IDH Signalling Pathway in Cholangiocarcinoma: From Biological Rationale to Therapeutic Targeting

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
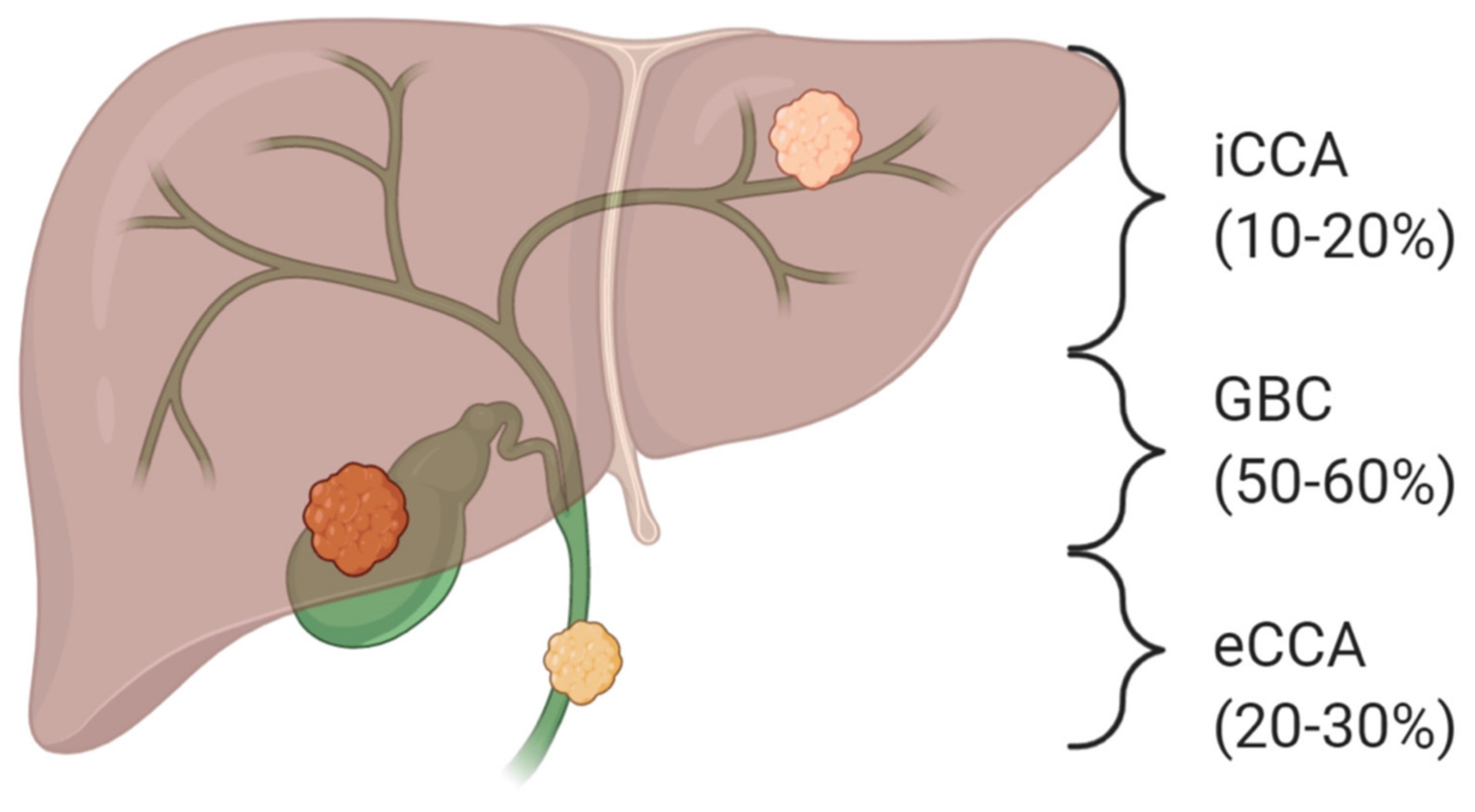
2. The Genomic Landscape of Cholangiocarcinoma
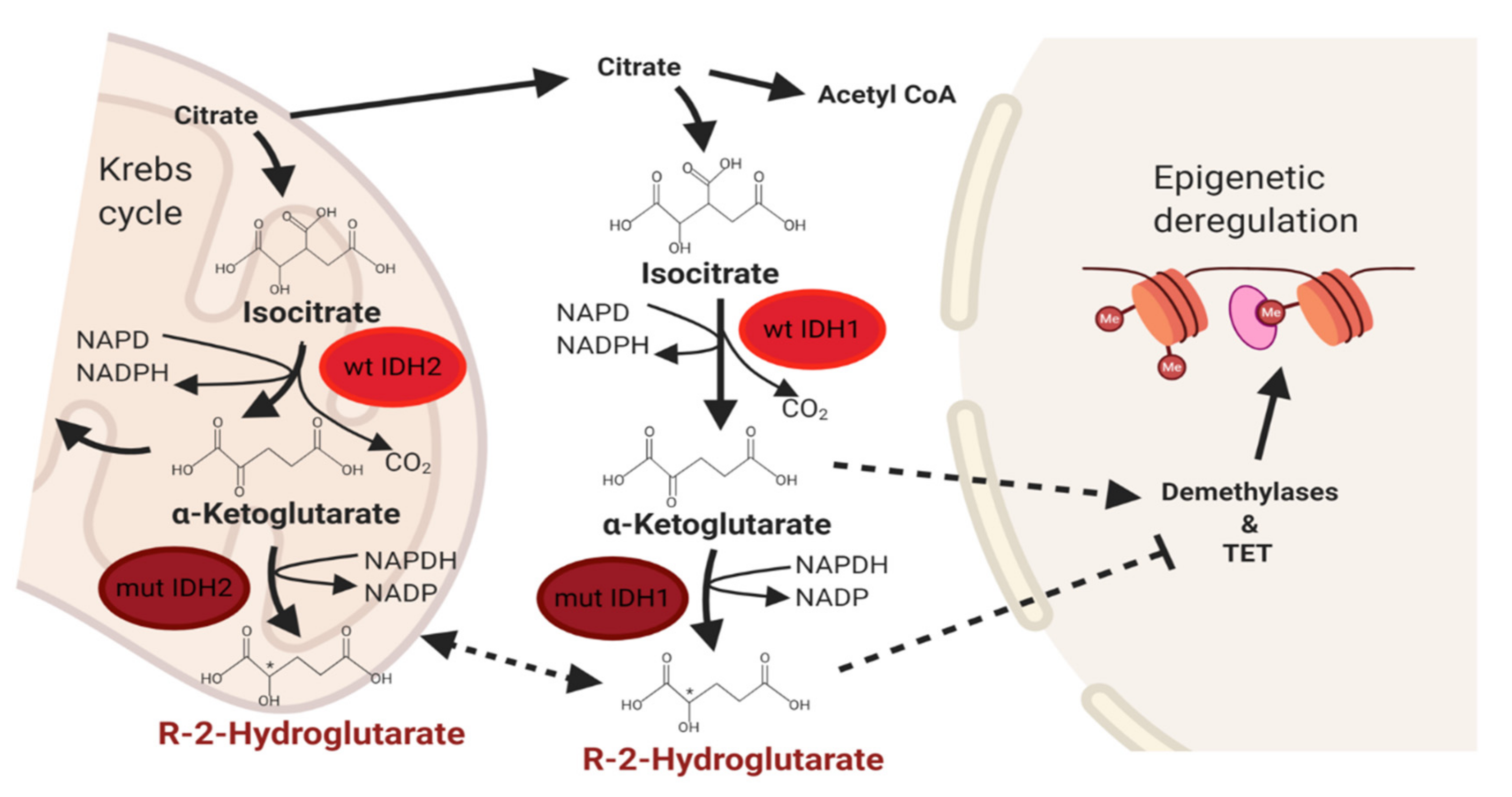
3. IDH Signaling Pathway in Cancer
4. Targeting IDH in Cancer
5. IDH Mutations in Cholangiocarcinoma
6. Clinical Development of IDH Inhibitors in Cholangiocarcinoma
7. Future Perspectives and Conclusions
Funding
Conflicts of Interest
References
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Davidson, B.R.; Goldin, R.D.; Heaton, N.; Karani, J.; Pereira, S.P.; Rosenberg, W.M.C.; Tait, P.; Taylor-Robinson, S.D.; Thillainayagam, A.V.; et al. Guidelines for the Diagnosis and Treatment of Cholangiocarcinoma: An Update. Gut 2012, 61, 1657–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.K.; Zhu, A.X.; Fuchs, C.S.; Brooks, G.A. Forty-year trends in cholangiocarcinoma incidence in the US: Intrahepatic disease on the rise. Oncologist 2016, 21, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeva, J.; Sangro, B.; Salati, M.; Edeline, J.; La Casta, A.; Bittoni, A.; Berardi, R.; Bruix, J.; Valle, J.W. Medical treatment for cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 123–142. [Google Scholar] [CrossRef] [Green Version]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Okusaka, T.; Nakachi, K.; Fukutomi, A.; Mizuno, N.; Ohkawa, S.; Funakoshi, A.; Nagino, M.; Kondo, S.; Nagaoka, S.; Funai, J.; et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: A comparative multicentre study in Japan. Br. J. Cancer 2010, 103, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. ABC-06: A randomised phase III, multi-centre, open-label study of active symptom control (ASC) alone or ASC with oxaliplatin/5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced/metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. Proc. Am. Soc. Clin. Oncol. 2019, 37, 4003. [Google Scholar]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational Landscape of Intrahepatic Cholangiocarcinoma. Nat. Commun. 2014, 5, 5596. [Google Scholar]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Churi, C.R.; Shro, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation Profiling in Cholangiocarcinoma: Prognostic and Therapeutic Implications. PLoS ONE 2014, 9, e115383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javle, M.; Bekaii-Saab, T.; Jain, A.; Wang, Y.; Kelley, R.K.; Wang, K.; Kang, H.C.; Catenacci, D.; Ali, S.; Krishnan, S.; et al. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer 2016, 122, 3838–3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, M.A.; Ptashkin, R.N.; Jordan, E.J.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El Dika, I.; Jarnagin, W.R.; et al. Comprehensive molecular profiling of intra- and extrahepatic cholangiocarcinomas: Potential targets for intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; ElZawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Chan-On, W.; Nairismägi, M.-L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.-C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Cancer Genome Atlas Network. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; Mclendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinardo, C.D.; Propert, K.J.; Loren, A.W.; Paietta, E.; Sun, Z.; Levine, R.L.; Straley, K.S.; Yen, K.; Patel, J.P.; Agresta, S.; et al. Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood 2013, 121, 4917–4924. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Fathi, A.T.; Wander, S.A.; Faramand, R.; Emadi, A. Biochemical, epigenetic, and metabolic approaches to target IDH mutations in acute myeloid leukemia. Semin. Hematol. 2015, 52, 165–171. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; Mclellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; Mcgrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Eng. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Yen, K.E.; Bittinger, M.A.; Su, S.M.; Fantin, V.R. Cancer-associated IDH mutations: Biomarker and therapeutic opportunities. Oncogene 2010, 29, 6409–6417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Seo, S.I.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int. J. Cancer 2009, 125, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef]
- Cairns, R.A.; Iqbal, J.; Kucuk, C.; de Leval, L.; Jais, J.; Parrens, M.; Martin, A.; Xerri, L.; Brousset, P.; Chan, L.C.; et al. Brief report IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef] [Green Version]
- Popovici-Muller, J.; Saunders, J.O.; Salituro, F.G.; Travins, J.M.; Yan, S.; Zhao, F.; Gross, S.; Dang, L.; Yen, K.E.; Yang, H.; et al. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 2012, 3, 850–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Schimmer, A.D.; Yee, K.W.L.; Hochhaus, A.; Kraemer, A.; Carvajal, R.D.; Janku, F.; Bedard, P.; Carpio, C.; Wick, A.; et al. A Phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 Mutations. Blood 2016, 128, 1073. [Google Scholar] [CrossRef]
- Heredia, V. AG-120, a novel IDH1 targeted molecule, inhibits invasion and migration of chondrosarcoma cells in vitro. Ann. Oncol. 2017, 28. [Google Scholar] [CrossRef]
- Nicolay, B.; Narayanaswamy, R.; Aguado, E.; Nagaraja, R.; Murtie, J.; Liu, G.; Ishii, Y. The IDH1 mutant inhibitor AG-120 shows strong inhibition of 2-HG production in an orthotopic IDH1 mutant glioma model in vivo. Neuro Oncol. 2017, 19 (Suppl. 6), vi86. [Google Scholar] [CrossRef] [Green Version]
- Yen, K.; Chopra, V.S.; Tobin, E.; Avanzino, B.; Mavrommatis, K.; Dimartino, J.; MacBeth, K.J. Functional characterization of the ivosidenib (AG-120) and azacitidine combination in a mutant IDH1 AML cell model. Cancer Res. 2018, 78 (Suppl. 13), 4956. [Google Scholar]
- Dhillon, S. Ivosidenib: First global approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Kim, E.S. Enasidenib: First global approval. Drugs 2017, 77, 1705–1711. [Google Scholar] [CrossRef]
- Yen, K.; Konteatis, Z.; Sui, Z.; Artin, E.; Dang, L.; Straley, K.; Tobin, E.; Campos, C.; Yang, H.; Nagaraja, R.; et al. AG-881, a brain penetrant, potent, pan-mutant IDH (mIDH) inhibitor for use in mIDH solid and hematologic malignancies. Mol. Cancer Ther. 2018, 17 (Suppl. 1), B126. [Google Scholar]
- Chen, J.; Yang, J.; Cao, P. The evolving landscape in the development of isocitrate dehydrogenase mutant inhibitors. Mini Rev. Med. Chem. 2016, 16, 1344–1358. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Yun, C.H. Crystal structures of pan-IDH inhibitor AG-881 in complex with mutant human IDH1 and IDH2. Biochem. Biophys. Res. Commun. 2018, 503, 2912–2917. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef]
- Choe, S.; Wang, H.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef]
- Grassian, A.R.; Pagliarini, R.; Chiang, D.Y. Mutations of isocitrate dehydrogenase 1 and 2 in intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2014, 30, 295–302. [Google Scholar] [CrossRef]
- Jain, A.; Kwong, L.N.; Javle, M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr. Treat. Opt. Oncol. 2016, 17, 58. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef]
- Lowery, M.A.; Burris, H.A.; Janku, F.; Shroff, R.T.; Cleary, J.M.; Azad, N.S.; Goyal, L.; Maher, E.A.; Gore, L.; Hollebecque, A.; et al. Safety and efficacy of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: A phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 711–720. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Biliary Tract Cancer: State of the Art and potential role of DNA Damage Repair. Cancer Treat. Rev. 2018, 70, 168–177. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Molecular targeted therapies: Ready for “prime time” in biliary tract cancer. J. Hepatol. 2020, 73, 170–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Gordan, J.D.; Kleinstiver, B.P.; Vu, P.; Najem, M.S.; Yeo, J.C.; Shi, L.; Kato, Y.; Levin, R.S.; Webber, J.T.; et al. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2016, 6, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Trial Number | Compound | Phase | Setting | Status |
|---|---|---|---|---|
| NCT02428855 | Dasatinib | II | Advanced iCCA IDH1/2 mut | Completed |
| NCT04088188 | Gem/Cis + ivosidenib or pemigatinib | I | Advanced CCA | Not yet recruiting |
| NCT03684811 | FT2102 | Ib/II | Advanced iCCA IDH1 mut | Active, not recruiting |
| NCT02273739 | Enasidenib | I/II | Advanced iCCA IDH2 mut | Completed |
| NCT02381886 | IDH305 | I | Advanced tumours IDH1 R132 mut | Active, not recruiting |
| NCT02481154 | AG-881 | I | Advanced tumours IDH1/2 mut | Active, not recruiting |
| NCT04056910 | Ivosidenib + nivolumab | II | Advanced tumours IDH1 mut | Not yet recruiting |
| NCT04521686 | LY3410738 | I | Advanced tumours IDH1 R132 mut | Recruiting |
| NCT02746081 | BAY 1436032 | II | Advanced tumours IDH1 R132X mut | Active, not recruiting |
| NCT02073994 | AG-120 | I | Advanced tumours IDH1 mut | Active, not recruiting |
| NCT03878095 | Olaparib + ceralasertib | II | Advanced tumours IDH1/2 mut | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salati, M.; Caputo, F.; Baldessari, C.; Galassi, B.; Grossi, F.; Dominici, M.; Ghidini, M. IDH Signalling Pathway in Cholangiocarcinoma: From Biological Rationale to Therapeutic Targeting. Cancers 2020, 12, 3310. https://doi.org/10.3390/cancers12113310
Salati M, Caputo F, Baldessari C, Galassi B, Grossi F, Dominici M, Ghidini M. IDH Signalling Pathway in Cholangiocarcinoma: From Biological Rationale to Therapeutic Targeting. Cancers. 2020; 12(11):3310. https://doi.org/10.3390/cancers12113310
Chicago/Turabian StyleSalati, Massimiliano, Francesco Caputo, Cinzia Baldessari, Barbara Galassi, Francesco Grossi, Massimo Dominici, and Michele Ghidini. 2020. "IDH Signalling Pathway in Cholangiocarcinoma: From Biological Rationale to Therapeutic Targeting" Cancers 12, no. 11: 3310. https://doi.org/10.3390/cancers12113310
APA StyleSalati, M., Caputo, F., Baldessari, C., Galassi, B., Grossi, F., Dominici, M., & Ghidini, M. (2020). IDH Signalling Pathway in Cholangiocarcinoma: From Biological Rationale to Therapeutic Targeting. Cancers, 12(11), 3310. https://doi.org/10.3390/cancers12113310