Chronic Active Epstein–Barr Virus Infection: Is It Immunodeficiency, Malignancy, or Both?

Abstract
:Simple Summary
Abstract
1. Introduction
2. Chronic Active Epstein–Barr Virus Infection and Related EBV-Positive T/NK-Cell LPDs and Leukemia/Lymphomas
2.1. Chronic Active EBV Infection
2.2. EBV-Positive T/NK-Cell LPDs and Leukemia/Lymphomas Related to CAEBV
3. CAEBV as a Malignant Neoplasm
4. CAEBV as Immunodeficiency
4.1. Analysis of Immune Functions in CAEBV
4.2. Clinical Manifestations Consistent with B-Cell-Type CAEBV in Patients with Primary Immunodeficiency
4.3. Primary Immunodeficiency Accompanied by EBV-Positive T/NK-Cell LPDs
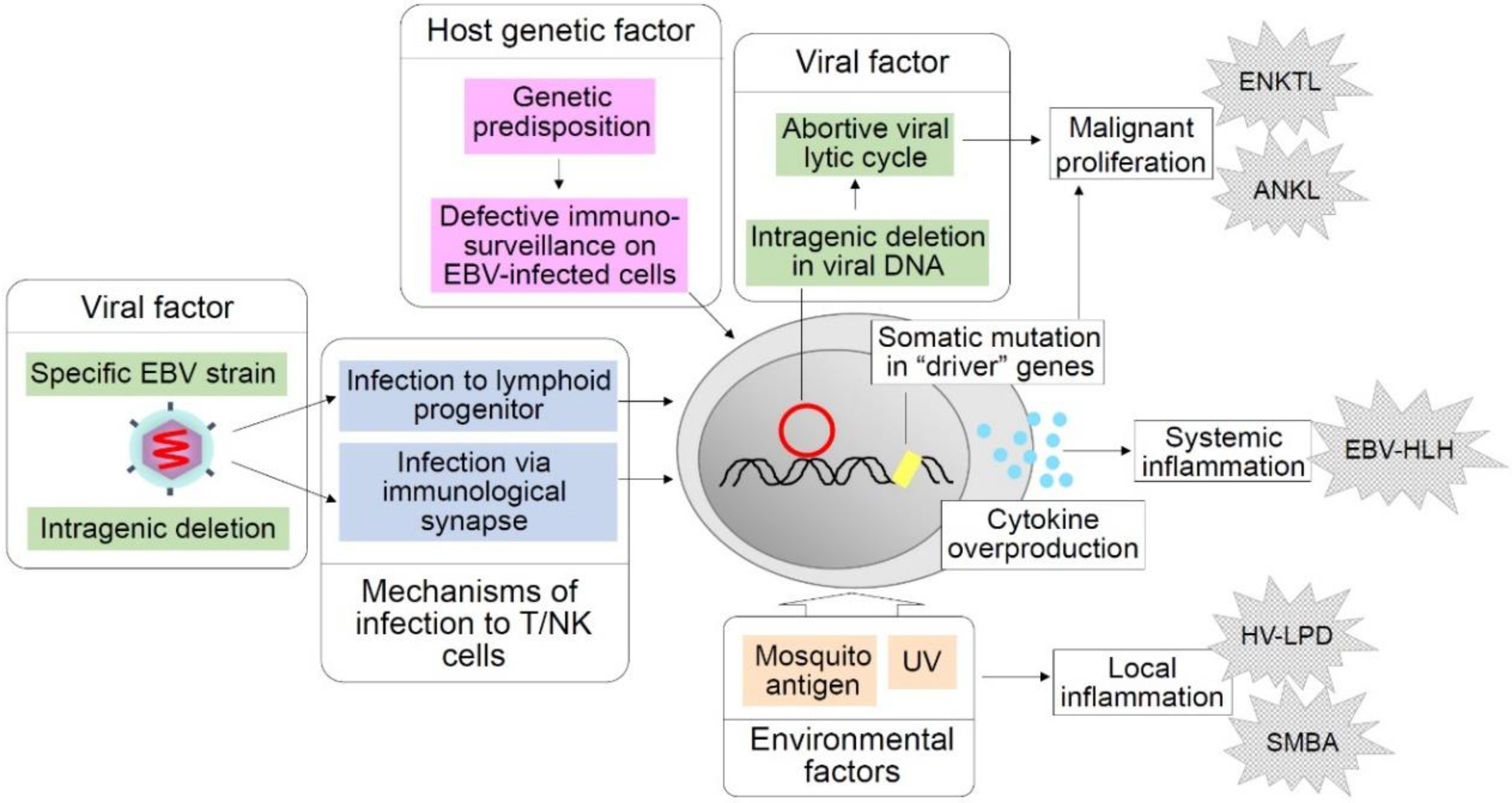
5. Unsolved Questions in CAEBV
5.1. Mechanisms of EBV Infection to T and NK Cells
5.2. Restricted Geographic Distribution
5.3. The Origins of Clinical Heterogeneity
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-Barr virus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume II, pp. 1898–1959. [Google Scholar]
- Dunmire, S.K.; Verghese, P.S.; Balfour, H.H., Jr. Primary Epstein-Barr virus infection. J. Clin. Virol. 2018, 102, 84–92. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [Green Version]
- Watry, D.; Hedrick, J.A.; Siervo, S.; Rhodes, G.; Lamberti, J.J.; Lambris, J.D.; Tsoukas, C.D. Infection of human thymocytes by Epstein-Barr virus. J. Exp. Med. 1991, 173, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Paterson, R.L.; Kelleher, C.; Amankonah, T.D.; Streib, J.E.; Xu, J.W.; Jones, J.F.; Gelfand, E.W. Model of Epstein-Barr virus infection of human thymocytes: Expression of viral genome and impact on cellular receptor expression in the T-lymphoblastic cell line, HPB-ALL. Blood 1995, 85, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, R. Chronic infectious mononucleosis. Blood 1948, 3, 858–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virelizier, J.L.; Lenoir, G.; Griscelli, C. Persistent Epstein-Barr virus infection in a child with hypergammaglobulinaemia and immunoblastic proliferation associated with a selective defect in immune interferon secretion. Lancet 1978, 2, 231–234. [Google Scholar] [CrossRef]
- Tobi, M.; Morag, A.; Ravid, Z.; Chowers, I.; Feldman-Weiss, V.; Michaeli, Y.; Ben-Chetrit, E.; Shalit, M.; Knobler, H. Prolonged atypical illness associated with serological evidence of persistent Epstein-Barr virus infection. Lancet 1982, 1, 61–64. [Google Scholar] [CrossRef]
- Jones, J.F.; Straus, S.E. Chronic Epstein-Barr virus infection. Annu. Rev. Med. 1987, 38, 195–209. [Google Scholar] [CrossRef]
- Straus, S.E. The chronic mononucleosis syndrome. J. Infect. Dis. 1988, 157, 405–412. [Google Scholar] [CrossRef]
- Rickinson, A.B. Chronic, symptomatic Epstein-Barr virus infection. Immunol. Today 1996, 7, 13–14. [Google Scholar] [CrossRef]
- Okano, M.; Matsumoto, S.; Osato, T.; Sakiyama, Y.; Thiele, G.M.; Purtilo, D.T. Severe chronic active Epstein-Barr virus infection syndrome. Clin. Microbiol. Rev. 1991, 4, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Carey, R.W.; Miller, G.; Henle, W.; Eastman, R.; Mark, E.J.; Kenyon, K.; Wheeler, E.O.; Rubin, R.H. Chronic Epstein-Barr virus infection associated with fever and interstitial pneumonitis. Clinical and serologic features and response to antiviral chemotherapy. Ann. Intern. Med. 1986, 104, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Kimura, H.; Imadome, K.; Arai, A.; Kodama, E.; Morio, T.; Shimizu, N.; Wakiguchi, H. Current research on chronic active Epstein-Barr virus infection in Japan. Pediatr. Int. 2014, 56, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, K.; Ito, Y.; Shibata-Watanabe, Y.; Kawada, J.; Takahashi, Y.; Yagasaki, H.; Kojima, S.; Nishiyama, Y.; Kimura, H. Clinical and virological characteristics of 15 patients with chronic active Epstein-Barr virus infection treated with hematopoietic stem cell transplantation. Clin. Infect. Dis. 2008, 46, 1525–1534. [Google Scholar] [CrossRef] [Green Version]
- Sawada, A.; Inoue, M.; Kawa, K. How we treat chronic active Epstein-Barr virus infection. Int. J. Hematol. 2017, 105, 406–418. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L.; Ko, Y.H.; Kimura, H.; Jaffe, E.S. EBV-positive T-cell and NK-cell lymphoproliferative diseases of childhood. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D.A., Hasserjian, R.P., Le Beau, M.M., et al., Eds.; IARC Press: Lyon, France, 2017; pp. 355–362. [Google Scholar]
- Okano, M.; Kawa, K.; Kimura, H.; Yachie, A.; Wakiguchi, H.; Maeda, A.; Imai, S.; Ohga, S.; Kanegane, H.; Tsuchiya, S.; et al. Proposed guidelines for diagnosing chronic active Epstein-Barr virus infection. Am. J. Hematol. 2005, 80, 64–69. [Google Scholar] [CrossRef]
- Cohen, J.I.; Jaffe, E.S.; Dale, J.K.; Pittaluga, S.; Heslop, H.E.; Rooney, C.M.; Gottschalk, S.; Bollard, C.M.; Rao, V.K.; Marques, A.; et al. Characterization and treatment of chronic active Epstein-Barr virus disease: A 28-year experience in the United States. Blood 2011, 117, 5835–5849. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Ito, Y.; Kawabe, S.; Gotoh, K.; Takahashi, Y.; Kojima, S.; Naoe, T.; Esaki, S.; Kikuta, A.; Sawada, A.; et al. EBV-associated T/NK-cell lymphoproliferative diseases in nonimmunocompromised hosts: Prospective analysis of 108 cases. Blood 2012, 119, 673–686. [Google Scholar] [CrossRef] [Green Version]
- Kikuta, H.; Taguchi, Y.; Tomizawa, K.; Kojima, K.; Kawamura, N.; Ishizaka, A.; Sakiyama, Y.; Matsumoto, S.; Imai, S.; Kinoshita, T.; et al. Epstein-Barr virus genome-positive T lymphocytes in a boy with chronic active EBV infection associated with Kawasaki-like disease. Nature 1988, 333, 455–457. [Google Scholar] [CrossRef]
- Jones, J.F.; Shurin, S.; Abramowsky, C.; Tubbs, R.R.; Sciotto, C.G.; Wahl, R.; Sands, J.; Gottman, D.; Katz, B.Z.; Sklar, J. T-cell lymphomas containing Epstein-Barr viral DNA in patients with chronic Epstein-Barr virus infections. N. Engl. J. Med. 1988, 318, 733–741. [Google Scholar] [CrossRef]
- Kawa-Ha, K.; Ishihara, S.; Ninomiya, T.; Yumura-Yagi, K.; Hara, J.; Murayama, F.; Tawa, A.; Hirai, K. CD3-negative lymphoproliferative disease of granular lymphocytes containing Epstein-Barr viral DNA. J. Clin. Investig. 1989, 84, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla-Martinez, L.; Kumar, S.; Fend, F.; Reyes, E.; Teruya-Feldstein, J.; Kingma, D.W.; Sorbara, L.; Raffeld, M.; Straus, S.E.; Jaffe, E.S. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: A distinct clinicopathologic syndrome. Blood 2000, 96, 443–451. [Google Scholar] [CrossRef]
- Kuis, W.; Roord, J.J.; Zegers, B.J.; Rickinson, A.B.; Kapsenberg, J.G.; The, H.; Stoop, J.W. Heterogeneity of immune defects in three children with a chronic active Epstein-Barr virus infection. J. Clin. Immunol. 1985, 5, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Morishima, T.; Kanegane, H.; Ohga, S.; Hoshino, Y.; Maeda, A.; Imai, S.; Okano, M.; Morio, T.; Yokota, S.; et al. Prognostic factors for chronic active Epstein-Barr virus infection. J. Infect. Dis. 2003, 187, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Takeoka, Y.; Nakao, Y.; Ueda, M.; Koh, K.R.; Aoyama, Y.; Nakamae, H.; Yamamura, R.; Ohta, K.; Takubo, T.; Yamane, T.; et al. A case of a long-time survivor with chronic active Epstein-Barr virus infection. Eur. J. Haematol. 2004, 72, 73–76. [Google Scholar] [CrossRef]
- Ohshima, K.; Kimura, H.; Yoshino, T.; Kim, C.W.; Ko, Y.H.; Lee, S.S.; Peh, S.C.; Chan, J.K.; Group, C.S. Proposed categorization of pathological states of EBV-associated T/natural killer-cell lymphoproliferative disorder (LPD) in children and young adults: Overlap with chronic active EBV infection and infantile fulminant EBV T-LPD. Pathol. Int. 2008, 58, 209–217. [Google Scholar] [CrossRef]
- Arai, A.; Imadome, K.; Watanabe, Y.; Yoshimori, M.; Koyama, T.; Kawaguchi, T.; Nakaseko, C.; Fujiwara, S.; Miura, O. Clinical features of adult-onset chronic active Epstein-Barr virus infection: A retrospective analysis. Int. J. Hematol. 2011, 93, 602–609. [Google Scholar] [CrossRef]
- Kawamoto, K.; Miyoshi, H.; Suzuki, T.; Kozai, Y.; Kato, K.; Miyahara, M.; Yujiri, T.; Choi, I.; Fujimaki, K.; Muta, T.; et al. A distinct subtype of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disorder: Adult patients with chronic active Epstein-Barr virus infection-like features. Haematologica 2018, 103, 1018–1028. [Google Scholar] [CrossRef] [Green Version]
- Iwatsuki, K.; Miyake, T.; Hirai, Y.; Yamamoto, T. Hydroa vacciniforme: A distinctive form of Epstein-Barr virus-associated T-cell lymphoproliferative disorders. Eur. J. Dermatol. 2019, 29, 21–28. [Google Scholar] [CrossRef]
- Tatsuno, K.; Fujiyama, T.; Matsuoka, H.; Shimauchi, T.; Ito, T.; Tokura, Y. Clinical categories of exaggerated skin reactions to mosquito bites and their pathophysiology. J. Dermatol. Sci. 2016, 82, 145–152. [Google Scholar] [CrossRef]
- Harabuchi, Y.; Takahara, M.; Kishibe, K.; Nagato, T.; Kumai, T. Extranodal Natural Killer/T-Cell Lymphoma, Nasal Type: Basic Science and Clinical Progress. Front. Pediatr. 2019, 7, 141. [Google Scholar] [CrossRef]
- Montes-Mojarro, I.A.; Kim, W.Y.; Fend, F.; Quintanilla-Martinez, L. Epstein—Barr virus positive T and NK-cell lymphoproliferations: Morphological features and differential diagnosis. Semin. Diagn. Pathol. 2020, 37, 32–46. [Google Scholar] [CrossRef]
- Ishida, F. Aggressive NK-Cell Leukemia. Front. Pediatr. 2018, 6, 292. [Google Scholar] [CrossRef] [Green Version]
- Hue, S.S.; Oon, M.L.; Wang, S.; Tan, S.Y.; Ng, S.B. Epstein-Barr virus-associated T- and NK-cell lymphoproliferative diseases: An update and diagnostic approach. Pathology 2020, 52, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, E.; Ohshima, K.; Kimura, H.; Hara, K.; Suzuki, R.; Kawa, K.; Eimoto, T.; Nakamura, S.; NK-cell Tumor Study Group. Clinicopathological analysis of the age-related differences in patients with Epstein-Barr virus (EBV)-associated extranasal natural killer (NK)/T-cell lymphoma with reference to the relationship with aggressive NK cell leukaemia and chronic active EBV infection-associated lymphoproliferative disorders. Histopathology 2011, 59, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, R.; Abe, Y.; Sada, E.; Kiyasu, J.; Ashikari, A.; Shiratsuchi, M.; Nishimura, J.; Takayanagi, R.; Ohshima, K. Adult patient with Epstein-Barr virus (EBV)-associated lymphoproliferative disorder: Chronic active EBV infection or de novo extranodal natural killer (NK)/T-cell lymphoma, nasal type? Intern. Med. 2009, 48, 471–474. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.T.; Wang, D.; Luo, H.; Xiao, M.; Zhou, H.S.; Liu, D.; Ling, S.P.; Wang, N.; Hu, X.L.; Luo, Y.; et al. Aggressive NK-cell leukemia: Clinical subtypes, molecular features, and treatment outcomes. Blood Cancer J. 2017, 7, 660. [Google Scholar] [CrossRef]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Tsai, M.H.; Shumilov, A.; Poirey, R.; Bannert, H.; Middeldorp, J.M.; Feederle, R.; Delecluse, H.J. The Epstein-Barr Virus BART miRNA Cluster of the M81 Strain Modulates Multiple Functions in Primary B Cells. PLoS Pathog. 2015, 11, e1005344. [Google Scholar] [CrossRef]
- Arvey, A.; Ojesina, A.I.; Pedamallu, C.S.; Ballon, G.; Jung, J.; Duke, F.; Leoncini, L.; De Falco, G.; Bressman, E.; Tam, W.; et al. The tumor virus landscape of AIDS-related lymphomas. Blood 2015, 125, e14–e22. [Google Scholar] [CrossRef] [Green Version]
- Walens, A.; DiMarco, A.V.; Lupo, R.; Kroger, B.R.; Damrauer, J.S.; Alvarez, J.V. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. Elife 2019, 8. [Google Scholar] [CrossRef]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Fujieda, M.; Wakiguchi, H.; Hisakawa, H.; Kubota, H.; Kurashige, T. Defective activity of Epstein-Barr virus (EBV) specific cytotoxic T lymphocytes in children with chronic active EBV infection and in their parents. Acta Paediatr. Jpn. 1993, 35, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, I.; Morishima, T.; Kimura, H.; Kuzushima, K.; Matsuoka, H. Impaired cytotoxic T lymphocyte response to Epstein-Barr virus-infected NK cells in patients with severe chronic active EBV infection. J. Med. Virol. 2001, 64, 141–148. [Google Scholar] [CrossRef]
- Sugaya, N.; Kimura, H.; Hara, S.; Hoshino, Y.; Kojima, S.; Morishima, T.; Tsurumi, T.; Kuzushima, K. Quantitative analysis of Epstein-Barr virus (EBV)-specific CD8+ T cells in patients with chronic active EBV infection. J. Infect. Dis. 2004, 190, 985–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakiguchi, H.; Fujieda, M.; Matsumoto, K.; Ohara, Y.; Wakiguchi, A.; Kurashige, T. Defective killer cell activity in patients with chronic active Epstein-Barr virus infection. Acta Med. Okayama 1988, 42, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Joncas, J.; Monczak, Y.; Ghibu, F.; Alfieri, C.; Bonin, A.; Ahronheim, G.; Rivard, G. Brief report: Killer cell defect and persistent immunological abnormalities in two patients with chronic active Epstein-Barr virus infection. J. Med. Virol. 1989, 28, 110–117. [Google Scholar] [CrossRef]
- Imai, S.; Sugiura, M.; Oikawa, O.; Koizumi, S.; Hirao, M.; Kimura, H.; Hayashibara, H.; Terai, N.; Tsutsumi, H.; Oda, T.; et al. Epstein-Barr virus (EBV)-carrying and -expressing T-cell lines established from severe chronic active EBV infection. Blood 1996, 87, 1446–1457. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, M.; Ishiguro, N.; Ishiko, H.; Ma, X.; Kikuta, H.; Kobayashi, K. Heterogeneous, restricted patterns of Epstein-Barr virus (EBV) latent gene expression in patients with chronic active EBV infection. J. Gen. Virol. 2001, 82, 2385–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H.; Hoshino, Y.; Hara, S.; Sugaya, N.; Kawada, J.; Shibata, Y.; Kojima, S.; Nagasaka, T.; Kuzushima, K.; Morishima, T. Differences between T cell-type and natural killer cell-type chronic active Epstein-Barr virus infection. J. Infect. Dis. 2005, 191, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Cohen, J.I. Chronic Active Epstein-Barr Virus Disease. Front. Immunol. 2017, 8, 1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.I. Primary Immunodeficiencies Associated with EBV Disease. Curr. Top. Microbiol. Immunol. 2015, 390, 241–265. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Latour, S. Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood 2020, 135, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Ali, M.A.; Patera, A.C.; Catalfamo, M.; Jaffe, E.S.; Kimura, H.; Dale, J.K.; Straus, S.E.; Cohen, J.I. Chronic active Epstein-Barr virus infection associated with mutations in perforin that impair its maturation. Blood 2004, 103, 1244–1252. [Google Scholar] [CrossRef]
- Rohr, J.; Beutel, K.; Maul-Pavicic, A.; Vraetz, T.; Thiel, J.; Warnatz, K.; Bondzio, I.; Gross-Wieltsch, U.; Schundeln, M.; Schutz, B.; et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica 2010, 95, 2080–2087. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Niemela, J.E.; Stoddard, J.L.; Pittaluga, S.; Heslop, H.; Jaffe, E.S.; Dowdell, K. Late-onset severe chronic active EBV in a patient for five years with mutations in STXBP2 (MUNC18-2) and PRF1 (perforin 1). J. Clin. Immunol. 2015, 35, 445–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, C.L.; Kuehn, H.S.; Zhao, F.; Niemela, J.E.; Deenick, E.K.; Palendira, U.; Avery, D.T.; Moens, L.; Cannons, J.L.; Biancalana, M.; et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat. Immunol. 2014, 15, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Li, F.Y.; Chaigne-Delalande, B.; Kanellopoulou, C.; Davis, J.C.; Matthews, H.F.; Douek, D.C.; Cohen, J.I.; Uzel, G.; Su, H.C.; Lenardo, M.J. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 2011, 475, 471–476. [Google Scholar] [CrossRef]
- Huck, K.; Feyen, O.; Niehues, T.; Ruschendorf, F.; Hubner, N.; Laws, H.J.; Telieps, T.; Knapp, S.; Wacker, H.H.; Meindl, A.; et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J. Clin. Investig. 2009, 119, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Dropulic, L.; Hsu, A.P.; Zerbe, C.S.; Krogmann, T.; Dowdell, K.; Hornung, R.L.; Lovell, J.; Hardy, N.; Hickstein, D.; et al. Association of GATA2 Deficiency With Severe Primary Epstein-Barr Virus (EBV) Infection and EBV-associated Cancers. Clin. Infect. Dis. 2016, 63, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Izawa, K.; Martin, E.; Soudais, C.; Bruneau, J.; Boutboul, D.; Rodriguez, R.; Lenoir, C.; Hislop, A.D.; Besson, C.; Touzot, F.; et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J. Exp. Med. 2017, 214, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, H.; Edwards, E.S.; Ikinciogullari, A.; Jing, H.; Borte, S.; Buggert, M.; Du, L.; Matsuda-Lennikov, M.; Romano, R.; Caridha, R.; et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J. Exp. Med. 2017, 214, 91–106. [Google Scholar] [CrossRef]
- Van Montfrans, J.M.; Hoepelman, A.I.; Otto, S.; van Gijn, M.; van de Corput, L.; de Weger, R.A.; Monaco-Shawver, L.; Banerjee, P.P.; Sanders, E.A.; Jol-van der Zijde, C.M.; et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J. Allergy Clin. Immunol. 2012, 129, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Kucuk, Z.Y.; Zhang, K.; Filipovich, L.; Bleesing, J.J. CTP Synthase 1 Deficiency in Successfully Transplanted Siblings with Combined Immune Deficiency and Chronic Active EBV Infection. J. Clin. Immunol. 2016, 36, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Bekker, V.; Scherpbier, H.; Beld, M.; Piriou, E.; van Breda, A.; Lange, J.; van Leth, F.; Jurriaans, S.; Alders, S.; Wertheim-van Dillen, P.; et al. Epstein-Barr virus infects B and non-B lymphocytes in HIV-1-infected children and adolescents. J. Infect. Dis. 2006, 194, 1323–1330. [Google Scholar] [CrossRef]
- Calattini, S.; Sereti, I.; Scheinberg, P.; Kimura, H.; Childs, R.W.; Cohen, J.I. Detection of EBV genomes in plasmablasts/plasma cells and non-B cells in the blood of most patients with EBV lymphoproliferative disorders by using Immuno-FISH. Blood 2010, 116, 4546–4559. [Google Scholar] [CrossRef] [Green Version]
- Mutsaers, P.G.; van de Loosdrecht, A.A.; Tawana, K.; Bodor, C.; Fitzgibbon, J.; Menko, F.H. Highly variable clinical manifestations in a large family with a novel GATA2 mutation. Leukemia 2013, 27, 2247–2248. [Google Scholar] [CrossRef]
- Alkhairy, O.K.; Perez-Becker, R.; Driessen, G.J.; Abolhassani, H.; van Montfrans, J.; Borte, S.; Choo, S.; Wang, N.; Tesselaar, K.; Fang, M.; et al. Novel mutations in TNFRSF7/CD27: Clinical, immunologic, and genetic characterization of human CD27 deficiency. J. Allergy Clin. Immunol. 2015, 136, 703–712. [Google Scholar] [CrossRef]
- Sekinaka, Y.; Mitsuiki, N.; Imai, K.; Yabe, M.; Yabe, H.; Mitsui-Sekinaka, K.; Honma, K.; Takagi, M.; Arai, A.; Yoshida, K.; et al. Common Variable Immunodeficiency Caused by FANC Mutations. J. Clin. Immunol. 2017, 37, 434–444. [Google Scholar] [CrossRef]
- Tanita, K.; Hoshino, A.; Imadome, K.I.; Kamiya, T.; Inoue, K.; Okano, T.; Yeh, T.W.; Yanagimachi, M.; Shiraishi, A.; Ishimura, M.; et al. Epstein-Barr Virus-Associated gammadelta T-Cell Lymphoproliferative Disorder Associated With Hypomorphic IL2RG Mutation. Front. Pediatr. 2019, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimura, M.; Eguchi, K.; Shiraishi, A.; Sonoda, M.; Azuma, Y.; Yamamoto, H.; Imadome, K.I.; Ohga, S. Systemic Epstein-Barr Virus-Positive T/NK Lymphoproliferative Diseases With SH2D1A/XIAP Hypomorphic Gene Variants. Front. Pediatr. 2019, 7, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, R.; Fournier, B.; Cordeiro, D.J.; Winter, S.; Izawa, K.; Martin, E.; Boutboul, D.; Lenoir, C.; Fraitag, S.; Kracker, S.; et al. Concomitant PIK3CD and TNFRSF9 deficiencies cause chronic active Epstein-Barr virus infection of T cells. J. Exp. Med. 2019, 216, 2800–2818. [Google Scholar] [CrossRef] [PubMed]
- Tabiasco, J.; Vercellone, A.; Meggetto, F.; Hudrisier, D.; Brousset, P.; Fournie, J.J. Acquisition of viral receptor by NK cells through immunological synapse. J. Immunol. 2003, 170, 5993–5998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohga, S.; Ishimura, M.; Yoshimoto, G.; Miyamoto, T.; Takada, H.; Tanaka, T.; Ohshima, K.; Ogawa, Y.; Imadome, K.; Abe, Y.; et al. Clonal origin of Epstein-Barr virus (EBV)-infected T/NK-cell subpopulations in EBV-positive T/NK-cell lymphoproliferative disorders of childhood. J. Clin Virol. 2011, 51, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Panzer-Grumayer, E.R.; Panzer, S.; Wolf, M.; Majdic, O.; Haas, O.A.; Kersey, J.H. Characterization of CD7+CD19+ lymphoid cells after Epstein-Barr virus transformation. J. Immunol. 1993, 151, 92–99. [Google Scholar]
- Ichigi, Y.; Naitoh, K.; Tokushima, M.; Haraoka, S.; Tagoh, H.; Kimoto, M.; Muraguchi, A. Generation of cells with morphological and antigenic properties of microglia from cloned EBV-transformed lymphoid progenitor cells derived from human fetal liver. Cell Immunol. 1993, 149, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.M.; Vera-Lozada, G.; Segges, P.; Hassan, R.; Niedobitek, G. Revisiting the Tissue Microenvironment of Infectious Mononucleosis: Identification of EBV Infection in T Cells and Deep Characterization of Immune Profiles. Front. Immunol. 2019, 10, 146. [Google Scholar] [CrossRef]
- Trempat, P.; Tabiasco, J.; Andre, P.; Faumont, N.; Meggetto, F.; Delsol, G.; Gascoyne, R.D.; Fournie, J.J.; Vivier, E.; Brousset, P. Evidence for early infection of nonneoplastic natural killer cells by Epstein-Barr virus. J. Virol. 2002, 76, 11139–11142. [Google Scholar] [CrossRef] [Green Version]
- Coleman, C.B.; Wohlford, E.M.; Smith, N.A.; King, C.A.; Ritchie, J.A.; Baresel, P.C.; Kimura, H.; Rochford, R. Epstein-Barr virus type 2 latently infects T cells, inducing an atypical activation characterized by expression of lymphotactic cytokines. J. Virol. 2015, 89, 2301–2312. [Google Scholar] [CrossRef] [Green Version]
- Smith, N.A.; Coleman, C.B.; Gewurz, B.E.; Rochford, R. CD21 (Complement Receptor 2) is the receptor for Epstein-Barr virus entry into T cells. J. Virol. 2020, 94, e00428-20. [Google Scholar] [CrossRef]
- Kimura, H. EBV in T-/NK-Cell Tumorigenesis. Adv. Exp. Med. Biol. 2018, 1045, 459–475. [Google Scholar] [CrossRef]
- Alfieri, C.; Ghibu, F.; Joncas, J.H. Lytic, nontransforming Epstein-Barr virus (EBV) from a patient with chronic active EBV infection. Can. Med. Assoc. J. 1984, 131, 1249–1252. [Google Scholar] [PubMed]
- Schwarzmann, F.; von Baehr, R.; Jager, M.; Prang, N.; Bohm, S.; Reischl, U.; Wolf, H.; Bieger, W.P. A case of severe chronic active infection with Epstein-Barr virus: Immunologic deficiencies associated with a lytic virus strain. Clin. Infect. Dis. 1999, 29, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Joncas, J.H. Biomolecular analysis of a defective nontransforming Epstein-Barr virus (EBV) from a patient with chronic active EBV infection. J. Virol. 1987, 61, 3306–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.X.; Hoshida, Y.; Yang, W.I.; Inohara, H.; Kubo, T.; Kim, G.E.; Yoon, J.H.; Kojya, S.; Bandoh, N.; Harabuchi, Y.; et al. Life-style and environmental factors in the development of nasal NK/T-cell lymphoma: A case-control study in East Asia. Int. J. Cancer 2007, 120, 406–410. [Google Scholar] [CrossRef]
- Ito, Y.; Suzuki, R.; Torii, Y.; Kawa, K.; Kikuta, A.; Kojima, S.; Kimura, H. HLA-A*26 and HLA-B*52 are associated with a risk of developing EBV-associated T/NK lymphoproliferative disease. Blood e-Lett. 2013, bloodjournal_el, 8085. [Google Scholar]
- Nishida, N.; Yang, X.; Takasaki, I.; Imai, K.; Kato, K.; Inoue, Y.; Imamura, T.; Miyashita, R.; Kato, F.; Yamaide, A.; et al. Dysgammaglobulinemia Associated With Glu349del, a Hypomorphic XIAP Mutation. J. Investig. Allergol. Clin. Immunol. 2015, 25, 205–213. [Google Scholar]
- Cohen, J.I.; Iwatsuki, K.; Ko, Y.H.; Kimura, H.; Manoli, I.; Ohshima, K.; Pittaluga, S.; Quintanilla-Martinez, L.; Jaffe, E.S. Epstein-Barr virus NK and T cell lymphoproliferative disease: Report of a 2018 international meeting. Leuk. Lymphoma 2020, 61, 808–819. [Google Scholar] [CrossRef]


{kind=link}
| Affected Gene | Type of Mutation/Variation | EBV-Related Manifestation | EBV-Unrelated Manifestation | Reference |
|---|---|---|---|---|
| GATA2 | Haploinsufficiency due to unialleleic expression | HV-like LPD HLH | Enterococcus faecium bacteremia, infections with Mycobacterium avium complex and histoplasma; neutropenia, lymphopenia, reduced numbers of B cells and NK cells, hypogammaglobulinemia | [64] |
| Heterozygous mutations (c.G28fs and p.H26P) | EBV-positive non-Hodgkin T-cell lymphoma | Pancytopenia | [71] | |
| CD27 | Homozygous mutation (c.G158A, p.C53Y) | EBV+ T-cell LPD developing into lymphoma | Oral ulcer, uveitis, recurrent non-EBV infections | [72] |
| FANCA | Homozygous mutation (c.190_191insT, p.E65RfsX5) | NK-cell-type CAEBV | Common variable immunodeficiency (hypogammaglobulinemia, sinusitis) | [73] |
| IL2RG | Hemizygous hypomorphic point mutation (c.C982T, p.R328*) | EBV-positive γδT-cell LPD | Recurrent respiratory infection, Yersinia enteritis, infection with Haemophilus influenzae Low T-cell count, complement deficiency (C2, C9), reduced mitogen-induced proliferation, dysgammaglobulinemia | [74] |
| SH2D1A | Hemizygous hypomorphic mutation/variation (c.G7T, p.A3S) | NK-cell-type CAEBV (indolent), photosensitivity, SMBA | No apparent immunodeficiency or other infections | [75] |
| XIAP | Hypomorphic mutation/variation (c.1045_1047delGAG, p.E349del) | NK/B-cell type CAEBV and SMBA in a hemizygous boy | No apparent immunodeficiency or other infections | [75] |
| EBV-HLH following primary infection in a hemizygous boy | No apparent immunodeficiency or other infections | |||
| NK/CD4+ T-cell type CAEBV and SMBA in a heterozygous woman | No apparent immunodeficiency or other infections | |||
| TNFRSF9/PIK3CD | Homozygous LOF mutation in TNFRSF9 (c.170delG, p.G57fsX91); homozygous LOF mutation in PIK3CD (c.2462G > A, p. R821H) | T-cell type CAEBV, HV-like LPD, EBV-HLH | Recurrent respiratory and skin infections (panaritium and boils), no apparent signs of immunodeficiency | [76] |
| TNFRSF9 | Homozygous LOF mutation in TNFRSF9 (c.170delG, p.G57fsX91) | EBV+ T-cell lymphoproliferation without symptoms in the healthy sister of the patient described in the row above | No apparent signs of immunodeficiency | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujiwara, S.; Nakamura, H. Chronic Active Epstein–Barr Virus Infection: Is It Immunodeficiency, Malignancy, or Both? Cancers 2020, 12, 3202. https://doi.org/10.3390/cancers12113202
Fujiwara S, Nakamura H. Chronic Active Epstein–Barr Virus Infection: Is It Immunodeficiency, Malignancy, or Both? Cancers. 2020; 12(11):3202. https://doi.org/10.3390/cancers12113202
Chicago/Turabian StyleFujiwara, Shigeyoshi, and Hiroyuki Nakamura. 2020. "Chronic Active Epstein–Barr Virus Infection: Is It Immunodeficiency, Malignancy, or Both?" Cancers 12, no. 11: 3202. https://doi.org/10.3390/cancers12113202
APA StyleFujiwara, S., & Nakamura, H. (2020). Chronic Active Epstein–Barr Virus Infection: Is It Immunodeficiency, Malignancy, or Both? Cancers, 12(11), 3202. https://doi.org/10.3390/cancers12113202