1. Introduction
Colorectal cancer (CRC) is a fatal malignancy that is frequently diagnosed worldwide [
1]. Both hereditary and environmental risk factors play a part in the development of CRC. Although genetic mutations have recently been found to play a key role in colon carcinogenesis, environmental factors also contribute to the development and progression of CRC [
2]. In a large number of experimental, clinical, and epidemiologic studies, the incidence of non-familial CRC has been inextricably associated with dietary and lifestyle factors, such as red meat consumption, the use of artificial preservatives, and smoking [
3].
Chronic inflammation has been found to play a key role in the progression of colon cancers. Numerous studies have shown that inflammatory bowel diseases (IBDs), including Crohn’s disease and ulcerative colitis (UC), are associated with the development and progression of CRC [
4,
5,
6]. It has been reported that 25% of patients with IBD have colon cancer [
7,
8]. Based on the evidence of a strong association between IBD and CRC, colitis-associated cancer (CAC) has been proposed and is currently recognized as a special subtype of CRC.
Parallelly, there has been an interest in defining gut microbiota that are linked to colon cancer [
9]. With advances in sequencing techniques, studies have reported correlations between microbiota dysbiosis and CRC, and pathogenic microbiota that accelerate colon cancer progression have been identified [
10]. For example, it has been reported that the enterotoxigenic
Bacteroides fragilis contribute to colon carcinogenesis by producing toxins [
11]. Together with numerous potential carcinogenic intestinal microorganisms,
Fusobacterium nucleatum (
F. nucleatum) has been reported to be a contributing factor to CRC progression based on its presence in CRC specimens [
12,
13,
14]. The importance of
F. nucleatum in CRC is further supported by reports that it increases the chemoresistance of colon cancer via modulating autophagy and enhances the proliferation and tumor development of CRC by activating Toll-like receptor 4 (TLR4) signaling [
9]. In addition, studies have demonstrated a relationship between
F. nucleatum and colitis by showing that
F. nucleatum aggravates colitis via damaging epithelial integrity and regulating M1 macrophage polarization [
15]. However, despite such a focus on clarifying the role of
F. nucleatum in the development and progression of colon diseases, including colitis and CRC, there are few studies exploring the correlation between them, and the mechanism involved remains unclear. Since
F. nucleatum has been shown to modulate the progression of both CRC and colitis, it has been hypothesized that
F. nucleatum may drive the progression of UC and the development of CAC. Here, we investigated the contribution of
F. nucleatum to CAC progression by assessing the phenotypic changes in CRC cells treated with dextran sulfate sodium (DSS), a colitis-inducing chemical, in the presence of
F. nucleatum. In addition, the effect of
F. nucleatum on azoxymethane (AOM)/DSS-induced ulcerative colitis colorectal cancer (UC-CRC) in a C57BL/6 mouse model was examined.
3. Discussion
F. nucleatum, a Gram-negative anaerobic bacterium, is one of the pathogens that cause periodontitis; it has been shown to be an important driver in both UC and CRC [
26,
27]. Previous studies have reported that
F. nucleatum contributes to the progression of colitis by damaging the integrity of colon epithelium and downregulating intercellular adhesion molecules, and that it also induces characteristic changes in macrophages in the colorectal tumor microenvironment [
28]. Studies on the development and progression of CRC have shown that
F. nucleatum promotes tumor formation as well as chemoresistance [
29]. Although numerous works have reported the importance of
F. nucleatum in the pathogenesis of UC and CRC, the role of
F. nucleatum in the pathogenesis of CAC [
30], which is the product of the close relationship between UC and CRC, remains unclear.
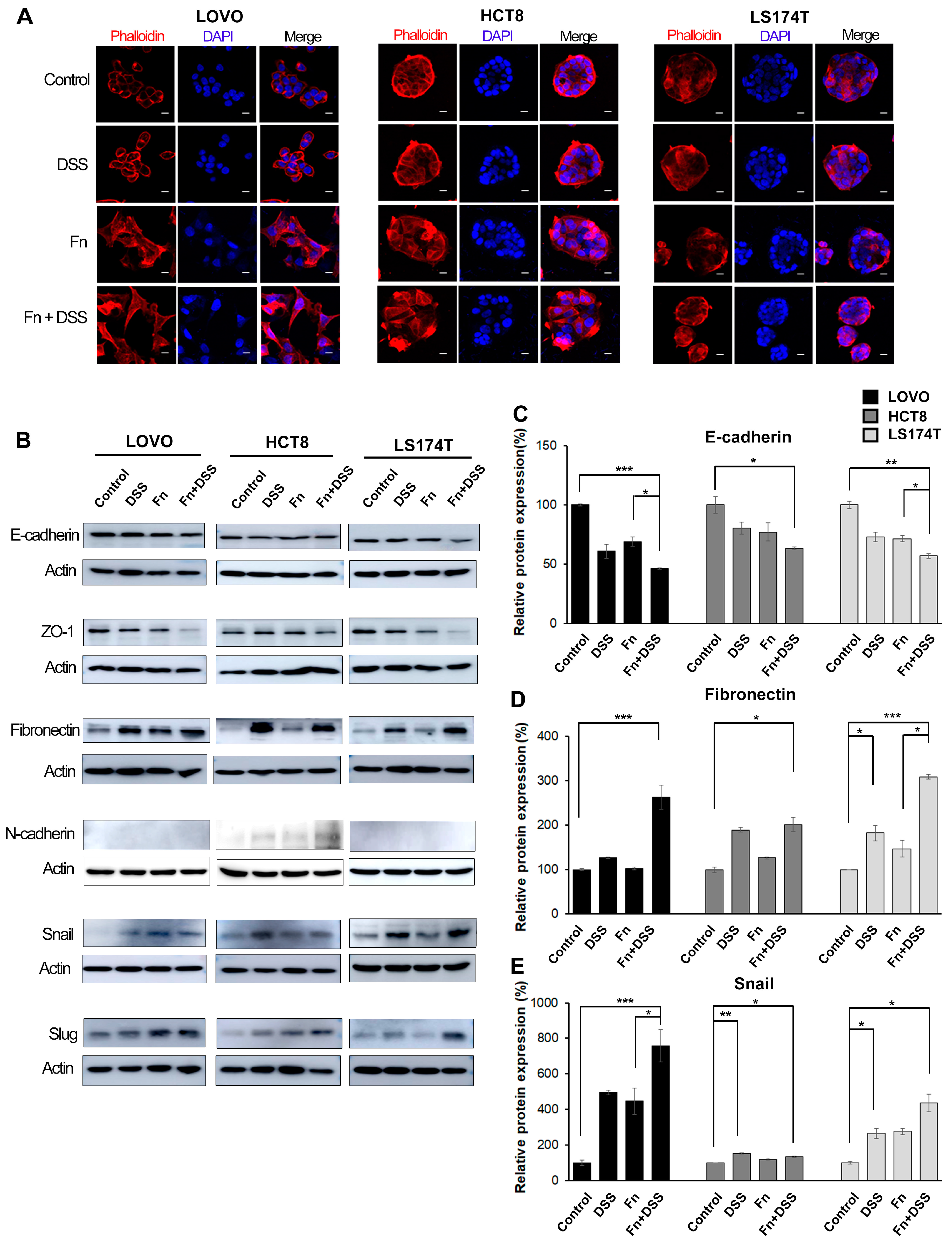
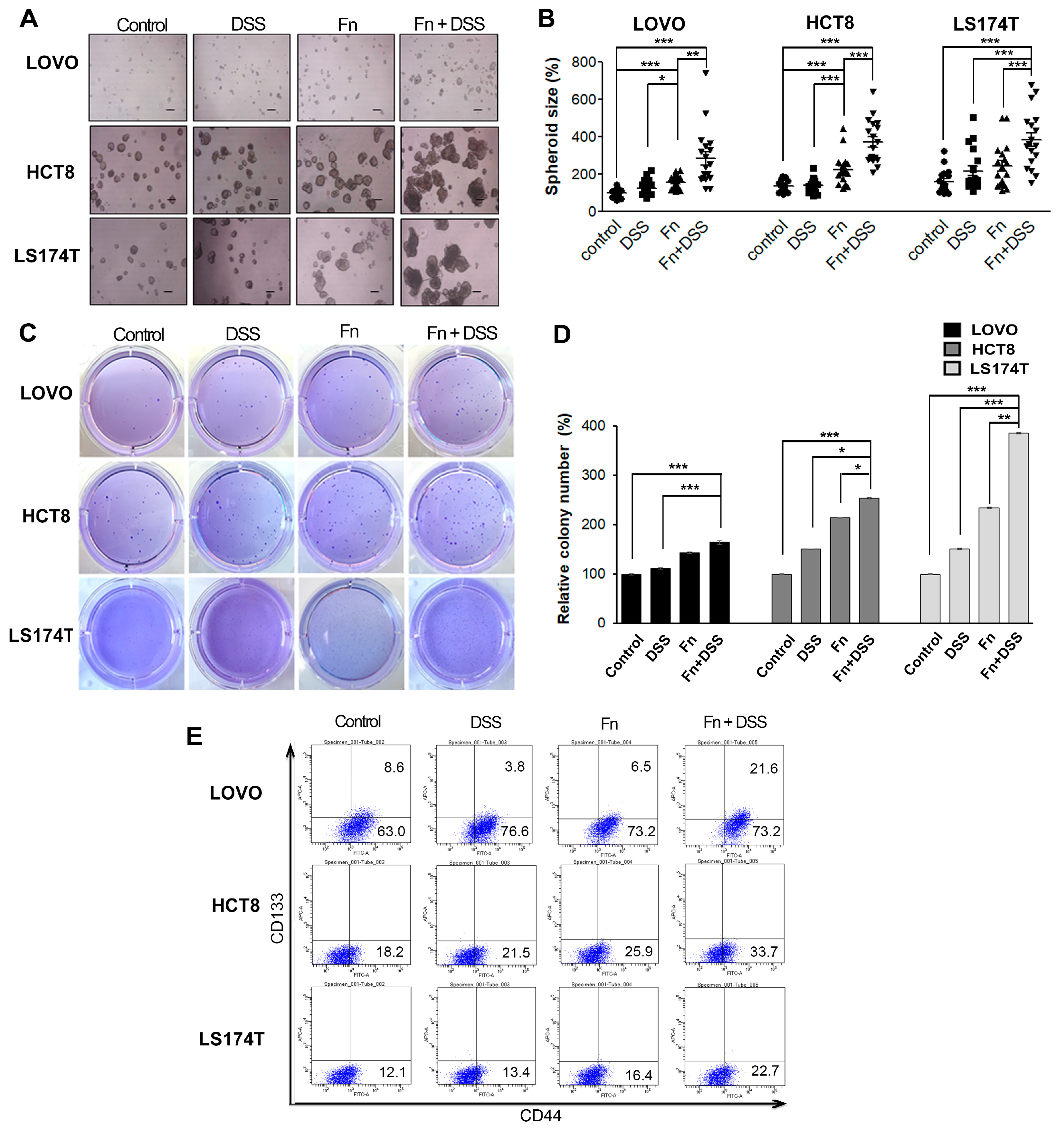
In the present study, we determined whether F. nucleatum could modify the EMT and development of CAC. We found that F. nucleatum accelerated the growth and tumorigenic potential of colitis-associated CRC cells, as evidenced by a higher proliferation rate and colony-forming ability in F. nucleatum-infected CRC cells with DSS treatment compared to those in cells treated with DSS or F. nucleatum infection alone. In addition, FN-infected/DSS-treated CRC cells formed more and larger tumor spheroids than cells treated with FN infection or DSS alone. These findings suggest the possibility that F. nucleatum infection plays an important role in the progression of CAC. The enhanced oncogenic potential of CAC following F. nucleatum infection was further supported by an in vivo experiment in an AOM/DSS-induced CAC mouse model, in which there was a larger number of tumor nodules in the group treated with AOM/DSS and F. nucleatum inoculation than in the group treated with AOM/DSS alone.
Among the numerous ways in which cancer cells display biological behaviors, including invasion and metastasis, EMT, a process involving the transition from an epithelial to a mesenchymal phenotype, is the most well-known [
31]. The prognostic importance of EMT markers was analyzed using Kaplan–Meier survival curves and a human protein atlas program. Based on the Kaplan–Meier survival curves, patients with high Snail expression had a significantly lower survival probability than those with low Snail expression, while the opposite was true for E-cadherin (
Figure S5). These results indicated that CRC was positively associated with Snail expression but negatively associated with E-cadherin expression. Previous studies on the role of
F. nucleatum in the progression of CRC reported that
F. nucleatum accelerates the metastatic potential of CRC and that the presence of
F. nucleatum is related to the advanced stages of CRC, suggesting that the increased aggressiveness of CRC induced by
F. nucleatum possibly depends on EMT. However, there have been few studies on EMT induction by
F. nucleatum, and only a very recent study showed a correlation between
F. nucleatum and metastasis [
32].
Although the effect of DSS on EMT has not been extensively studied, a study that investigated DSS-induced intestinal fibrosis found that DSS induces the expression of EMT-like features in mice, including reducing E-cadherin and significantly inducing Snail [
33]. Here, we found that
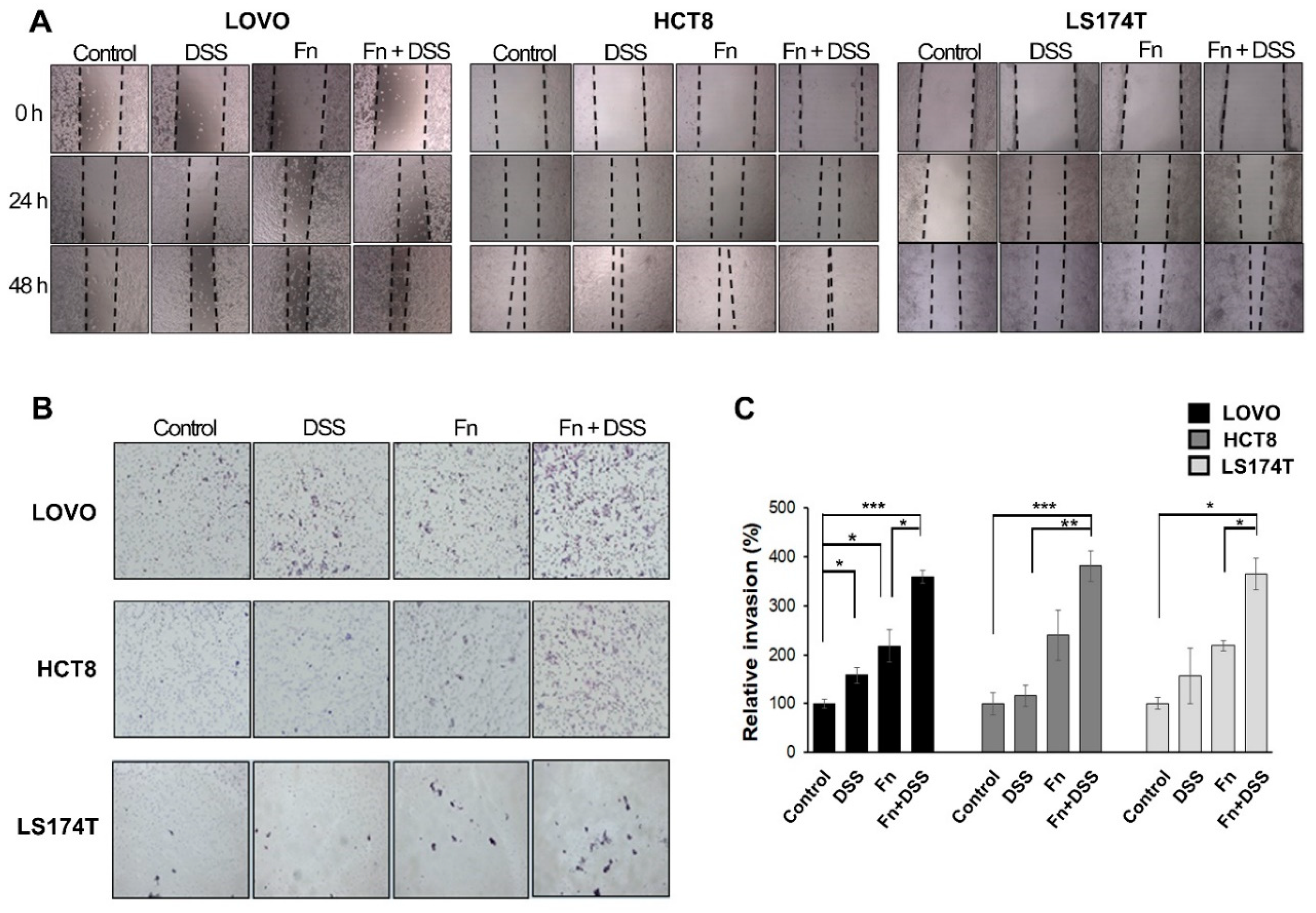
F. nucleatum promoted the aggressiveness of CRC and CAC cells based on the increased migratory and invasive capabilities in FN-infected/DSS-treated CRC cells and FN-infected cells. Furthermore, DSS and
F. nucleatum synergistically modulated the expression of EMT-related transcription factors and EMT markers in both in vitro and in vivo experiments, indicating that the increased aggressiveness of CAC induced by
F. nucleatum is acquired through promoting EMT. Notably,
F. nucleatum has been suggested to be a risk factor mainly in CRC, while
Porphyromonas gingivalis, another major periodontopathic bacterium, has been extensively studied as an important pathogen in the pathogenesis of other types of cancers such as oral, esophageal, and pancreatic carcinomas.
P. gingivalis has been reported to play a role in EMT in a few studies on oral cancer [
34]. In
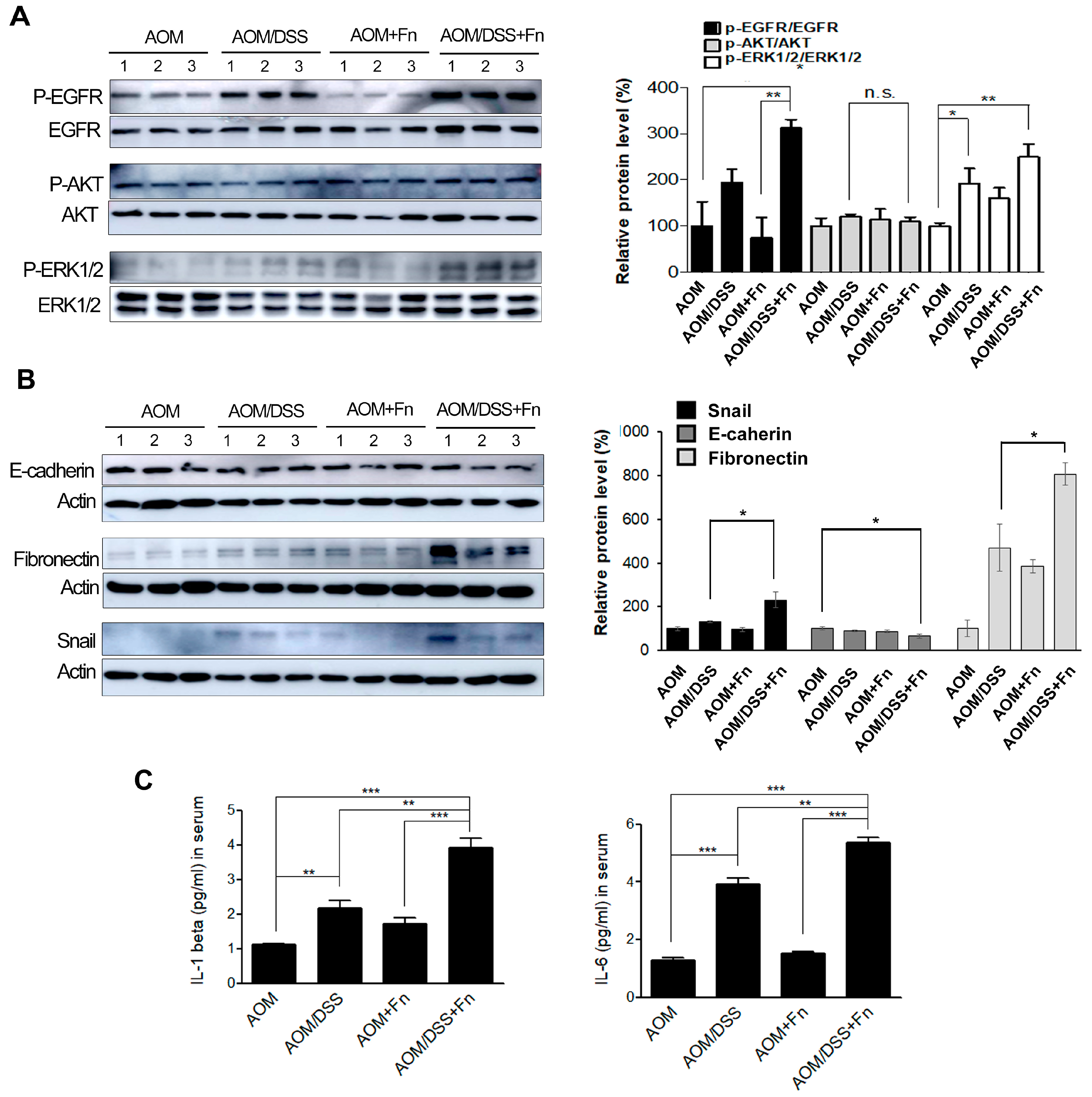
Figure 7A, protein expression of AKT and ERK tended to decrease in the DSS +
F. nucleatum group. The degradation of AKT and ERK can be due to ubiquitination, caspase cleavage, and other factors. The decrease in protein expression of AKT and ERK is thought to be degradation due to toxicity to DSS and
F. nucleatum depending on the cell condition. It is assumed that the activation of EGFR signals by
F. nucleatum is possible both in the cell membrane and inside the cytoplasm. In general, it is expected that oncogenic signals are activated by LPS secreted by
F. nucleatum. However, it remains uncertain how directly virulence is linked to host cell invasion. New studies have shown that
F. nucleatum can bind to host colon cells and stimulate directly oncogenic signaling by interactions between fusobacterial FadA and host E-cadherin [
35]. It is presumed that
F. nucleatum within the cytoplasm directly and indirectly (
F. nucleatum-secreting LPS) activates carcinogenic signals. We believe that EMT promotion through EGFR activation by
F. nucleatum may have a mechanism similar to other carcinogenic signals. In this study, we found no changes in the levels of EMT-related factors in
P. gingivalis-infected CRC cells (data not shown), while
F. nucleatum dramatically affected EMT-related molecules. In addition, EMT is known to be closely associated with stemness properties, and these two phenomena are intertwined [
36]. FN-infected/DSS-treated CRC cells were found to have the strongest stemness characteristics, indicating that
F. nucleatum promotes the aggressiveness of CAC via increasing stemness as well as EMT. Since EMT is possibly a major phenomenon that leads to a poor prognosis for patients with cancer, we investigated ways to reverse the EMT that is induced by
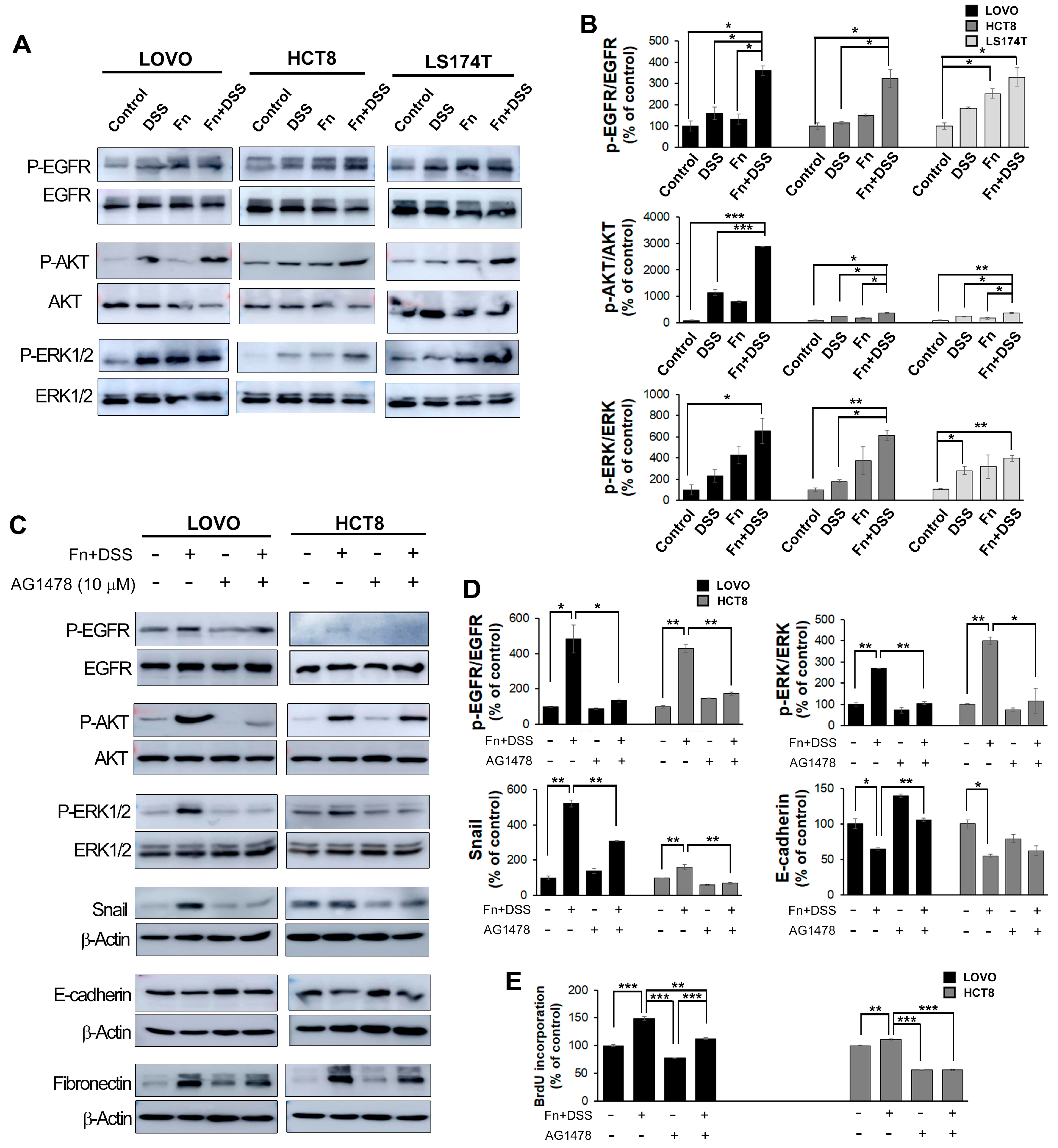
F. nucleatum infection and DSS treatment. The EGFR signaling pathway has been reported to be one of the mechanisms by which EMT-like features are induced in various cancers. A recent study on salivary gland cancer revealed that EGFR activation induces EMT in a Snail-dependent manner, and a study using CRC cells found a high expression of EGFR and the occurrence of EMT following EGFR activation through EGF binding to EGFR [
37]. In this study, we observed the activation of EGFR, Akt, and Erk, an increase in EMT-related transcription factors and markers in DSS-treated cells also infected with
F. nucleatum, and an EGFR inhibitor that modulated the EMT program by inactivating these kinases. Taken together, these findings suggested that
F. nucleatum is a novel predictive biomarker for EMT, and targeting
F. nucleatum may be an effective approach to prevent the development of CRC.
4. Materials and Methods
4.1. Cell Culture
Three colon cancer cell lines (LOVO, HCT8, LS174T) were used in this study. LOVO cells were grown in Harm’s F12K medium (Welegene, Daegu, Korea), LS174T cells (ATCC, Manassas, VA, USA) were grown in MEM (Welegene, Daegu, Korea), and HCT8 cells (ATCC, Manassas, VA, USA) were grown in RPMI 1640 medium (Welegene, Daegu, Korea); all media contained 10% fetal bovine serum (FBS; Hyclone, South Logan, UT, USA), and all cells were grown at 37 °C in a humidified environment with 5% CO2 and 95% O2. For this study, the CRC cells were infected with live F. nucleatum at a MOI (multiplicity of infection) of 1:500 for 4 h at 37 °C in 5% CO2. Then, the cells were washed with phosphate-buffered saline (PBS, Welegene, Daegu, Korea) and the media was replaced with fresh media containing 2.5% 50,000 Da DSS (MP bio, Santa Ana, CA, USA) until the cells were harvested. Controls were subjected to similar media changes and wash conditions but without bacterial inoculation.
4.2. Bacterial Culture and Bacterial Internalization into CRC Cells
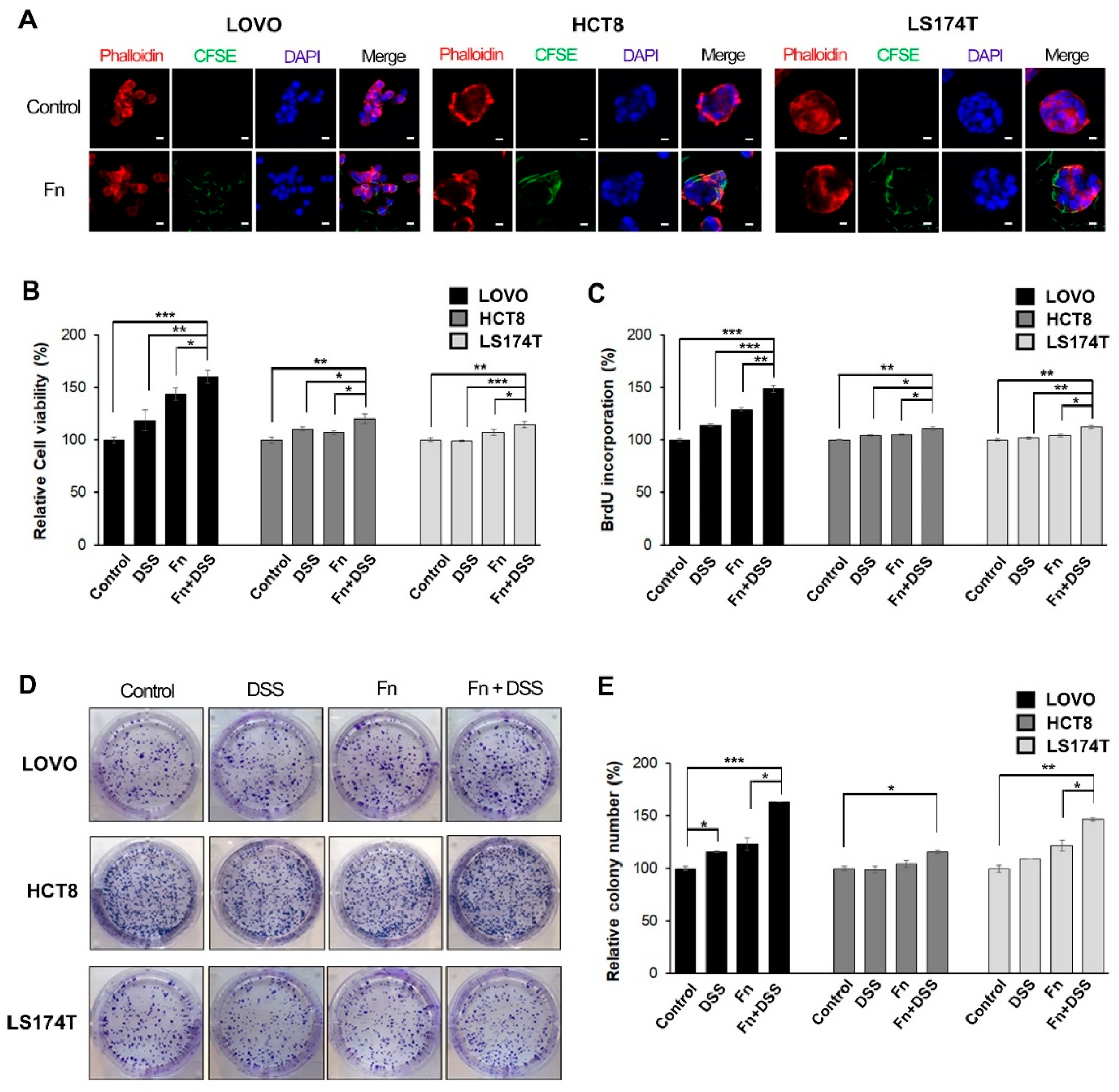
F. nucleatum was cultured anaerobically and grown in a GAM broth (Nissui Pharmaceutical, Tokyo, Japan) at 37 °C overnight. Colon cancer cells were infected with the CFSE-labeled bacteria at an MOI of 500 for 3 h. For confocal microscopy, the infected cells were fixed, permeabilized, and then stained with rhodamine–phalloidin (Invitrogen, Taastrup, Denmark) and DAPI (Invitrogen). Mounted slides were imaged using a confocal microscope (Carl Zeiss, Oberkochen, Germany).
4.3. Cell Proliferation Assay
Cell proliferation was performed using MTT assay by an EZ-Cytox cell viability assay kit (Daeillab service, Seoul, Korea) and a BrdU proliferation assay kit (Calbiochem, San Diego, CA, USA) according to the manufacturer’s protocols. The results are expressed as percentages after comparing the absorbance at 450 nm with that in the uninfected control cells.
4.4. Western Blot Analysis
To analyze protein expression, Western blotting was performed using specific antibodies. Briefly, the cells were washed once with cold PBS and lysed with 1X RIPA buffer (Cell signaling Technology, Danvers, MA, USA) containing a protease inhibitor cocktail and phosphatase inhibitors. The lysates were centrifuged at 13,000× g for 10 min. Subsequently, the supernatants were collected. Using the Bradford method, the protein concentration was determined to be 20 µg. Samples were separated on 8–12% SDS-polyacrylamide gels (Bio-Rad, Hercules, CA, USA). Then, the SDS-gels were transferred electrophoretically onto polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA, USA) using a wet transfer kit. The transferred membranes were blocked with 5% skim milk in TBST (BD Biosciences, San Jose, CA, USA) containing 0.1% Tween-20 (Bio-Rad, Richmond, CA, USA) for 1 h at room temperature. The membranes were incubated overnight at 4 °C with the primary antibodies against E-cadherin (Cell Signaling, #3195, Danvers, MA, USA), fibronectin (BD Bioscience, #610077 San Jose, CA, USA and Santa Cruz, #SC-9068, CA, USA), Snail (Cell Signaling, #3879, Danvers, MA, USA), p-EGFR (Invitrogen, #18-2463, Carlsbad, CA, USA), EGFR (BD Bioscience, Santa Cruz, #SC-03, CA, USA), p-AKT (Cell Signaling, #9271, Danvers, MA, USA), AKT (Cell Signaling, #9272, Danvers, MA, USA), p-ERK1/2 (BD Bioscience, Santa Cruz, #SC-7976, CA, USA), ERK1/2 (BD Biosciences, Santa Cruz, #SC-94, CA, USA), and β-actin (BD Bioscience, Santa Cruz, #SC-47778, CA, USA). The membranes were washed three times with 1x TBST buffer, and HRP-conjugated secondary antibodies (ENZO, #ADI-SAB-300-J, and #ADI-SAB-100-J, 1:8000, NY, USA) were applied for 2 h at room temperature. The antigen-antibody complexes were detected using a SuperSignal West–Femto reagent (Thermo Fisher, Waltham, MA, USA).
4.5. Colony Formation Assay
For the colony formation assay, single cells (1000 cells/well) were plated in 6-well plates. The cells were allowed to grow for 7–11 d, and the media was changed every 2 d. The colonies were fixed with 4% paraformaldehyde for 30 min. After washing three times with PBS, colonies were stained with 0.1% crystal violet (Sigma-Aldrich, St. Louis, MO, USA) for 15 min. After they were washed with distilled water, the cells were photographed and analyzed to assess their proliferation and colony-forming efficiency. All experiments were performed at least two times.
4.6. Colon Spheroid Formation
Colon cancer cells were grown as spheroids. Briefly, single-cell suspensions were seeded in 24-well culture plates at a density of 5 × 103 cells/well in a tumor sphere medium (medium supplemented with B27 (Invitrogen, Karlsruhe, Germany) containing 10% FBS), pre-coated with a poly-2-hydroxyethyl methacrylate (polyhema) solution (Sigma-Aldrich, St. Louis, MO, USA). The spheroids were incubated for 3–6 days; then, the spheres in each well were counted. The total number of tumor spheroids formed in each well was plotted, and representative images were taken.
4.7. Soft Agar Assay
Treated cells were harvested and pipetted to form a single-cell suspension. The cells were seeded at a density of 1.5 × 103–3 × 103 cells/well in growth medium containing 0.25% soft agar (top layer) on top of 0.5% soft agar (base layer) in 6-well plates and cultured for an additional 2–3 weeks. The colonies that appeared were observed using microscopy, and the colony number was determined after staining with 0.05% (w/v) crystal violet (in 5% methanol).
4.8. Wound-Healing Assay
Wound-healing assays were performed to evaluate cell-migration ability. Briefly, colon cancer cells were seeded at a density of 1 × 106 cells/mL in 6-well plates and incubated in FBS-containing medium. When they reached 80% confluence, the cells were infected with F. nucleatum with the addition of 1 mM thymidine (Sigma-Aldrich, St. Louis, MO, USA) for the indicated times. After 4 h, a wound was made in the middle of the well using a sterile 200 µL pipette tip. The wells were washed three times with PBS; then, the colon-cancer cells were incubated with media containing 1% FBS and 2.5% DSS for 24 h. Images were captured using a microscope (Nikon Eclipse TS100, Tokyo, Japan). The rates of the healing of the wound area were calculated as a percentage of the remaining wound area compared to the initial wound area.
4.9. Transwell Invasion Assay
Cell invasion capacity was assessed using 8 µm pore size 24-insert transwell chambers (Corning, NY, USA). For the invasion assay, the upper chambers were pre-coated with Matrigel (BD Biosciences, San Jose, CA, USA). Medium containing 10% fetal bovine serum (700 µL) was added to the lower chamber. After 48 h, cells on the bottom of the inserts were fixed in 4% paraformaldehyde and stained with hematoxylin (YD CORP., Gyeonggi-do, Korea) and eosin (Sigma-Aldrich, St Louis, MO, USA). Then, cells that invaded the lower surface were counted in at least three fields using microscopy (Nikon Eclipse TS100, Tokyo, Japan). Each experiment was performed at least two times.
4.10. Flow Cytometry
The expression of the CSC markers CD133 and CD44 was detected using flow cytometry. Briefly, colon cancer cells were harvested with 0.05% trypsin and washed with phosphate-buffered saline (PBS). Cells were incubated with FITC mouse anti-human CD44 (BD Biosciences, Franklin Lakes, NJ, USA) and CD133/2-APC human antibodies (Miltenyi Biotec, Bergisch Gladbach, Germany) in the dark at 4 °C. After 30 min, the cells were washed and analyzed using an FACS system (BD, San Jose, CA, USA).
4.11. Animals and Establishment of an AOM/DSS Mouse Model
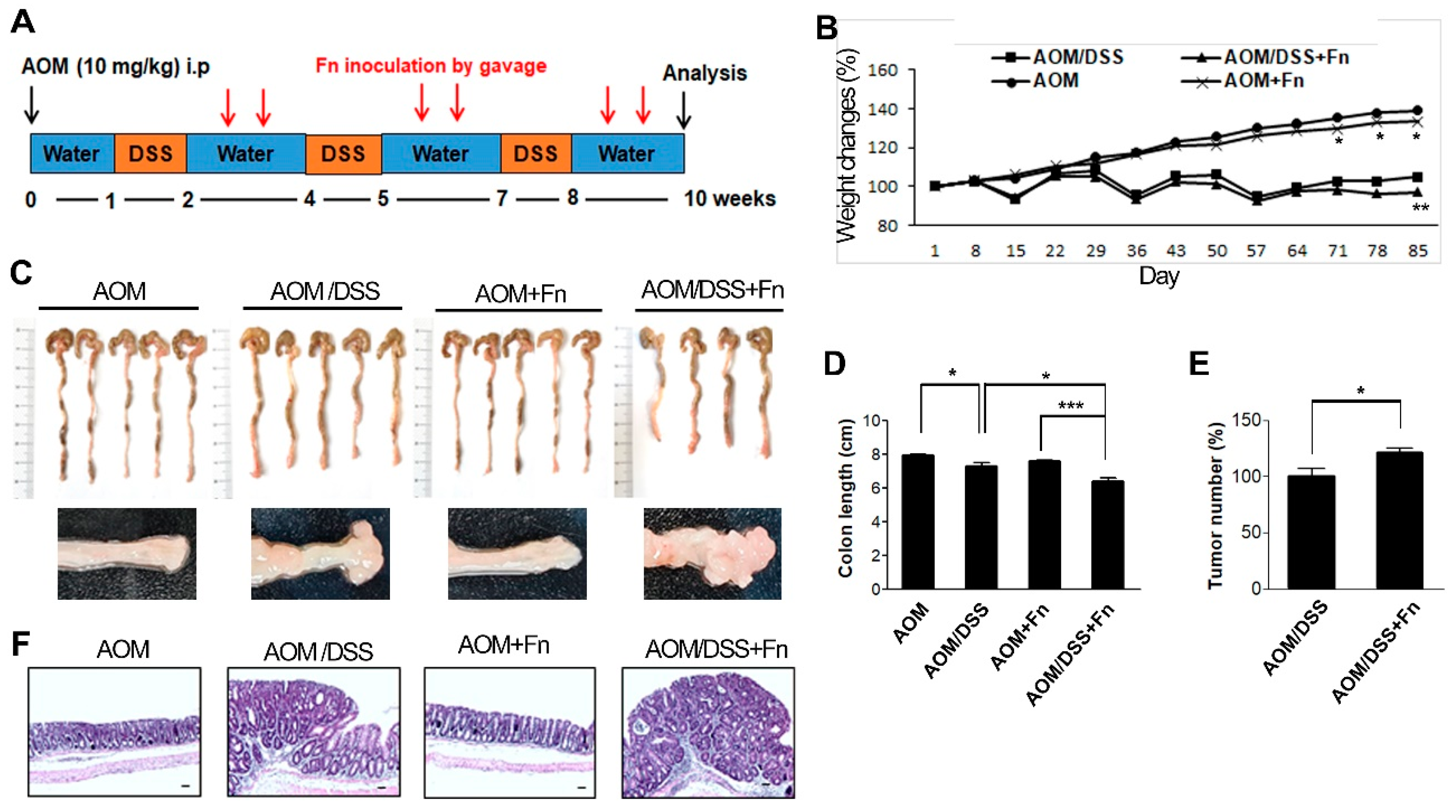
Female C57BL/6 mice (6 weeks old) were purchased from Orient Bio Inc (Seungnam, Korea). The care of animals and experimental procedures were approved by the Institutional Animal Care and Use Committee of Pusan National University (Protocol No. PNU-2019-2207, June 2019). All animals were housed under controlled SPF conditions (temperature 22 ± 1 °C, 12 h dark/light cycle) with free access to a standard diet and water. The procedure used for the establishment of the UC-CRC model using AOM and DSS is shown in
Figure 5A. The mice were randomly divided into four groups: the AOM group (
n = 5), AOM/DSS group (
n = 5), AOM + Fn group (
n = 5), and AOM/DSS + Fn group (
n = 5). To develop the UC-CRC model, 7-week-old female C57BL/6 mice were given a single intraperitoneal injection of azoxymethane (Sigma-Aldrich, St.Louis, MO, USA) at a dose of 10 mg/kg. One week later, the animals were given 2.5% DSS in their drinking water for 7 d followed by 14 d of normal drinking water for recovery, and this cycle was repeated three times.
F. nucleatum (1 × 10
9/CFU) or the vehicle was ad, ministrated by gavage six times until the end of the 12-week study period. During the experimental period, body weights were measured every week. At the end of the experiment, blood was collected for ELISA, the mice were sacrificed, and colon tissues were removed. After the weights and lengths of the tissues were measured, the colons were slit open longitudinally along the main axis and washed with PBS (pH 7.4). The number of tumors in the colons was recorded. Subsequently, some colon tissues were fixed in 4% paraformaldehyde buffer for further histopathological examination and immunohistochemical analysis, while others were flash-frozen in liquid nitrogen and kept at −80 °C for Western blot analysis.
4.12. Hematoxylin and Eosin (H & E) Staining
Large intestine tissue samples were fixed in 4% paraformaldehyde solution and embedded in paraffin. For histopathology analysis, paraformaldehyde-fixed colonic tissues were dehydrated in a gradient alcohol series, embedded in paraffin, and cut into 5 μm-thick serial sections. Then, tissue sections were stained with H & E to examine tissue morphology and observed using optical microscopy.
4.13. Immunohistochemistry
Tissue samples were dehydrated, embedded in paraffin, and cut into 5 μm-thick sections for immunohistochemistry (IHC). Prior to being subjected to immunostaining, 4% paraformaldehyde-fixed paraffin-embedded tumors were excised from mouse flanks (5 μm), deparaffinized, and rehydrated. Tissue slides were heated in 10 mM sodium acetate (pH 6.0, Duksan Chemical Co., Seoul, Korea) for 10 min at 121 °C for antigen retrieval and then bathed in a 0.3% H2O2-PBS solution (Welegene, Deagu, Korea) for 15 min at room temperature in the dark to quench endogenous peroxidase. After samples were washed with 0.5% Tris-HCl-Tween (Welegene, Deagu, Korea), tissue sections (5 μm) were blocked with 5% FBS (Hyclone, South Logan, UT, USA) in PBS (Welegene, Deagu, Korea) with 0.1% Tween 20 (PBST), incubated in a 1:250 dilution of the primary antibodies overnight at 4°C, and repeatedly washed with PBST. Samples were incubated with horseradish peroxidase-conjugated secondary antibody (ENZO, #ADI-SAB-300-J, 1:250, New York, NY, USA) for 2 h at room temperature, after which they were subjected to repeated washing with PBST. Then, the bound antibodies were determined using freshly prepared substrate buffer (0.05% diaminobenzidine (DAB; Sigma-Aldrich), 0.015% H2O2 in PBS) for 2 min. After a final wash in PBS and distilled water, the slides were counterstained with Mayer’s hematoxylin (MUTO, Tokyo, Japan) for 4 min and then dehydrated in a graded alcohol series (50%, 70%, 80%, 90%, 95%, 100%, and 100%, Samjun, Seoul, Korea). Sections were subsequently examined at various magnifications using fluorescence microscopy (OLYMPUS, Tokyo, Japan).
4.14. Fusobacterium Nucleatum-Specific Detection Using RNA Extraction and Real-Time PCR
Total RNA was extracted from cells using an RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The isolated DNA was used as a template and detected using F. nucleatum 16s rRNA-specific primers. F. nucleatum 16s rRNA was detected using two-step quantitative real-time PCR with a QuantiTect reverse transcription kit (Qiagen, Hilden, Germany) and TOPreal SYBR Green PCR Kit (Enzynomics, Seoul, Korea) in an ABI 7500 real-time PCR detection system (Applied Biosystems, Foster City, CA, USA). Reactions were carried out for 45 cycles. Reactions were duplicated, and the average mRNA level of each gene was determined using the 2-ΔΔCt method. The following primers were used for real-time PCR: 5′-GGG-CTC-AAC-TCT-GTA-TTG-CG-3′ and 5′-CTG-TTT-GCT-ACC-CAC-GCT-TT-3′.
4.15. Cytokine Analysis
Blood was retro-orbitally collected to measure cytokines. Serum was separated using centrifugation and stored at − 80 °C until measured. Samples were diluted 1:1, and serum IL-1β and IL-6 pre-inflammatory cytokine levels were measured using Affymetrix mouse ELISA kits (Invitrogen, San Diego, CA, USA) according to the manufacturers’ instructions.
4.16. Statistical Analyses
Statistical analyses were performed using GraphPad Prism version 5.01 (GraphPad Software, Inc., San Diego, CA, USA, website:
https://www.graphpad.com/scientific-software/prism/). For the comparative analysis of two groups of data, Student’s
t-test was performed.
p-values < 0.05 were considered statistically significant. All data are shown as the mean and standard error of the mean (SEM).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}