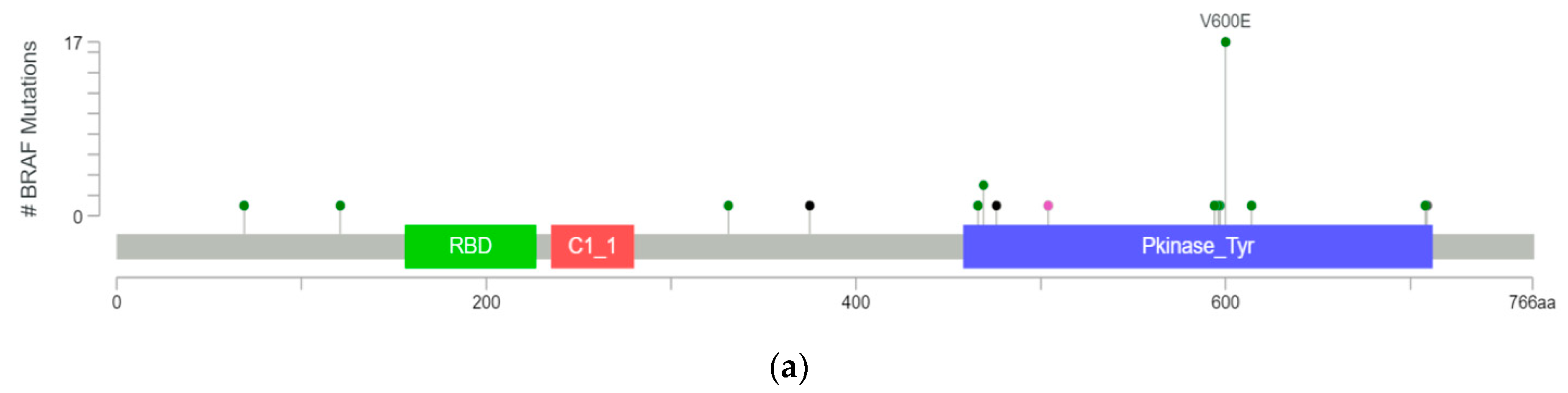
2. BRAF Mutation Classes
2.1. Class I Mutations
2.2. Class II Mutations
2.3. Class III Mutations
3. RAF and MEK Inhibitors
3.1. Type I RAF Inhibitors
3.2. Paradox Breakers
3.3. Dimer Disrupters
3.4. MEK Inhibitors
3.5. ERK Inhibitors
4. BRAF Mutations in Brain Tumor Subtypes and Sensitivity to Targeted Therapy
4.1. Pilocytic Astrocytoma
4.2. Pediatric Astrocytoma
4.3. Adult Astrocytoma
4.4. PXA
4.5. Ganglioglioma
4.6. Craniopharyngioma
5. Resistance to RAF-Targeted Therapy in Brain Tumors
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.E., Jr.; Stephens, R.M.; Saracino, M.R.; Morrison, D.K. Autoregulation of the Raf-1 serine/threonine Kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 9214–9219. [Google Scholar] [CrossRef] [PubMed]
- Daum, G.; Eisenmann-Tappe, I.; Fries, H.W.; Troppmair, J.; Rapp, U.R. The ins and outs of Raf kinases. Trends Biochem. Sci. 1994, 19, 474–480. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Solit, D.B. Therapeutic Strategies for Targeting BRAF in Human Cancer. Rev. Recent. Clin. Trials 2007, 2, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-Mutated Metastatic Melanoma: A Multicentre, Open-Label, Phase 3 Randomised Controlled Trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Kim, K.B.; Kefford, R.; Pavlick, A.C.; Infante, J.R.; Ribas, A.; Sosman, J.A.; Fecher, L.A.; Millward, M.; McArthur, G.A.; Hwu, P.; et al. Phase II Study of the MEK1/MEK2 Inhibitor Trametinib in Patients with Metastatic BRAF-Mutant Cutaneous Melanoma Previously Treated with or without a BRAF Inhibitor. J. Clin. Oncol. 2013, 31, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition Versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall Survival in Patients with BRAF-Mutant Melanoma Receiving Encorafenib Plus Binimetinib Versus Vemurafenib Or Encorafenib (COLUMBUS): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.; Kitchen, N.; Mulholland, P. Dabrafenib and Trametinib in BRAFV600E Mutated Glioma. CNS Oncol. 2017, 6, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Aguiar, D.; Vargas, M.I.; Lobrinus, A.; Dietrich, P.Y. BRAF/MEK Double Blockade in Refractory Anaplastic Pleomorphic Xanthoastrocytoma. Neurology 2017, 88, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Schreck, K.C.; Guajardo, A.; Lin, D.D.M.; Eberhart, C.G.; Grossman, S.A. Concurrent BRAF/MEK Inhibitors in BRAF V600-Mutant High-Grade Primary Brain Tumors. J. Natl. Compr. Cancer Netw. 2018, 16, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Ferguson, C.J.; Grierson, P.M.; Dahiya, S.; Ansstas, G. Rapid Clinical and Radiographic Response with Combined Dabrafenib and Trametinib in Adults with BRAF-Mutated High-Grade Glioma. J. Natl. Compr. Cancer Netw. 2018, 16, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Kaley, T.; Touat, M.; Subbiah, V.; Hollebecque, A.; Rodon, J.; Lockhart, A.C.; Keedy, V.; Bielle, F.; Hofheinz, R.D.; Joly, F.; et al. BRAF Inhibition in BRAF(V600)-Mutant Gliomas: Results from the VE-BASKET Study. J. Clin. Oncol. 2018, 36, 3477. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer. Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine their Sensitivity to Pharmacologic Inhibition. Cancer. Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef] [PubMed]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. (V600E)BRAF is Associated with Disabled Feedback Inhibition of RAF-MEK Signaling and Elevated Transcriptional Output of the Pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates their Activity in BRAFV600E Melanomas. Cancer. Cell 2012, 22, 668–682. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with Class 3 BRAF Mutants are Sensitive to the Inhibition of Activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E Mutation in 1,320 Nervous System Tumors Reveals High Mutation Frequencies in Pleomorphic Xanthoastrocytoma, Ganglioglioma and Extra-Cerebellar Pilocytic Astrocytoma. Acta Neuropathol. 2011, 121, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, S.; Emnett, R.J.; Haydon, D.H.; Leonard, J.R.; Phillips, J.J.; Perry, A.; Gutmann, D.H. BRAF-V600E Mutation in Pediatric and Adult Glioblastoma. Neuro Oncol. 2014, 16, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Behling, F.; Barrantes-Freer, A.; Skardelly, M.; Nieser, M.; Christians, A.; Stockhammer, F.; Rohde, V.; Tatagiba, M.; Hartmann, C.; Stadelmann, C.; et al. Frequency of BRAF V600E Mutations in 969 Central Nervous System Neoplasms. Diagn. Pathol. 2016, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt-DeMasters, B.K.; Aisner, D.L.; Foreman, N.K. BRAF VE1 Immunoreactivity Patterns in Epithelioid Glioblastomas Positive for BRAF V600E Mutation. Am. J. Surg. Pathol. 2015, 39, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Hatae, R.; Hata, N.; Suzuki, S.O.; Yoshimoto, K.; Kuga, D.; Murata, H.; Akagi, Y.; Sangatsuda, Y.; Iwaki, T.; Mizoguchi, M.; et al. A Comprehensive Analysis Identifies BRAF Hotspot Mutations Associated with Gliomas with Peculiar Epithelial Morphology. Neuropathology 2017, 37, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Taylor-Weiner, A.; Manley, P.E.; Jones, R.T.; Dias-Santagata, D.; Thorner, A.R.; Lawrence, M.S.; Rodriguez, F.J.; Bernardo, L.A.; Schubert, L.; et al. Exome Sequencing Identifies BRAF Mutations in Papillary Craniopharyngiomas. Nat. Genet. 2014, 46, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors Across all CNS Compartments, Histopathological Grades, and Age Groups. Cancer. Cell 2015, 27, 728–743. [Google Scholar] [CrossRef] [PubMed]
- Pratt, D.; Camelo-Piragua, S.; McFadden, K.; Leung, D.; Mody, R.; Chinnaiyan, A.; Koschmann, C.; Venneti, S. BRAF activating mutations involving the β3-αC loop in V600E-negative anaplastic pleomorphic xanthoastrocytoma. Acta Neuropathol. Commun. 2018, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Buchanan, S.; Yadav, V.; Mochalkin, I.; Wong, S.S.; Yue, Y.G.; Huber, L.; et al. Oncogenic BRAF Deletions that Function as Homodimers and are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer. Discov. 2016, 6, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.A.; Whalen, D.M.; Ozen, A.; Wongchenko, M.J.; Yin, J.; Yen, I.; Schaefer, G.; Mayfield, J.D.; Chmielecki, J.; Stephens, P.J.; et al. Activation Mechanism of Oncogenic Deletion Mutations in BRAF, EGFR, and HER2. Cancer. Cell 2016, 29, 477–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, E.E.; Lin, A.; Tihan, T.; Burger, P.C.; Eberhart, C.G. Frequent Gains at Chromosome 7q34 Involving BRAF in Pilocytic Astrocytoma. J. Neuropathol. Exp. Neurol. 2008, 67, 878–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievert, A.J.; Lang, S.S.; Boucher, K.L.; Madsen, P.J.; Slaunwhite, E.; Choudhari, N.; Kellet, M.; Storm, P.B.; Resnick, A.C. Paradoxical Activation and RAF Inhibitor Resistance of BRAF Protein Kinase Fusions Characterizing Pediatric Astrocytomas. Proc. Natl. Acad. Sci. USA 2013, 110, 5957–5962. [Google Scholar] [CrossRef] [PubMed]
- Sievert, A.J.; Jackson, E.M.; Gai, X.; Hakonarson, H.; Judkins, A.R.; Resnick, A.C.; Sutton, L.N.; Storm, P.B.; Shaikh, T.H.; Biegel, J.A. Duplication of 7q34 in Pediatric Low-Grade Astrocytomas Detected by High-Density Single-Nucleotide Polymorphism-Based Genotype Arrays Results in a Novel BRAF Fusion Gene. Brain Pathol. 2009, 19, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Backlund, L.M.; Ichimura, K.; Collins, V.P. Tandem Duplication Producing a Novel Oncogenic BRAF Fusion Gene Defines the Majority of Pilocytic Astrocytomas. Cancer Res. 2008, 68, 8673–8677. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Gronych, J.; Lichter, P.; Witt, O.; Pfister, S.M. MAPK Pathway Activation in Pilocytic Astrocytoma. Cell Mol. Life Sci. 2012, 69, 1799–1811. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, M.; Badiali, M.; Moi, L.; Buttarelli, F.R.; Baldi, C.; Massimino, M.; Sanson, M.; Giangaspero, F. KIAA1549:BRAF Fusion Gene in Pediatric Brain Tumors of various Histogenesis. Pediatr. Blood Cancer 2015, 62, 724–727. [Google Scholar] [CrossRef]
- Pekmezci, M.; Villanueva-Meyer, J.E.; Goode, B.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Bastian, B.C.; Chamyan, G.; Maher, O.M.; Khatib, Z.; et al. The Genetic Landscape of Ganglioglioma. Acta Neuropathol. Commun. 2018, 6, 47. [Google Scholar] [CrossRef]
- Miller, K.E.; Kelly, B.; Fitch, J.; Ross, N.; Avenarius, M.R.; Varga, E.; Koboldt, D.C.; Boue, D.R.; Magrini, V.; Coven, S.L.; et al. Genome Sequencing Identifies Somatic BRAF Duplication c.1794_1796dupTAC;p.Thr599dup in Pediatric Patient with Low-Grade Ganglioglioma. Cold Spring Harb Mol. Case Stud. 2018, 4. [Google Scholar] [CrossRef]
- Chmielecki, J.; Bailey, M.; He, J.; Elvin, J.; Vergilio, J.A.; Ramkissoon, S.; Suh, J.; Frampton, G.M.; Sun, J.X.; Morley, S.; et al. Genomic Profiling of a Large Set of Diverse Pediatric Cancers Identifies Known and Novel Mutations Across Tumor Spectra. Cancer Res. 2017, 77, 509–519. [Google Scholar] [CrossRef]
- Ryall, S.; Arnoldo, A.; Krishnatry, R.; Mistry, M.; Khor, K.; Sheth, J.; Ling, C.; Leung, S.; Zapotocky, M.; Guerreiro Stucklin, A.; et al. Multiplex Detection of Pediatric Low-Grade Glioma Signature Fusion Transcripts and Duplications using the NanoString nCounter System. J. Neuropathol. Exp. Neurol. 2017, 76, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. Whole-Genome Sequencing Identifies Genetic Alterations in Pediatric Low-Grade Gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [PubMed]
- Helgager, J.; Lidov, H.G.; Mahadevan, N.R.; Kieran, M.W.; Ligon, K.L.; Alexandrescu, S. A Novel GIT2-BRAF Fusion in Pilocytic Astrocytoma. Diagn. Pathol. 2017, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.J.; Karajannis, M.A.; Diolaiti, D.; Mansukhani, M.M.; Bender, J.G.; Kung, A.L.; Garvin, J.H., Jr. A Novel, Potentially Targetable TMEM106B-BRAF Fusion in Pleomorphic Xanthoastrocytoma. Cold Spring Harb Mol. Case Stud. 2017, 3, a001396. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; Kawakami, T.; et al. Functional Analysis of Mutations within the Kinase Activation Segment of B-Raf in Human Colorectal Tumors. Cancer Res. 2003, 63, 8132–8137. [Google Scholar] [PubMed]
- Summers, M.G.; Smith, C.G.; Maughan, T.S.; Kaplan, R.; Escott-Price, V.; Cheadle, J.P. BRAF and NRAS Locus-Specific Variants have Different Outcomes on Survival to Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2742–2749. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Tseng, L.H.; Chen, G.; Haley, L.; Illei, P.; Gocke, C.D.; Eshleman, J.R.; Lin, M.T. Clinical Detection and Categorization of Uncommon and Concomitant Mutations Involving BRAF. BMC Cancer 2015, 15, 779. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF Inhibitors Transactivate RAF Dimers and ERK Signalling in Cells with Wild-Type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Zelboraf [Package Insert]; Genentech USA, Inc.: South San Francisco, CA, USA, 2017.
- Tafinlar [Package Insert]; Novartis Pharmaceuticals Corporation: East Hanover, NJ, USA, 2018.
- Braftovi [Package Insert]; Array BioPharma Inc.: Boulder, CO, USA, 2019.
- Nicolaides, T.P.; Li, H.; Solomon, D.A.; Hariono, S.; Hashizume, R.; Barkovich, K.; Baker, S.J.; Paugh, B.S.; Jones, C.; Forshew, T.; et al. Targeted Therapy for BRAFV600E Malignant Astrocytoma. Clin. Cancer Res. 2011, 17, 7595–7604. [Google Scholar] [CrossRef] [PubMed]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF Inhibitors Prime Wild-Type RAF to Activate the MAPK Pathway and Enhance Growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Karajannis, M.A.; Legault, G.; Fisher, M.J.; Milla, S.S.; Cohen, K.J.; Wisoff, J.H.; Harter, D.H.; Goldberg, J.D.; Hochman, T.; Merkelson, A.; et al. Phase II Study of Sorafenib in Children with Recurrent Or Progressive Low-Grade Astrocytomas. Neuro Oncol. 2014, 16, 1408–1416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF Inhibitors that Evade Paradoxical MAPK Pathway Activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Lavoie, H.; Sahmi, M.; David, M.; Hilt, C.; Hammell, A.; Therrien, M. RAF Inhibitors Promote RAS-RAF Interaction by Allosterically Disrupting RAF Autoinhibition. Nat. Commun. 2017, 8, 1211. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, R.A.; Lin, L.; Olivas, V.; Chan, E.; Markegard, E.; Rymar, A.; Neel, D.; Chen, X.; Hemmati, G.; Bollag, G.; et al. Preclinical Efficacy of a RAF Inhibitor that Evades Paradoxical MAPK Pathway Activation in Protein Kinase BRAF-Mutant Lung Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 13456–13461. [Google Scholar] [CrossRef] [PubMed]
- Basile, K.J.; Le, K.; Hartsough, E.J.; Aplin, A.E. Inhibition of Mutant BRAF Splice Variant Signaling by Next-Generation, Selective RAF Inhibitors. Pigment Cell. Melanoma Res. 2014, 27, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Gao, Y.; Su, W.; Yaeger, R.; Tao, J.; Na, N.; Zhang, Y.; Zhang, C.; Rymar, A.; Tao, A.; et al. RAF Inhibitor PLX8394 Selectively Disrupts BRAF Dimers and RAS-Independent BRAF-Mutant-Driven Signaling. Nat. Med. 2019, 25, 284–291. [Google Scholar] [CrossRef]
- Hong, D.S.; Hollebecque, A.; Gordon, M.S.; Flaherty, K.T.; Shapiro, G.; Rodon, J.; Millward, M.; Ramdas, N.; Zhang, W.; Gao, L.; et al. A First-in-Human Dose Phase 1 Study of LY3009120 in Advanced Cancer Patients. JCO 2017, 35, 2507. [Google Scholar] [CrossRef]
- Girotti, M.R.; Lopes, F.; Preece, N.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Whittaker, S.; Saturno, G.; Viros, A.; Pedersen, M.; et al. Paradox-Breaking RAF Inhibitors that also Target SRC are Effective in Drug-Resistant BRAF Mutant Melanoma. Cancer. Cell 2015, 27, 85–96. [Google Scholar] [CrossRef]
- Wang, J.; Yao, Z.; Jonsson, P.; Allen, A.N.; Qin, A.C.R.; Uddin, S.; Dunkel, I.J.; Petriccione, M.; Manova, K.; Haque, S.; et al. A Secondary Mutation in BRAF Confers Resistance to RAF Inhibition in a BRAF V600E-Mutant Brain Tumor. Cancer. Discov. 2018, 8, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fangusaro, J.; Onar-Thomas, A.; Young Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Goldman, S.; et al. Selumetinib in Paediatric Patients with BRAF-Aberrant Or Neurofibromatosis Type 1-Associated Recurrent, Refractory, Or Progressive Low-Grade Glioma: A Multicentre, Phase 2 Trial. Lancet Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Robison, N.; Pauly, J.; Malvar, J.; Gruber-Filbin, M.; de Mola, R.L.; Dorris, K.; Bendel, A.; Bowers, D.; Bornhorst, M.; Gauvain, K.; et al. LGG-44. A phase I dose escalation trial of the MEK1/2 inhibitor MEK162 (binimetinib) in children with low-grade gliomas and other Ras/Raf pathway-activated tumors. Neuro-Oncology 2018, 20, i114. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer. Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; Severson, E.; Gay, L.; Vergilio, J.A.; Elvin, J.; Suh, J.; Daniel, S.; Covert, M.; Frampton, G.M.; Hsu, S.; et al. Comprehensive Genomic Profiling of 282 Pediatric Low- and High-Grade Gliomas Reveals Genomic Drivers, Tumor Mutational Burden, and Hypermutation Signatures. Oncologist 2017, 22, 1478–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.P.; Scapulatempo-Neto, C.; Carloni, A.C.; Paulino, A.; Sheren, J.; Aisner, D.L.; Musselwhite, E.; Clara, C.; Machado, H.R.; Oliveira, R.S.; et al. KIAA1549: BRAF Gene Fusion and FGFR1 Hotspot Mutations are Prognostic Factors in Pilocytic Astrocytomas. J. Neuropathol. Exp. Neurol. 2015, 74, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.; Walker, E.; Mohamed, N.; Zhang, C.; Jacob, K.; Shirinian, M.; Alon, N.; Kahn, D.; Fried, I.; Scheinemann, K.; et al. BRAF-KIAA1549 Fusion Predicts Better Clinical Outcome in Pediatric Low-Grade Astrocytoma. Clin. Cancer Res. 2011, 17, 4790–4798. [Google Scholar] [CrossRef] [PubMed]
- Lassaletta, A.; Zapotocky, M.; Mistry, M.; Ramaswamy, V.; Honnorat, M.; Krishnatry, R.; Guerreiro Stucklin, A.; Zhukova, N.; Arnoldo, A.; Ryall, S.; et al. Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J. Clin. Oncol. 2017, 35, 2934. [Google Scholar] [CrossRef] [PubMed]
- Drobysheva, A.; Klesse, L.J.; Bowers, D.C.; Rajaram, V.; Rakheja, D.; Timmons, C.F.; Wang, J.; Koral, K.; Gargan, L.; Ramos, E.; et al. Targeted MAPK Pathway Inhibitors in Patients with Disseminated Pilocytic Astrocytomas. J. Natl. Compr. Cancer Netw. 2017, 15, 978–982. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Mobley, B.C.; Gordish-Dressman, H.; VandenBussche, C.J.; Mason, G.E.; Bornhorst, M.; Esbenshade, A.J.; Tehrani, M.; Orr, B.A.; LaFrance, D.R.; et al. A Clinicopathologic Study of Diencephalic Pediatric Low-Grade Gliomas with BRAF V600 Mutation. Acta Neuropathol. 2015, 130, 575–585. [Google Scholar] [CrossRef]
- Kieran, M.W.; Bouffet, E.; Tabori, U.; Broniscer, A.; Cohen, K.; Hansford, J.; Geoerger, B.; Hingorani, P.; Dunkel, I.; Russo, M.; et al. The First Study of Dabrafenib in Pediatric Patients with BRAF V600-Mutant Relapsed or Refractory Low-Grade Gliomas. Ann. Oncol. 2016, 27, vi552–vi587. [Google Scholar] [CrossRef]
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Young Poussaint, T.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A Phase I Trial of the MEK Inhibitor Selumetinib (AZD6244) in Pediatric Patients with Recurrent or Refractory Low-Grade Glioma: A Pediatric Brain Tumor Consortium (PBTC) Study. Neuro Oncol. 2017, 19, 1135–1144. [Google Scholar] [CrossRef]
- Korshunov, A.; Ryzhova, M.; Hovestadt, V.; Bender, S.; Sturm, D.; Capper, D.; Meyer, J.; Schrimpf, D.; Kool, M.; Northcott, P.A.; et al. Integrated Analysis of Pediatric Glioblastoma Reveals a Subset of Biologically Favorable Tumors with Associated Molecular Prognostic Markers. Acta Neuropathol. 2015, 129, 669–678. [Google Scholar] [CrossRef]
- Ballester, L.Y.; Fuller, G.N.; Powell, S.Z.; Sulman, E.P.; Patel, K.P.; Luthra, R.; Routbort, M.J. Retrospective Analysis of Molecular and Immunohistochemical Characterization of 381 Primary Brain Tumors. J. Neuropathol. Exp. Neurol. 2017, 76, 179–188. [Google Scholar] [CrossRef]
- Wen, P.; Alexander, S.; Yung-Jue, B.; van den Bent, M.J.; Gazzah, A.; Dietrich, S.; de Vos, F.; van Linde, M.E.; Lai, A.; Chi, A.; et al. Efficacy and Safety of Dabrafenib + Trametinib in Patients with recurrent/refractory BRAF V60E-Mutated High-Grade Glioma (HGG). Neuro Oncol. 2018, 20, vi238. [Google Scholar]
- Pages, M.; Beccaria, K.; Boddaert, N.; Saffroy, R.; Besnard, A.; Castel, D.; Fina, F.; Barets, D.; Barret, E.; Lacroix, L.; et al. Co-Occurrence of Histone H3 K27M and BRAF V600E Mutations in Paediatric Midline Grade I Ganglioglioma. Brain Pathol. 2018, 28, 103–111. [Google Scholar] [CrossRef]
- Meletath, S.K.; Pavlick, D.; Brennan, T.; Hamilton, R.; Chmielecki, J.; Elvin, J.A.; Palma, N.; Ross, J.S.; Miller, V.A.; Stephens, P.J.; et al. Personalized Treatment for a Patient with a BRAF V600E Mutation using Dabrafenib and a Tumor Treatment Fields Device in a High-Grade Glioma Arising from Ganglioglioma. J. Natl. Compr. Cancer Netw. 2016, 14, 1345–1350. [Google Scholar] [CrossRef]
- Touat, M.; Gratieux, J.; Condette Auliac, S.; Sejean, K.; Aldea, S.; Savatovsky, J.; Perkins, G.; Blons, H.; Ligon, K.L.; Idbaih, A.; et al. Vemurafenib and Cobimetinib Overcome Resistance to Vemurafenib in BRAF-Mutant Ganglioglioma. Neurology 2018, 91, 523–525. [Google Scholar] [CrossRef]
- Garnier, L.; Ducray, F.; Verlut, C.; Mihai, M.I.; Cattin, F.; Petit, A.; Curtit, E. Prolonged Response Induced by Single Agent Vemurafenib in a BRAF V600E Spinal Ganglioglioma: A Case Report and Review of the Literature. Front. Oncol. 2019, 9, 177. [Google Scholar] [CrossRef]
- Bautista, F.; Paci, A.; Minard-Colin, V.; Dufour, C.; Grill, J.; Lacroix, L.; Varlet, P.; Valteau-Couanet, D.; Geoerger, B. Vemurafenib in Pediatric Patients with BRAFV600E Mutated High-Grade Gliomas. Pediatr. Blood Cancer 2014, 61, 1101–1103. [Google Scholar] [CrossRef]
- del Bufalo, F.; Carai, A.; Figa-Talamanca, L.; Pettorini, B.; Mallucci, C.; Giangaspero, F.; Antonelli, M.; Badiali, M.; Moi, L.; Bianco, G.; et al. Response of Recurrent BRAFV600E Mutated Ganglioglioma to Vemurafenib as Single Agent. J. Transl. Med. 2014, 12, 356. [Google Scholar] [CrossRef]
- Rush, S.; Foreman, N.; Liu, A. Brainstem Ganglioglioma Successfully Treated with Vemurafenib. J. Clin. Oncol. 2013, 31, e159–e160. [Google Scholar] [CrossRef]
- Aguilera, D.; Janss, A.; Mazewski, C.; Castellino, R.C.; Schniederjan, M.; Hayes, L.; Brahma, B.; Fogelgren, L.; MacDonald, T.J. Successful Retreatment of a Child with a Refractory Brainstem Ganglioglioma with Vemurafenib. Pediatr. Blood Cancer 2016, 63, 541–543. [Google Scholar] [CrossRef]
- Beland, B.; Tsang, R.Y.; Sutherland, G. Unprecedented Response to Combination BRAF and MEK Inhibitors in Adult Anaplastic Ganglioglioma. J. Neurooncol. 2018, 137, 667–669. [Google Scholar] [CrossRef]
- Toll, S.A.; Tran, H.N.; Cotter, J.; Judkins, A.R.; Tamrazi, B.; Biegel, J.A.; Dhall, G.; Robison, N.J.; Waters, K.; Patel, P.; et al. Sustained Response of Three Pediatric BRAF(V600E) Mutated High-Grade Gliomas to Combined BRAF and MEK Inhibitor Therapy. Oncotarget 2019, 10, 551–557. [Google Scholar] [CrossRef]
- Marks, A.M.; Bindra, R.S.; DiLuna, M.L.; Huttner, A.; Jairam, V.; Kahle, K.T.; Kieran, M.W. Response to the BRAF/MEK Inhibitors dabrafenib/trametinib in an Adolescent with a BRAF V600E Mutated Anaplastic Ganglioglioma Intolerant to Vemurafenib. Pediatr. Blood Cancer 2018, 65, e26969. [Google Scholar] [CrossRef]
- Kumar, A.; Pathak, P.; Purkait, S.; Faruq, M.; Jha, P.; Mallick, S.; Suri, V.; Sharma, M.C.; Suri, A.; Sarkar, C. Oncogenic KIAA1549-BRAF Fusion with Activation of the MAPK/ERK Pathway in Pediatric Oligodendrogliomas. Cancer Genet. 2015, 208, 91–95. [Google Scholar] [CrossRef]
- Mistry, M.; Zhukova, N.; Merico, D.; Rakopoulos, P.; Krishnatry, R.; Shago, M.; Stavropoulos, J.; Alon, N.; Pole, J.D.; Ray, P.N.; et al. BRAF Mutation and CDKN2A Deletion Define a Clinically Distinct Subgroup of Childhood Secondary High-Grade Glioma. J. Clin. Oncol. 2015, 33, 1015–1022. [Google Scholar] [CrossRef]
- Del Bufalo, F.; Ceglie, G.; Cacchione, A.; Alessi, I.; Colafati, G.S.; Carai, A.; Diomedi-Camassei, F.; De Billy, E.; Agolini, E.; Mastronuzzi, A.; et al. BRAF V600E Inhibitor (Vemurafenib) for BRAF V600E Mutated Low Grade Gliomas. Front. Oncol. 2018, 8, 526. [Google Scholar] [CrossRef] [Green Version]
- Vuong, H.G.; Altibi, A.M.A.; Duong, U.N.P.; Ngo, H.T.T.; Pham, T.Q.; Fung, K.M.; Hassell, L. BRAF Mutation is Associated with an Improved Survival in Glioma-a Systematic Review and Meta-Analysis. Mol. Neurobiol. 2017, 55, 3718–3724. [Google Scholar] [CrossRef]
- Ferguson, S.D.; Xiu, J.; Weathers, S.P.; Zhou, S.; Kesari, S.; Weiss, S.E.; Verhaak, R.G.; Hohl, R.J.; Barger, G.R.; Reddy, S.K.; et al. GBM-Associated Mutations and Altered Protein Expression are More Common in Young Patients. Oncotarget 2016, 7, 69466–69478. [Google Scholar] [CrossRef]
- Zhang, R.Q.; Shi, Z.; Chen, H.; Chung, N.Y.; Yin, Z.; Li, K.K.; Chan, D.T.; Poon, W.S.; Wu, J.; Zhou, L.; et al. Biomarker-Based Prognostic Stratification of Young Adult Glioblastoma. Oncotarget 2016, 7, 5030–5041. [Google Scholar] [CrossRef]
- Schreck, K.; Vera, E.; Aboud, O.; Acquaye, A.; Boris, L.; Briceno, N.; Brown, M.; Chung, H.; Crandon, S.; Garren, N.; et al. PATH-28. The natural history of braf v600e-mutated glioblastomas in adults. Neuro-Oncology 2018, 20, vi164. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Salvage Therapy with BRAF Inhibitors for Recurrent Pleomorphic Xanthoastrocytoma: A Retrospective Case Series. J. Neurooncol. 2013, 114, 237–240. [Google Scholar] [CrossRef]
- Ida, C.M.; Rodriguez, F.J.; Burger, P.C.; Caron, A.A.; Jenkins, S.M.; Spears, G.M.; Aranguren, D.L.; Lachance, D.H.; Giannini, C. Pleomorphic Xanthoastrocytoma: Natural History and Long-Term Follow-Up. Brain Pathol. 2015, 25, 575–586. [Google Scholar] [CrossRef]
- Tabouret, E.; Bequet, C.; Denicolai, E.; Barrie, M.; Nanni, I.; Metellus, P.; Dufour, H.; Chinot, O.; Figarella-Branger, D. BRAF Mutation and Anaplasia may be Predictive Factors of Progression-Free Survival in Adult Pleomorphic Xanthoastrocytoma. Eur. J. Surg. Oncol. 2015, 41, 1685–1690. [Google Scholar] [CrossRef]
- Usubalieva, A.; Pierson, C.R.; Kavran, C.A.; Huntoon, K.; Kryvenko, O.N.; Mayer, T.G.; Zhao, W.; Rock, J.; Ammirati, M.; Puduvalli, V.K.; et al. Primary Meningeal Pleomorphic Xanthoastrocytoma with Anaplastic Features: A Report of 2 Cases, One with BRAF(V600E) Mutation and Clinical Response to the BRAF Inhibitor Dabrafenib. J. Neuropathol. Exp. Neurol. 2015, 74, 960–969. [Google Scholar] [CrossRef]
- Lee, E.Q.; Ruland, S.; LeBoeuf, N.R.; Wen, P.Y.; Santagata, S. Successful Treatment of a Progressive BRAF V600E-Mutated Anaplastic Pleomorphic Xanthoastrocytoma with Vemurafenib Monotherapy. J. Clin. Oncol. 2016, 34, e87–e89. [Google Scholar] [CrossRef]
- Leaver, K.M.; Zhang, N.; Ziskin, J.L.; Vogel, H.; Recht, L.; Thomas, R.P. Response of Metastatic Glioma to Vemurafenib. Neuro-Oncol. Pract. 2016, 3, 268–271. [Google Scholar] [CrossRef]
- Burger, M.C.; Ronellenfitsch, M.W.; Lorenz, N.I.; Wagner, M.; Voss, M.; Capper, D.; Tzaridis, T.; Herrlinger, U.; Steinbach, J.P.; Stoffels, G.; et al. Dabrafenib in Patients with Recurrent, BRAF V600E Mutated Malignant Glioma and Leptomeningeal Disease. Oncol. Rep. 2017, 38, 3291–3296. [Google Scholar] [CrossRef]
- Dahiya, S.; Haydon, D.H.; Alvarado, D.; Gurnett, C.A.; Gutmann, D.H.; Leonard, J.R. BRAF(V600E) Mutation is a Negative Prognosticator in Pediatric Ganglioglioma. Acta Neuropathol. 2013, 125, 901–910. [Google Scholar] [CrossRef]
- Louis, D.N.; Giannini, C.; Capper, D.; Paulus, W.; Figarella-Branger, D.; Lopes, M.B.; Batchelor, T.T.; Cairncross, J.G.; van den Bent, M.; Wick, W.; et al. CIMPACT-NOW Update 2: Diagnostic Clarifications for Diffuse Midline Glioma, H3 K27M-Mutant and Diffuse astrocytoma/anaplastic Astrocytoma, IDH-Mutant. Acta Neuropathol. 2018, 135, 639–642. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Shankar, G.M.; Gill, C.M.; Taylor-Weiner, A.; Nayyar, N.; Panka, D.J.; Sullivan, R.J.; Frederick, D.T.; Abedalthagafi, M.; Jones, P.S.; et al. Dramatic Response of BRAF V600E Mutant Papillary Craniopharyngioma to Targeted Therapy. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef]
- Aylwin, S.J.; Bodi, I.; Beaney, R. Pronounced Response of Papillary Craniopharyngioma to Treatment with Vemurafenib, a BRAF Inhibitor. Pituitary 2016, 19, 544–546. [Google Scholar] [CrossRef]
- Himes, B.T.; Ruff, M.W.; Van Gompel, J.J.; Park, S.S.; Galanis, E.; Kaufmann, T.J.; Uhm, J.H. Recurrent Papillary Craniopharyngioma with BRAF V600E Mutation Treated with Dabrafenib: Case Report. J. Neurosurg. 2018, 130, 1299–1303. [Google Scholar]
- Rostami, E.; Witt Nystrom, P.; Libard, S.; Wikstrom, J.; Casar-Borota, O.; Gudjonsson, O. Recurrent Papillary Craniopharyngioma with BRAFV600E Mutation Treated with Neoadjuvant-Targeted Therapy. Acta Neurochir. 2017, 159, 2217–2221. [Google Scholar] [CrossRef]
- Roque, A.; Odia, Y. BRAF-V600E Mutant Papillary Craniopharyngioma Dramatically Responds to Combination BRAF and MEK Inhibitors. CNS Oncol. 2017, 6, 95–99. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of Feedback Inhibition of HER3 Transcription by RAF and MEK Inhibitors Attenuates their Antitumor Effects in BRAF-Mutant Thyroid Carcinomas. Cancer. Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of Colon Cancer to BRAF(V600E) Inhibition through Feedback Activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas Acquire Resistance to B-RAF(V600E) Inhibition by RTK or N-RAS Upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in Cutaneous Melanoma is Associated with RAS Activation and MEK Dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF Inhibitor Resistance is Mediated by Dimerization of Aberrantly Spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and Strategies to Overcome Resistance to Molecularly Targeted Therapy for Melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
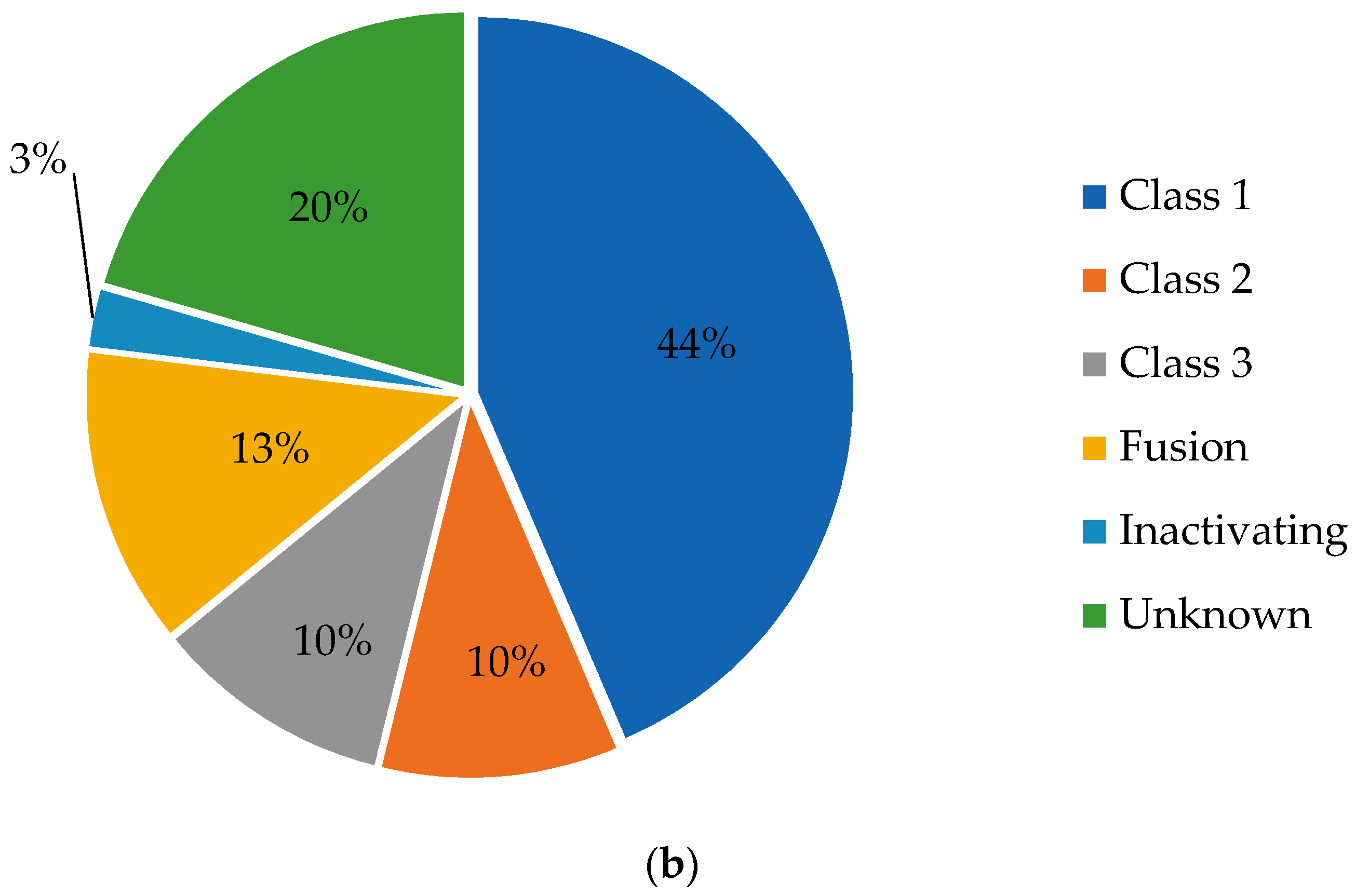{kind=link}
| Class I | Class II | Class III |
|---|---|---|
| V600E | G469A | G466E |
| G469R | D594G | |
| L597R | G596D | |
| T599_W604ins [38] | ||
| T599dup [39] | ||
| KIAA1549–BRAF fusion | ||
| BCAS1–BRAF fusion [40] | ||
| CCDC6–BRAF fusion | ||
| CDC42BPB–BRAF fusion [38] | ||
| ERC2–RAF1 fusion [38] | ||
| FAM131B–BRAF fusion [41] | ||
| FXR1–BRAF fusion [42] | ||
| GIT2–BRAF [43] | ||
| KLHL7–BRAF fusion [38] | ||
| RNF130–BRAF fusion [41] | ||
| TEMEM106B–BRAF fusion [40,44] |
| RAF Inhibitors | |||
| Generation | Drug Name | Manufacturer | FDA Phase |
| 1st | Sorafenib (Nexavar) | Bayer/Onyx Pharmaceuticals | Approved for hepatocellular and renal cell carcinoma |
| 2nd | Vemurafenib (Zelboraf) | Genentech | Approved for BRAF V600E advanced melanoma and Erdheim–Chester Disease |
| 2nd | Dabrafenib (Tafinlar) | Novartis | Approved for BRAF V600E/K melanoma or metastatic non-small cell lung cancer |
| 2nd | Encorafenib (Braftovi) | Array BioPharma | Approved for BRAF V600E/K advanced melanoma |
| 3rd | TAK-580 | Millennium Pharmaceuticals | Phase I/II ongoing |
| 3rd | PLX8394 | Plexxikon | Phase I/IIa ongoing |
| 3rd | BGB283 | BeiGene | Phase 1 ongoing |
| 3rd | LY3009120 | Eli Lilly | Phase I terminated |
| 3rd | BAL3833 (CCT3833) | Basilea | Phase 1 completed |
| MEK Inhibitors | |||
| Drug Name | Manufacturer | FDA Phase | |
| Cobimetinib (Cotellic) | Genentech | Approved for BRAF V600E advanced melanoma | |
| Trametinib (Mekinist) | Novartis | Approved for BRAF V600E/K melanoma or metastatic non-small cell lung cancer | |
| Binimetinib (Mektovi) | Array BioPharma | Approved for BRAF V600E/K advanced melanoma | |
| Selumetinib | AstraZeneca | Breakthrough Therapy Designation; Phase II trials ongoing | |
| RO5126766 | Chugai Pharmaceutical | Phase I ongoing | |
| HL-085 | Shanghai Kechow Pharma | Phase I ongoing | |
| ERK Inhibitors | |||
| Drug Name | Manufacturer | FDA Phase | |
| Ulixertinib | Merck | Phase I/IIa completed | |
| LY3214996 | Eli Lilly & Company | Phase I ongoing | |
| LTT462 | Novartis | Phase Ib ongoing | |
| Inhibitor | |||||||
|---|---|---|---|---|---|---|---|
| Brain Tumor Type | Mutation | Incidence | Type I RAF | Type II RAF | RAF Dimer | MEK | RAF + MEK |
| Pilocytic astrocytoma | KIAA1549–BRAF | 60–70% [42,69] | Not active [33] | TAK-580 (NCT03429803) | Selumetinib [66] Binimetinib [67] (NCT02285439) | ||
| V600E | 10% [22,42,72] | Dabrafenib/Trametinib case series [73] | |||||
| Pediatric low-grade astrocytoma | V600E | 20–35% [72,74] | Dabrafenib [75] Vemurafenib (NCT01748149, NCT03220035) | PLX8394 (NCT02428712) | TAK-580 (NCT03429803) | Trametinib [76] (NCT02124772) | Dabrafenib/Trametinib (NCT02684058; NCT02124772) |
| KIAA1549–BRAF | Preclinical activity [33] | ||||||
| Pediatric high-grade astrocytoma | V600E | 10–20% [23,77] | Vemurafenib (NCT01748149, NCT03220035) | Dabrafenib/Trametinib (NCT02684058) | |||
| Adult low-grade astrocytoma | V600E | 5–15% [24] | [15] | Dabrafenib/Trametinib (NCT02034110) | |||
| Adult high-grade astrocytoma | V600E | 3% [22,24,78] | Vemurafenib [15] | Dabrafenib/Trametinib [79] (NCT02034110) Encorafenib/Binimetinib (NCT03973918) | |||
| Pleomorphic xanthoastrocytoma | V600E | 70% [22] | Vemurafenib [15] | Dabrafenib/Trametinib [79] (NCT02034110) Encorafenib/Binimetinib (NCT03973918) | |||
| Ganglioglioma | V600E | 50% [72,80] | Vemurafenib case reports [15,81,82,83,84,85,86,87] Dabrafenib case [81] | Case reports [82,88,89,90] | |||
| Papillary craniopharyngioma | V600E | 95% [27] | Dabrafenib/Trametinib (NCT03224767) | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262. https://doi.org/10.3390/cancers11091262
Schreck KC, Grossman SA, Pratilas CA. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers. 2019; 11(9):1262. https://doi.org/10.3390/cancers11091262
Chicago/Turabian StyleSchreck, Karisa C., Stuart A. Grossman, and Christine A. Pratilas. 2019. "BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors" Cancers 11, no. 9: 1262. https://doi.org/10.3390/cancers11091262
APA StyleSchreck, K. C., Grossman, S. A., & Pratilas, C. A. (2019). BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers, 11(9), 1262. https://doi.org/10.3390/cancers11091262