In-house Implementation of Tumor Mutational Burden Testing to Predict Durable Clinical Benefit in Non-small Cell Lung Cancer and Melanoma Patients

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Description of Patient Cohort
2.2. Deamination in FFPE Samples
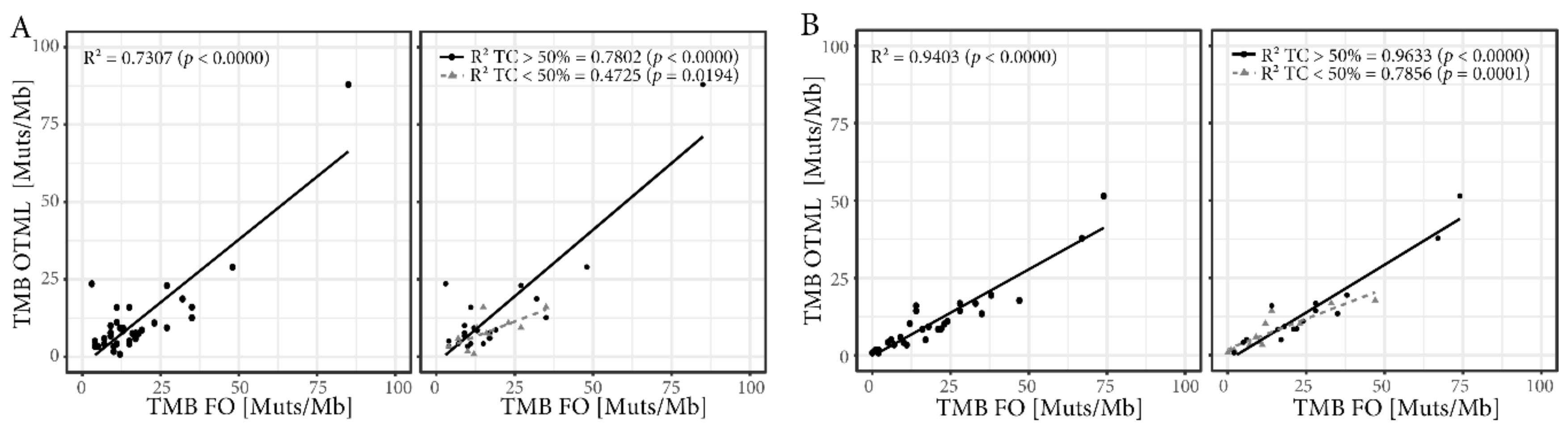
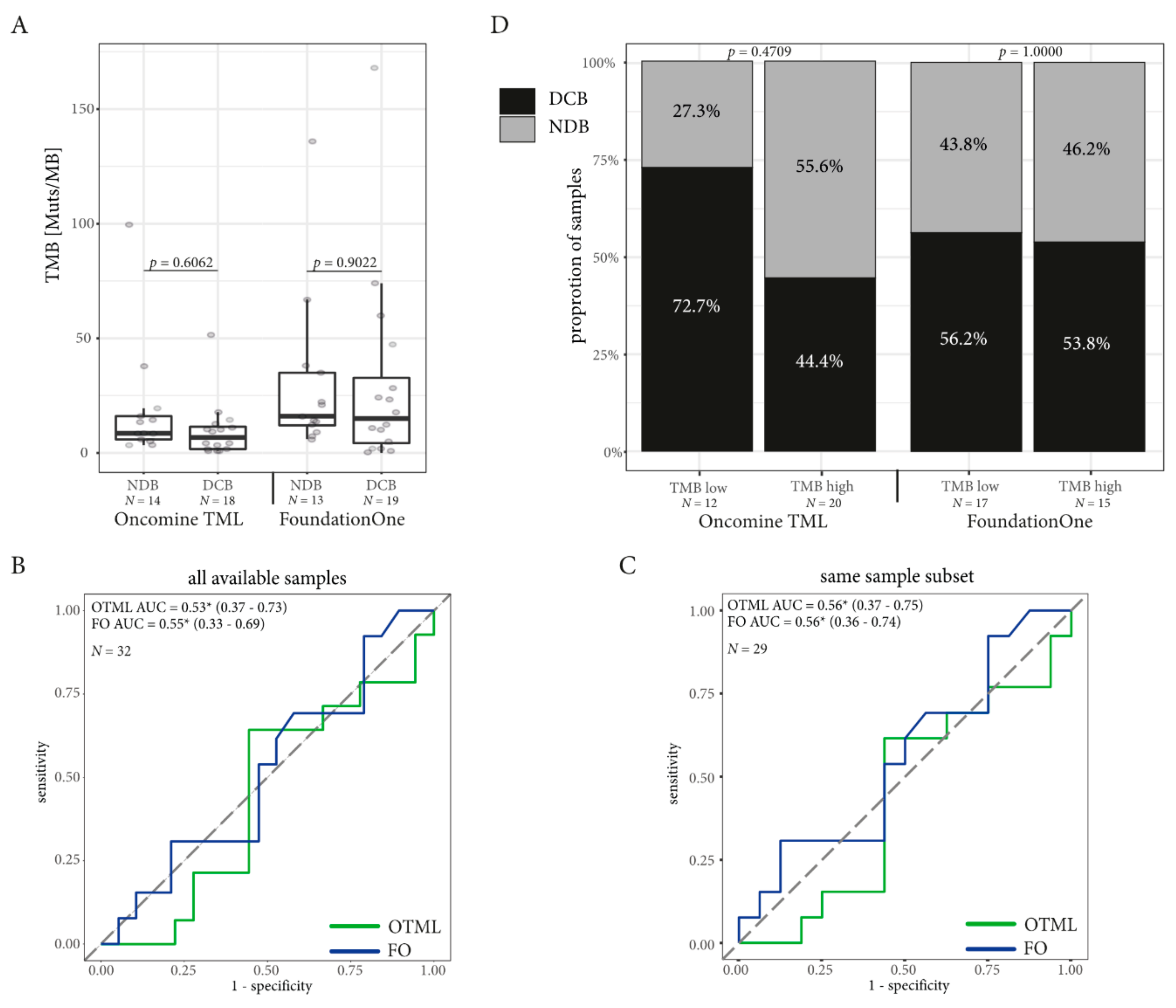
2.3. Tumor Mutational Burden (TMB) Assessed by Targeted Sequencing Panels are Well Correlated
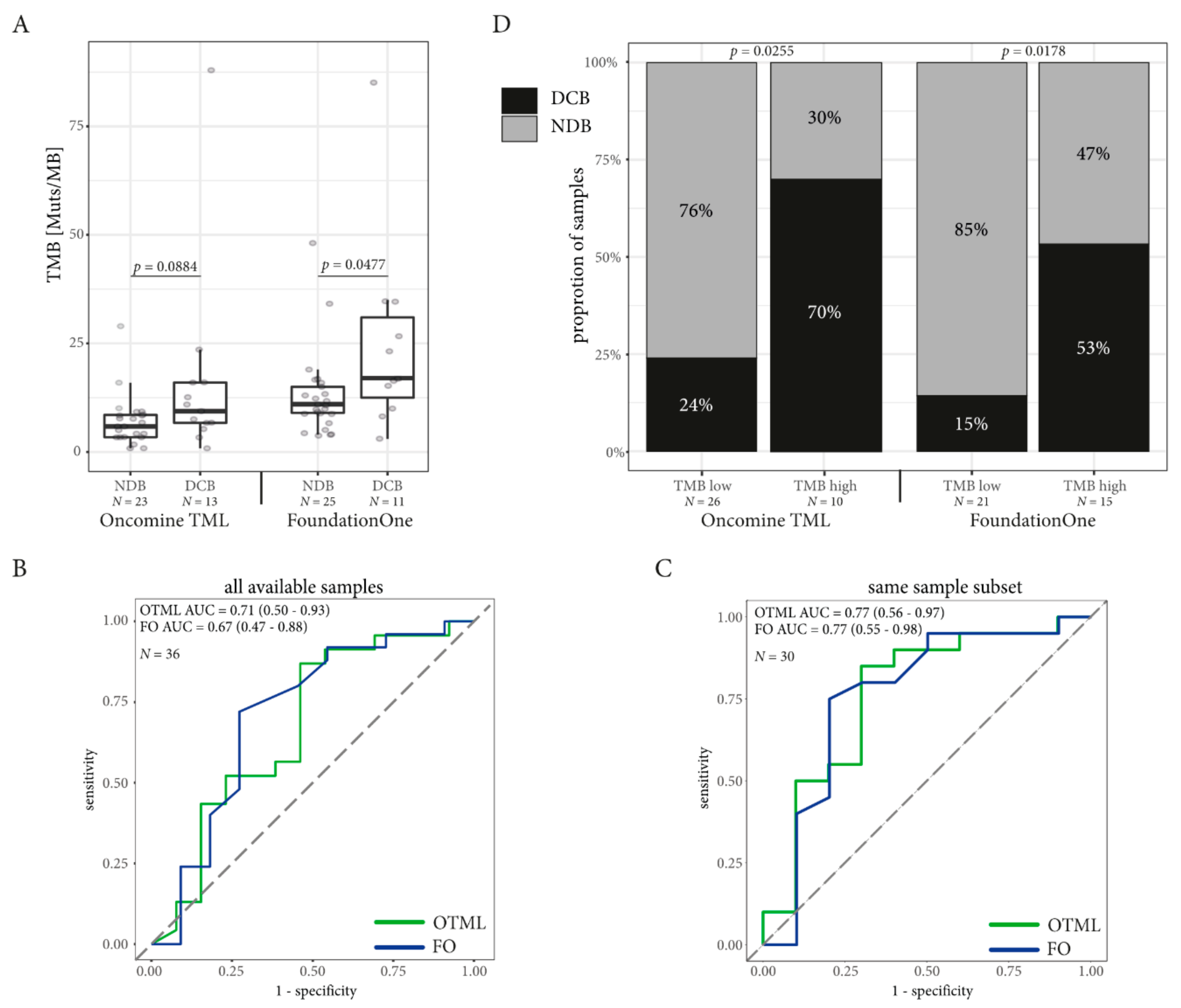
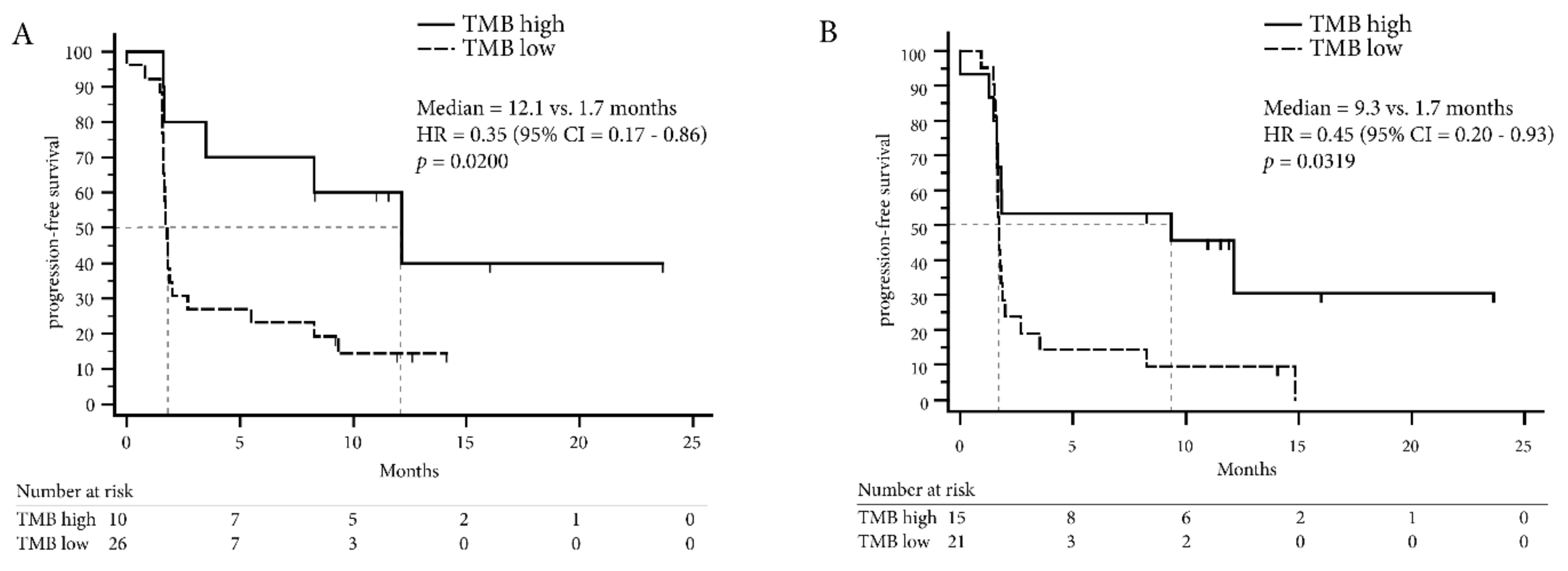
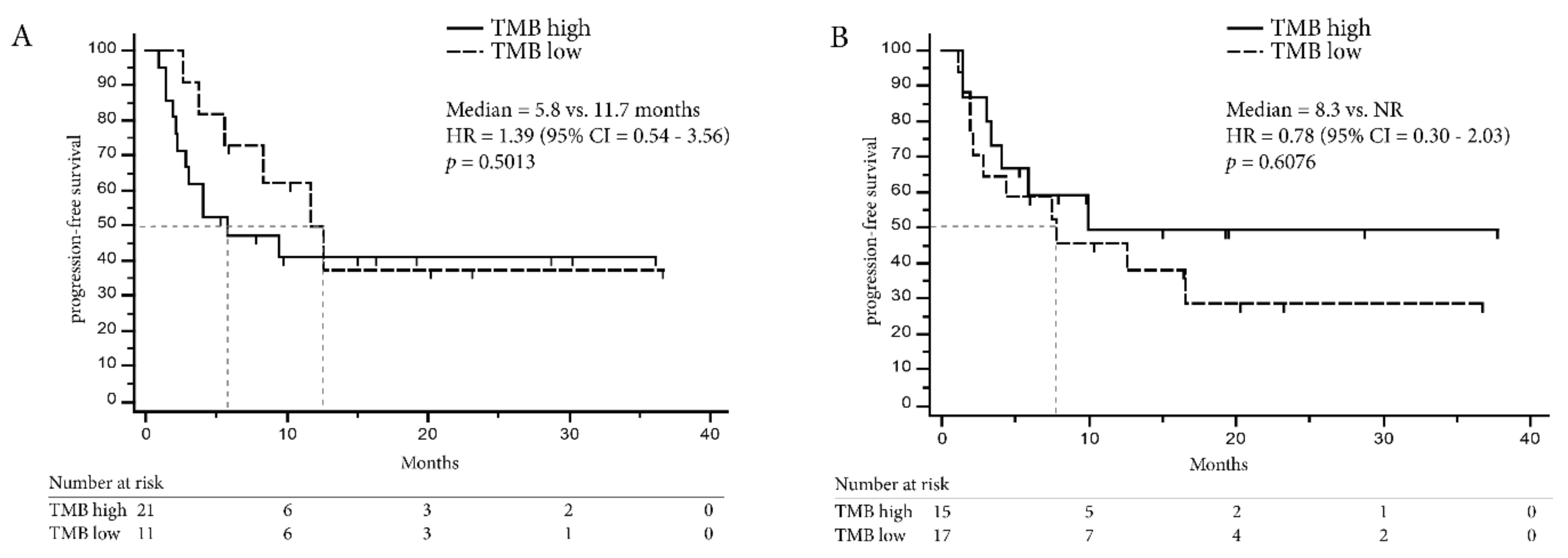
2.4. TMB Assessed by Targeted Sequencing Panels in the Same Patients is Associated with DCB in NSCLC but not in Melanoma
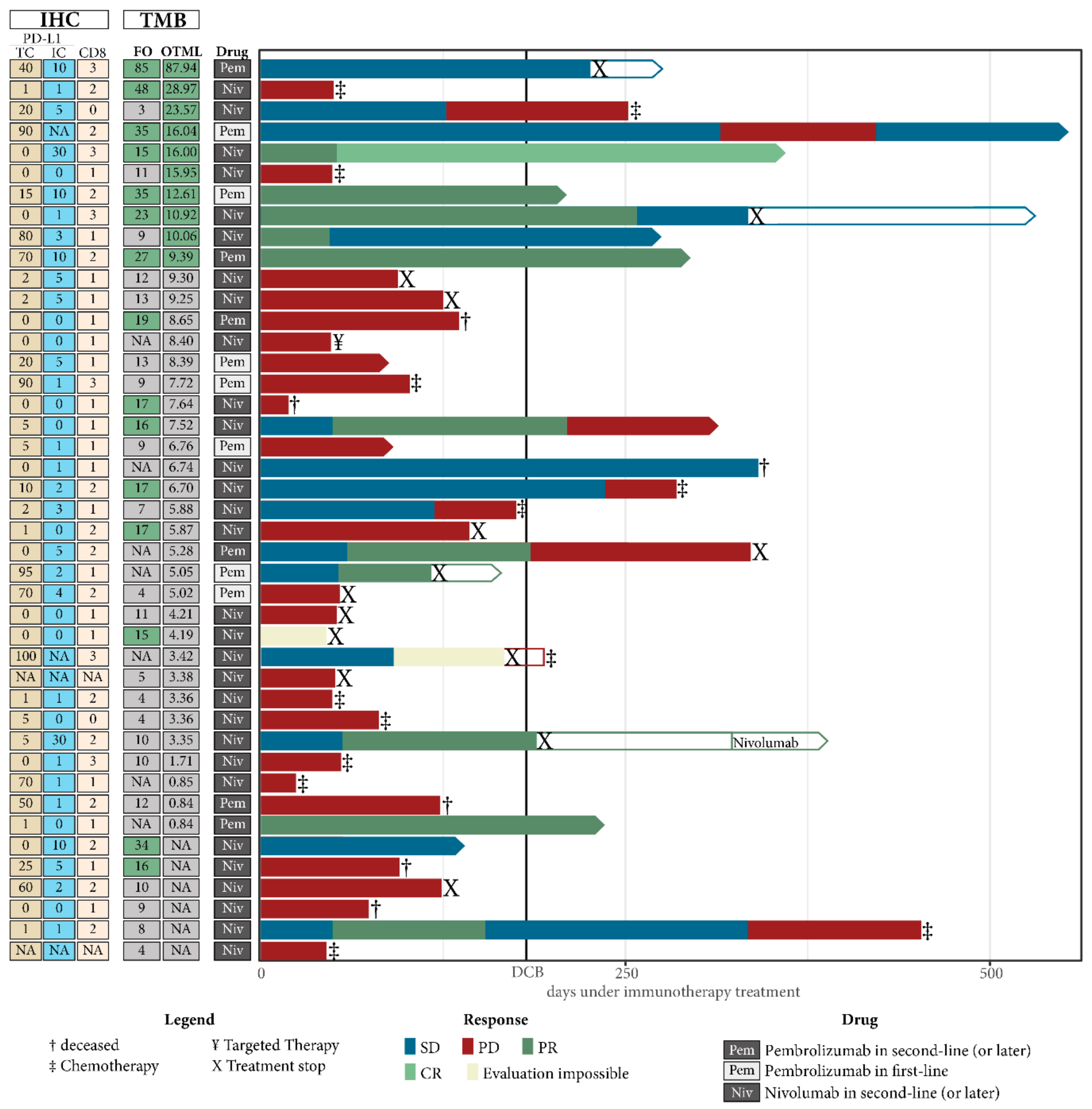
2.5. Combining TMB with PD-L1 Expression in Immune Cells Can Improve Prediction of DCB in NSCLC
3. Discussion
4. Materials and Methods
4.1. Patient Selection
4.2. Sample Preparation
4.3. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hofman, P.; Heeke, S.; Alix-Panabières, C.; Pantel, K. Liquid biopsy in the era of immune-oncology. Is it ready for prime-time use for cancer patients? Ann. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Doebele, R.C.; Kerr, K.M. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 2019, 16, 341–355. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.; Han, G.; Schalper, K.A.; Carvajal-Hausdorf, D.; Pelekanou, V.; Rehman, J.; Velcheti, V.; Herbst, R.; LoRusso, P.; Rimm, D.L. Quantitative Assessment of the Heterogeneity of PD-L1 Expression in Non–Small-Cell Lung Cancer. JAMA Oncol. 2016, 2, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Grigg, C.; Rizvi, N.A. PD-L1 biomarker testing for non-small cell lung cancer: Truth or fiction? J Immunother Cancer. J. Immunother. Cancer 2016, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Kowanetz, M.; Zou, W.; Gettinger, S.N.; Koeppen, H.; Kockx, M.; Schmid, P.; Kadel, E.E.; Wistuba, I.; Chaft, J.; Rizvi, N.A.; et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti–PD-L1). Proc. Natl. Acad. Sci. USA 2018, 115, E10119–E10126. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-H.; Ho, H.-L.; Lai, C.-R.; Luo, Y.-H.; Tseng, Y.-C.; Whang-Peng, J.; Lin, Y.; Chou, T.-Y.; Chen, Y.-M. PD-L1 Expression of Tumor Cells, Macrophages, and Immune Cells in Non–Small Cell Lung Cancer Patients with Malignant Pleural Effusion. J. Thorac. Oncol. 2018, 13, 447–453. [Google Scholar] [CrossRef]
- Mazzaschi, G.; Madeddu, D.; Falco, A.; Bocchialini, G.; Goldoni, M.; Sogni, F.; Armani, G.; Lagrasta, C.A.; Lorusso, B.; Mangiaracina, C.; et al. Low PD-1 Expression in Cytotoxic CD8 + Tumor-Infiltrating Lymphocytes Confers an Immune-Privileged Tissue Microenvironment in NSCLC with a Prognostic and Predictive Value. Clin. Cancer. Res. 2018, 24, 407–419. [Google Scholar] [CrossRef]
- Kitano, S.; Nakayama, T.; Yamashita, M. Biomarkers for Immune Checkpoint Inhibitors in Melanoma. Front Oncol. 2018, 8, 270. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Törngren, T.; Kvist, A. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.; Wang, H.; Sunshine, J.; Kaunitz, G.J.; Cottrell, T.R.; Xu, H.; Esandrio, J.; Anders, R.A.; Cope, L.; Pardoll, D.M.; et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc. Natl. Acad. Sci. USA 2016, 113, E7769–E7777. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J. Clin. Oncol. 2019, 37, 992–1000. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.-E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.B.; Liu, C. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 1–14. [Google Scholar] [CrossRef]
- Chaudhary, R.; Quagliata, L.; Martin, J.P.; Alborelli, I.; Cyanam, D.; Mittal, V.; Tom, W.; Au-Young, J.; Sadis, S.; Hyland, F. A scalable solution for tumor mutational burden from formalin-fixed, paraffin-embedded samples using the Oncomine Tumor Mutation Load Assay. Transl. Lung Cancer Res. 2018, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Barroso-sousa, R.; Jimenez, L.; Camargo, A.A. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget 2015, 6, 34221–34227. [Google Scholar] [CrossRef] [PubMed]
- Leichsenring, J.; Rempel, E.; Lier, A.; Penzel, R.; Fröhling, S.; Christopoulos, P.; Neumann, O.; Allgäuer, M.; Schirmacher, P.; Volckmar, A.-L.; et al. Implementing tumor mutational burden (TMB) analysis in routine diagnostics—A primer for molecular pathologists and clinicians. Transl. Lung Cancer Res. 2018, 7, 703–715. [Google Scholar] [CrossRef]
- Budczies, J.; Allgäuer, M.; Litchfield, K.; Rempel, E.; Christopoulos, P.; Kazdal, D.; Endris, V.; Thomas, M.; Fröhling, S.; Peters, S.; et al. Optimizing panel-based tumor mutational burden (TMB) measurement. Ann. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Heeke, S.; Hofman, P. Tumor mutational burden assessment as a predictive biomarker for immunotherapy in lung cancer patients: Getting ready for prime-time or not? Transl. Lung Cancer Res. 2018, 7, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, K.; Nagashima, T.; Urakami, K.; Ohshima, K.; Serizawa, M.; Ohnami, S.; Shimoda, Y.; Ohnami, S.; Maruyama, K.; Naruoka, A.; et al. Tumor mutational burden analysis of 2,000 Japanese cancer genomes using whole exome and targeted gene panel sequencing. Biomed. Res. 2018, 39, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tom, W.; Chaudhary, R.; Mittal, V.; Cyanam, D.; Casuga, I.; Wong-Ho, E.; Bennett, R.; Hyland, F.; Sadis, S.; Au-Young, J. Improvement of tumor mutation burden measurement by removal of deaminated bases in FFPE DNA. In Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019; p. 1701, Abstract 1701. [Google Scholar] [CrossRef]
- Serizawa, M.; Yokota, T.; Hosokawa, A.; Kusafuka, K.; Sugiyama, T.; Tsubosa, Y.; Yasui, H.; Nakajima, T.; Koh, Y. The efficacy of uracil DNA glycosylase pretreatment in amplicon-based massively parallel sequencing with DNA extracted from archived formalin-fixed paraffin-embedded esophageal cancer tissues. Cancer Genet. 2015, 208, 415–427. [Google Scholar] [CrossRef] [Green Version]
- Gorden, E.M.; Sturk-Andreaggi, K.; Marshall, C. Repair of DNA damage caused by cytosine deamination in mitochondrial DNA of forensic case samples. Forensic Sci. Int. Genet. 2018, 34, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Samorodnitsky, E.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Lyon, E.; Damodaran, S.; Bhatt, D.; Reeser, J.W.; Datta, J.; et al. Evaluation of Hybridization Capture Versus Amplicon-Based Methods for Whole-Exome Sequencing. Hum. Mutat. 2015, 36, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Kazdal, D.; Endris, V.; Allgäuer, M.; Kriegsmann, M.; Leichsenring, J.; Volckmar, A.-L.; Harms, A.; Kirchner, M.; Kriegsmann, K.; Neumann, O.; et al. Spatial and temporal heterogeneity of panel-based tumor mutational burden (TMB) in pulmonary adenocarcinoma: Separating biology from technical artifacts. J. Thorac. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Endris, V.; Buchhalter, I.; Allgäuer, M.; Rempel, E.; Lier, A.; Volckmar, A.; Kirchner, M.; Winterfeld, M.; Leichsenring, J.; Neumann, O.; et al. Measurement of tumor mutational burden (TMB) in routine molecular diagnostics: In silico and real-life analysis of three larger gene panels. Int. J. Cancer. 2019, 144, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-X.; Wang, Z.-X.; Zhao, Q.; Wang, F.; Xu, R.-H. Designing gene panels for tumor mutational burden estimation: The need to shift from ‘correlation’ to ‘accuracy’. J. Immunother. Cancer 2019, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weißgraeber, S.; Han, C.-T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma - results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Ilié, M.; Szafer-Glusman, E.; Hofman, V.; Chamorey, E.; Lalvée, S.; Selva, E.; Leroy, S.; Marquette, C.-H.; Kowanetz, M.; Hedge, P.; et al. Detection of PD-L1 in circulating tumor cells and white blood cells from patients with advanced non-small-cell lung cancer. Ann. Oncol. 2018, 29, 193–199. [Google Scholar] [CrossRef]
- Ilie, M.; Falk, A.T.; Butori, C.; Chamorey, E.; Bonnetaud, C.; Long, E.; Lassalle, S.; Zahaf, K.; Vénissac, N.; Mouroux, J.; et al. PD-L1 expression in basaloid squamous cell lung carcinoma: Relationship to PD-1 + and CD8 + tumor-infiltrating T cells and outcome. Mod. Pathol. 2016, 29, 1552–1564. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn Res. 2011, 12, 2825–2830. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heeke, S.; Benzaquen, J.; Long-Mira, E.; Audelan, B.; Lespinet, V.; Bordone, O.; Lalvée, S.; Zahaf, K.; Poudenx, M.; Humbert, O.; et al. In-house Implementation of Tumor Mutational Burden Testing to Predict Durable Clinical Benefit in Non-small Cell Lung Cancer and Melanoma Patients. Cancers 2019, 11, 1271. https://doi.org/10.3390/cancers11091271
Heeke S, Benzaquen J, Long-Mira E, Audelan B, Lespinet V, Bordone O, Lalvée S, Zahaf K, Poudenx M, Humbert O, et al. In-house Implementation of Tumor Mutational Burden Testing to Predict Durable Clinical Benefit in Non-small Cell Lung Cancer and Melanoma Patients. Cancers. 2019; 11(9):1271. https://doi.org/10.3390/cancers11091271
Chicago/Turabian StyleHeeke, Simon, Jonathan Benzaquen, Elodie Long-Mira, Benoit Audelan, Virginie Lespinet, Olivier Bordone, Salomé Lalvée, Katia Zahaf, Michel Poudenx, Olivier Humbert, and et al. 2019. "In-house Implementation of Tumor Mutational Burden Testing to Predict Durable Clinical Benefit in Non-small Cell Lung Cancer and Melanoma Patients" Cancers 11, no. 9: 1271. https://doi.org/10.3390/cancers11091271
APA StyleHeeke, S., Benzaquen, J., Long-Mira, E., Audelan, B., Lespinet, V., Bordone, O., Lalvée, S., Zahaf, K., Poudenx, M., Humbert, O., Montaudié, H., Dugourd, P. -M., Chassang, M., Passeron, T., Delingette, H., Marquette, C. -H., Hofman, V., Stenzinger, A., Ilié, M., & Hofman, P. (2019). In-house Implementation of Tumor Mutational Burden Testing to Predict Durable Clinical Benefit in Non-small Cell Lung Cancer and Melanoma Patients. Cancers, 11(9), 1271. https://doi.org/10.3390/cancers11091271