Autophagic Inhibition via Lysosomal Integrity Dysfunction Leads to Antitumor Activity in Glioma Treatment

 , , and
, , and
Abstract
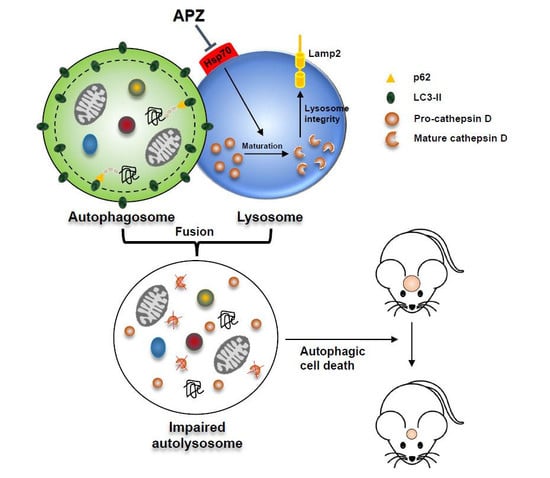
:
1. Introduction
2. Results
2.1. Autophagonizer Functions Not by Inducing Autophagy, but by Inhibiting Autophagy
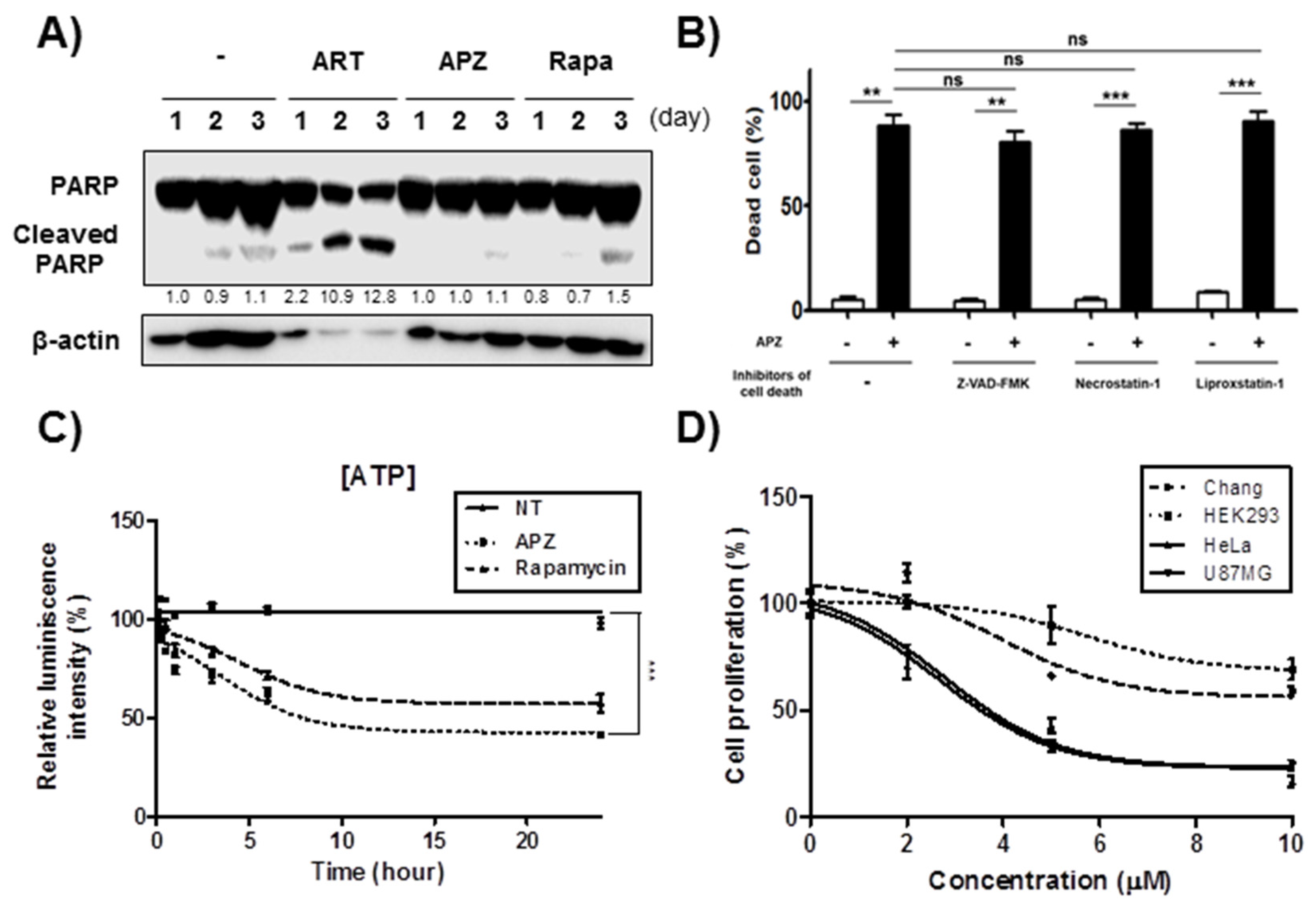
2.2. APZ Induces Autophagic Cell Death in Cancer Cell Lines
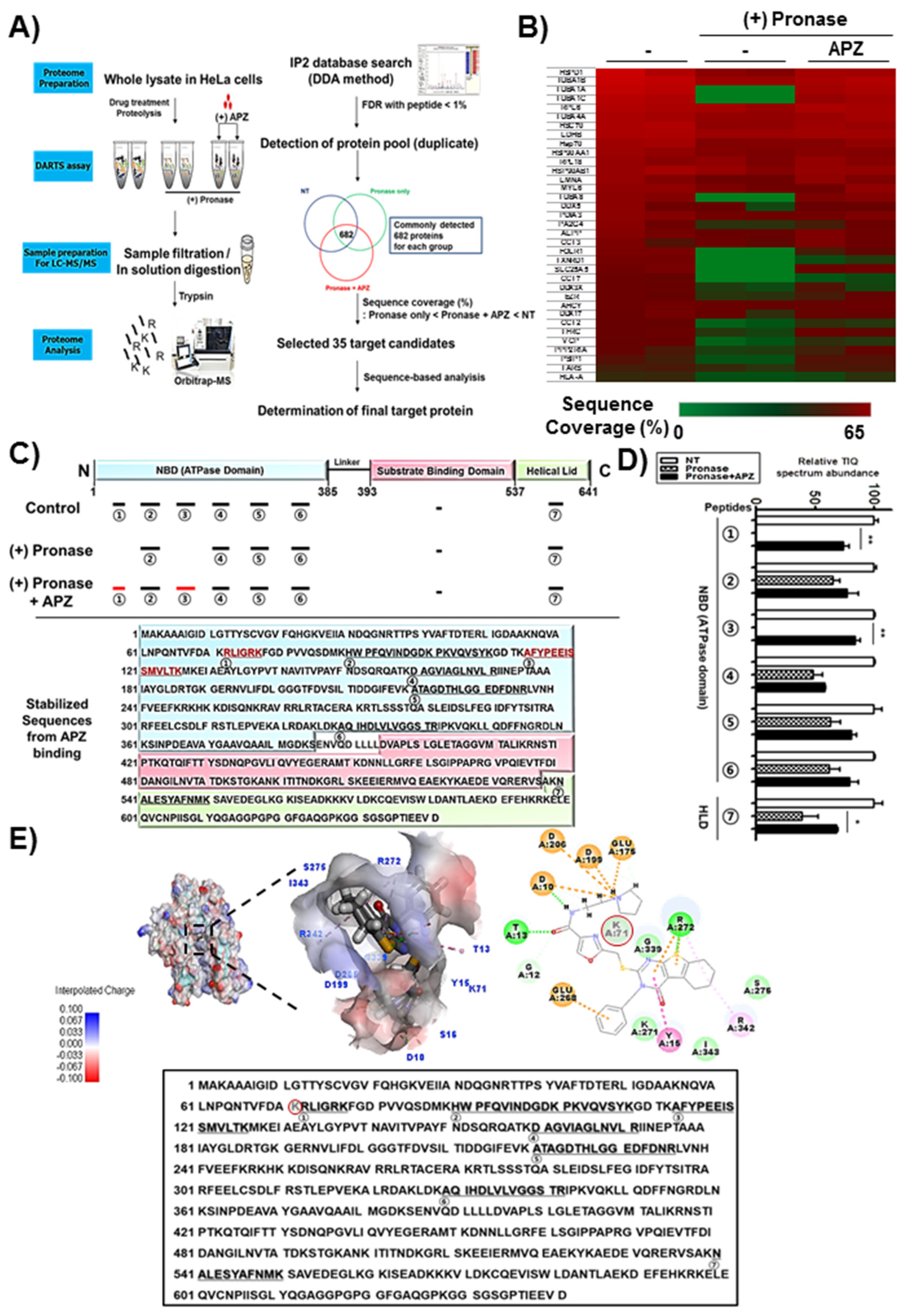
2.3. DARTS and LC–MS/MS Analysis for Identification of APZ Protein Targets
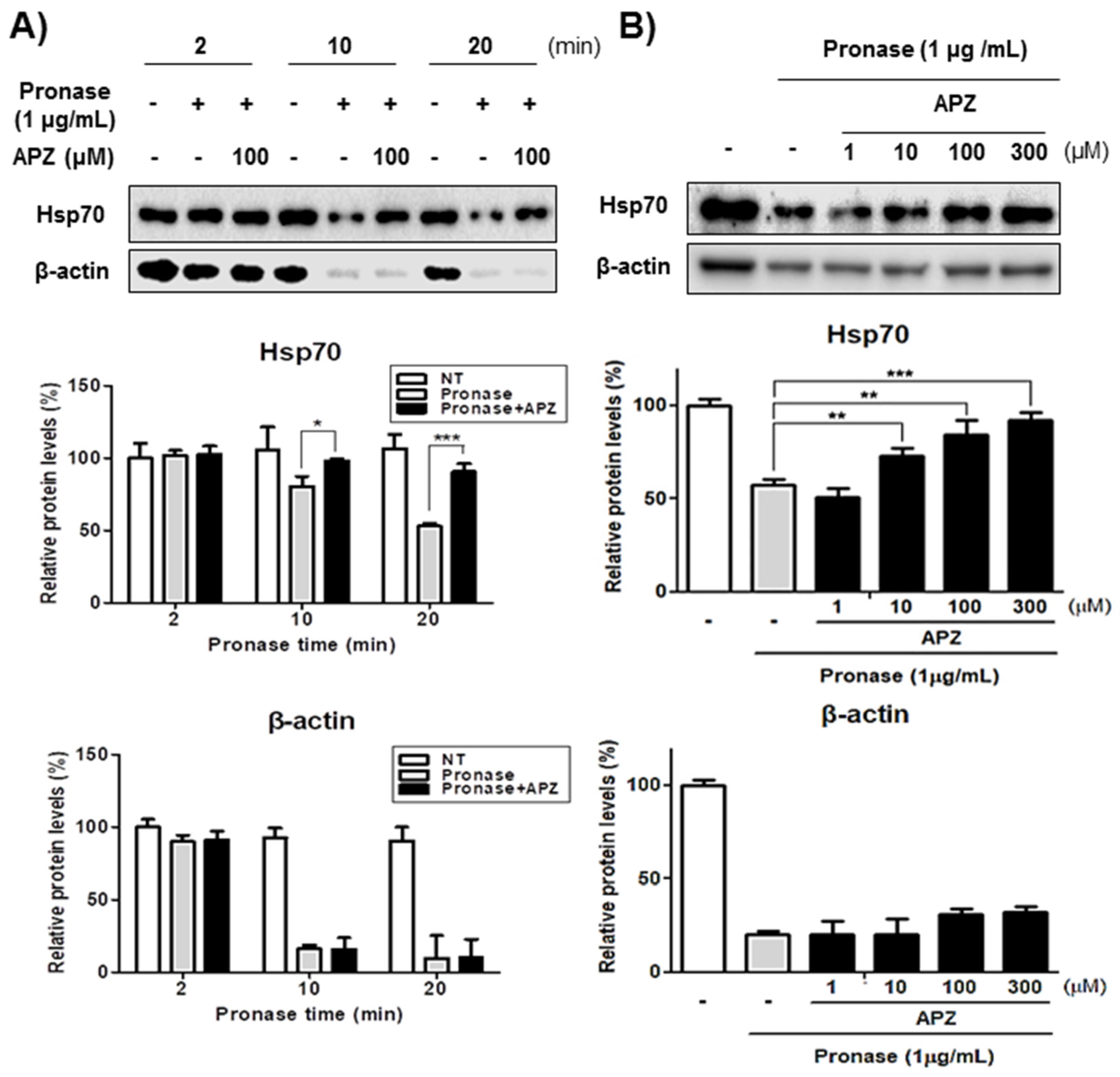
2.4. Characterization of Hsp70 as a Potential Protein Target of APZ
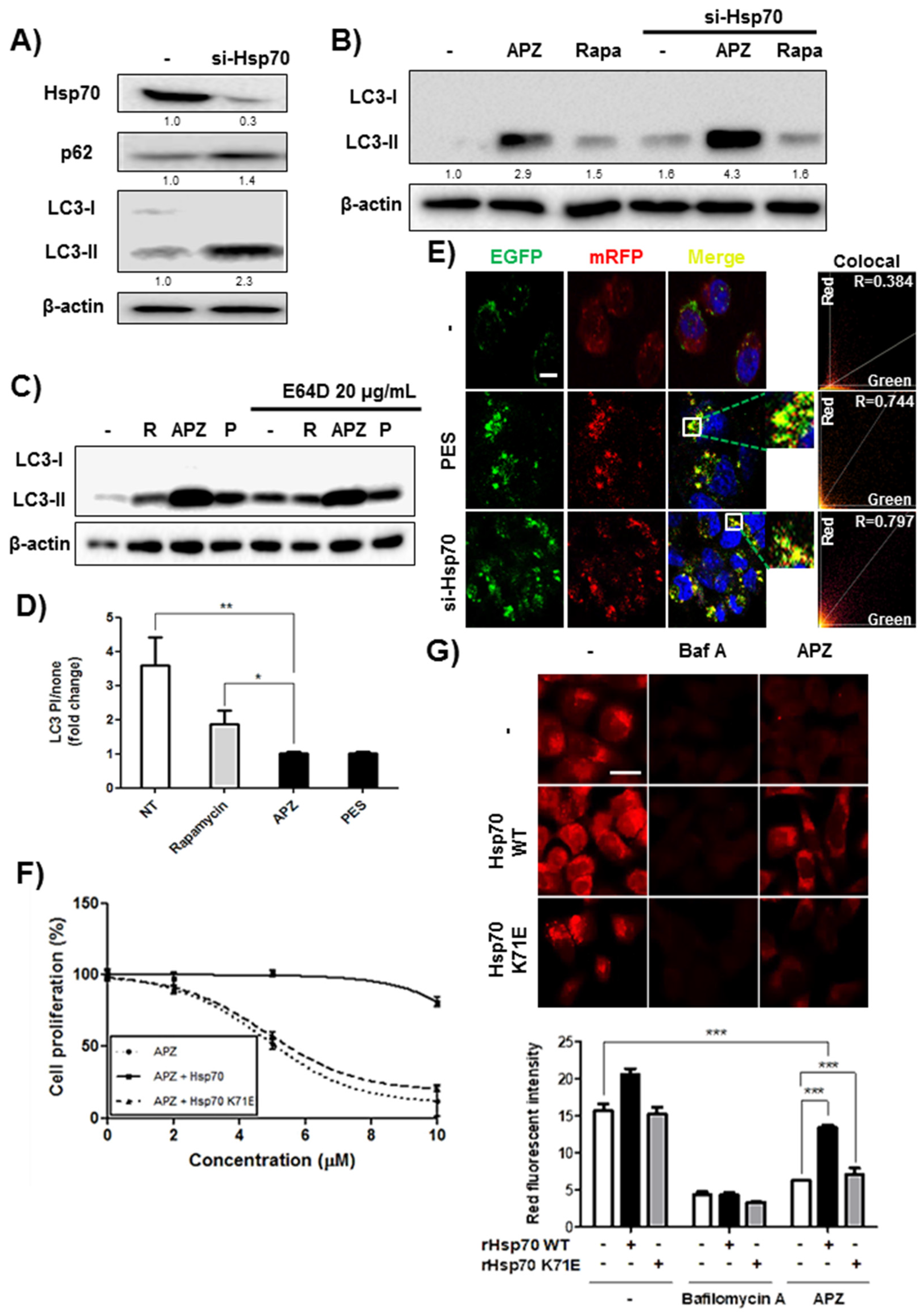
2.5. Validation of Hsp70 as a Protein Target of APZ
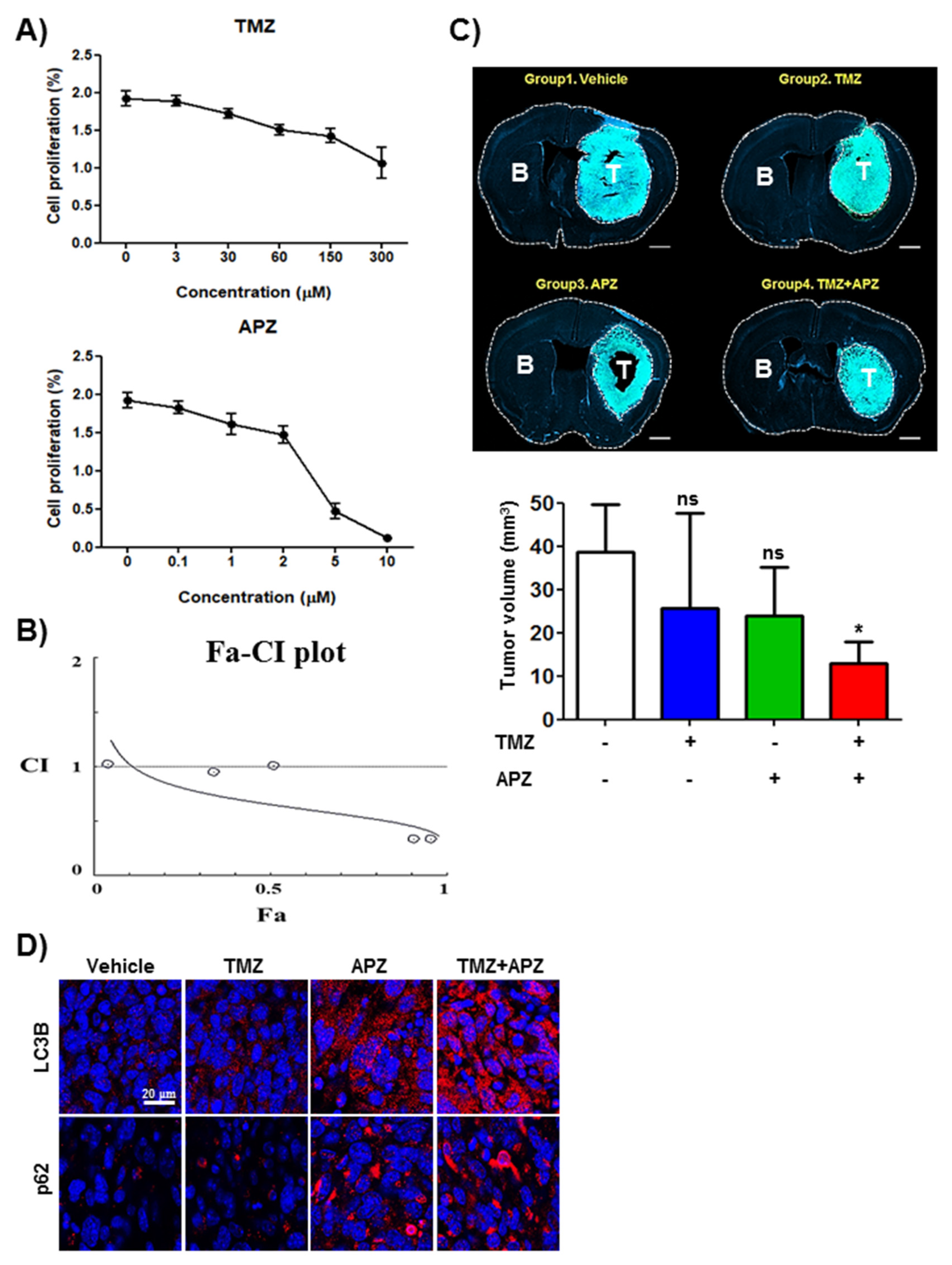
2.6. APZ Inhibits Lysosomal Integrity by Antagonizing Hsp70
3. Discussion
4. Materials and Methods
4.1. Compounds and Antibodies
4.2. Cell Culture
4.3. Hsp70 RNA Interference by Reverse Transcription Polymerase Chain Reaction and Recombinant Hsp70 Plasmid Transfection Analysis
4.4. In Silico Docking Study
4.5. Immunoblotting
4.6. Immunocytochemistry Staining
4.7. ATP-Monitoring Luminescence Assay
4.8. Cell Proliferation Assay
4.9. Cell Death Assay
4.10. Transmission Electron Microscopy
4.11. DARTS Assay
4.12. In Vivo Mouse Tumor Xenograft Assay
4.13. Acridine Orange Staining
4.14. mRFP-GFP-LC3B Plasmid Transfection
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Tasdemir, E.; Galluzzi, L.; Maiuri, M.C.; Criollo, A.; Vitale, I.; Hangen, E.; Modjtahedi, N.; Kroemer, G. Methods for assessing autophagy and autophagic cell death. Methods Mol. Biol. 2008, 445, 29–76. [Google Scholar]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer. Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [Green Version]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Han, Y.; Visconte, V.; Phillips, J.; Nawrocki, S.T. Disruption of Autophagic Degradation with ROC-325 Antagonizes Renal Cell Carcinoma Pathogenesis. Clin. Cancer. Res. 2017, 23, 2869–2879. [Google Scholar] [CrossRef] [Green Version]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.Y.; Cho, S.M.; Kwon, H.J. Approaches for discovering novel bioactive small molecules targeting autophagy. Expert Opin. Drug Discov. 2017, 12, 909–923. [Google Scholar] [CrossRef]
- Choi, I.K.; Cho, Y.S.; Jung, H.J.; Kwon, H.J. Autophagonizer, a novel synthetic small molecule, induces autophagic cell death. Biochem. Biophys. Res. Commun. 2010, 393, 849–854. [Google Scholar] [CrossRef]
- Kim, D.; Hwang, H.Y.; Kim, J.Y.; Lee, J.Y.; Yoo, J.S.; Marko-Varga, G.; Kwon, H.J. FK506, an Immunosuppressive Drug, Induces Autophagy by Binding to the V-ATPase Catalytic Subunit A in Neuronal Cells. J. Proteome Res. 2017, 16, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.S.; Kwon, H.J. Control of autophagy with small molecules. Arch. Pharm. Res. 2010, 33, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Pan, W.; Wang, X.P.; Chen, T.S. Artesunate induces apoptosis via a Bak-mediated caspase-independent intrinsic pathway in human lung adenocarcinoma cells. J. Cell. Physiol. 2012, 227, 3778–3786. [Google Scholar] [CrossRef]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzetti, E.; Huang, Z.J.; Shi, Y.X.; Xie, K.; Deng, X.J.; Li, J.P.; Li, Q.R.; Yang, W.Y.; Zeng, W.N.; Casartelli, M.; et al. Autophagy precedes apoptosis during the remodeling of silkworm larval midgut. Apoptosis 2012, 17, 305–324. [Google Scholar] [CrossRef]
- Xu, R.H.; Pelicano, H.; Zhang, H.; Giles, F.J.; Keating, M.J.; Huang, P. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia 2005, 19, 2153–2158. [Google Scholar] [CrossRef]
- Calzolari, A.; Larocca, L.M.; Deaglio, S.; Finisguerra, V.; Boe, A.; Raggi, C.; Ricci-Vitani, L.; Pierconti, F.; Malavasi, F.; De Maria, R.; et al. Transferrin receptor 2 is frequently and highly expressed in glioblastomas. Transl. Oncol. 2010, 3, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Kerber, M.; Reiss, Y.; Wickersheim, A.; Jugold, M.; Kiessling, F.; Heil, M.; Tchaikovski, V.; Waltenberger, J.; Shibuya, M.; Plate, K.H.; et al. Flt-1 signaling in macrophages promotes glioma growth in vivo. Cancer Res. 2008, 68, 7342–7351. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.K.; Kim, K.; Lee, E.; Oh, D.S.; Park, C.S.; Park, S.; Yang, J.M.; Kim, J.H.; Kim, H.S.; Shima, D.T.; et al. Sox7 promotes high-grade glioma by increasing VEGFR2-mediated vascular abnormality. J. Exp. Med. 2018, 215, 963–983. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Kim, Y.; Kwon, H.J. Advances in identification and validation of protein targets of natural products without chemical modification. Nat. Prod. Rep. 2016, 33, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Park, G.W.; Kim, J.Y.; Hwang, H.; Lee, J.Y.; Ahn, Y.H.; Lee, H.K.; Ji, E.S.; Kim, K.H.; Jeong, H.K.; Yun, K.N.; et al. Integrated GlycoProteome Analyzer (I-GPA) for Automated Identification and Quantitation of Site-Specific N-Glycosylation. Sci. Rep. 2016, 6, 21175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, P.C.; Xu, T.; Han, X.; Cociorva, D.; Barbosa, V.C.; Yates, J.R., 3rd. YADA: A tool for taking the most out of high-resolution spectra. Bioinformatics 2009, 25, 2734–2736. [Google Scholar] [CrossRef] [Green Version]
- Tabb, D.L.; McDonald, W.H.; Yates, J.R., 3rd. DTASelect and Contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 2002, 1, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Chittaranjan, S.; Bortnik, S.; Gorski, S.M. Monitoring Autophagic Flux by Using Lysosomal Inhibitors and Western Blotting of Endogenous MAP1LC3B. Cold Spring Harb. Protoc. 2015, 2015, 743–750. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef] [Green Version]
- Porter, K.; Nallathambi, J.; Lin, Y.; Liton, P.B. Lysosomal basification and decreased autophagic flux in oxidatively stressed trabecular meshwork cells: Implications for glaucoma pathogenesis. Autophagy 2013, 9, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; Sharma, P.K.; Tiwari, R.; Rayavarapu, R.G.; Shankar, J.; Chauhan, L.K.S.; Pandey, A.K. Impaired lysosomal activity mediated autophagic flux disruption by graphite carbon nanofibers induce apoptosis in human lung epithelial cells through oxidative stress and energetic impairment. Part. Fibre Toxicol. 2017, 14, 15. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gierasch, L.M.; Bahar, I. Role of Hsp70 ATPase domain intrinsic dynamics and sequence evolution in enabling its functional interactions with NEFs. PLoS Comput. Biol. 2010, 6, e1000931. [Google Scholar] [CrossRef] [Green Version]
- Kaye, F.J.; Modi, S.; Ivanovska, I.; Koonin, E.V.; Thress, K.; Kubo, A.; Kornbluth, S.; Rose, M.D. A family of ubiquitin-like proteins binds the ATPase domain of Hsp70-like Stch. FEBS Lett. 2000, 467, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Yamaji, T.; Ueno, T.; Ishiura, S.; Kominami, E.; Hanada, K. Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls. Autophagy 2008, 4, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leu, J.I.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Sekihara, K.; Harashima, N.; Tongu, M.; Tamaki, Y.; Uchida, N.; Inomata, T.; Harada, M. Pifithrin-mu, an inhibitor of heat-shock protein 70, can increase the antitumor effects of hyperthermia against human prostate cancer cells. PLoS ONE 2013, 8, e78772. [Google Scholar] [CrossRef] [Green Version]
- Rajapandi, T.; Wu, C.; Eisenberg, E.; Greene, L. Characterization of D10S and K71E mutants of human cytosolic hsp70. Biochemistry 1998, 37, 7244–7250. [Google Scholar] [CrossRef]
- Tapper, H.; Sundler, R. Bafilomycin A1 inhibits lysosomal, phagosomal, and plasma membrane H(+)-ATPase and induces lysosomal enzyme secretion in macrophages. J. Cell. Physiol. 1995, 163, 137–144. [Google Scholar] [CrossRef]
- Miura, E.; Hasegawa, T.; Konno, M.; Suzuki, M.; Sugeno, N.; Fujikake, N.; Geisler, S.; Tabuchi, M.; Oshima, R.; Kikuchi, A.; et al. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 2014, 71, 1–13. [Google Scholar] [CrossRef]
- Sarkar, C.; Zhao, Z.; Aungst, S.; Sabirzhanov, B.; Faden, A.I.; Lipinski, M.M. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 2014, 10, 2208–2222. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Zhang, L.; Qiang, P.; Yu, J.; Miao, Y.; Chen, Z.; Qu, J.; Zhao, Q.; Chen, Z.; Liu, Y.; Yao, X.; et al. Identification of compound CA-5f as a novel late-stage autophagy inhibitor with potent anti-tumor effect against non-small cell lung cancer. Autophagy 2019, 15, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taira, K.; Benkovic, S.J. Evaluation of the importance of hydrophobic interactions in drug binding to dihydrofolate reductase. J. Med. Chem. 1988, 31, 129–137. [Google Scholar] [CrossRef]
- Schlecht, R.; Scholz, S.R.; Dahmen, H.; Wegener, A.; Sirrenberg, C.; Musil, D.; Bomke, J.; Eggenweiler, H.M.; Mayer, M.P.; Bukau, B. Functional analysis of Hsp70 inhibitors. PLoS ONE 2013, 8, e78443. [Google Scholar] [CrossRef]
- Ko, S.K.; Kim, J.; Na, D.C.; Park, S.; Park, S.H.; Hyun, J.Y.; Baek, K.H.; Kim, N.D.; Kim, N.K.; Park, Y.N.; et al. A small molecule inhibitor of ATPase activity of HSP70 induces apoptosis and has antitumor activities. Chem. Biol. 2015, 22, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, W.; Liu, W.; Shao, Y.; Chen, L. VER-155008, a small molecule inhibitor of HSP70 with potent anti-cancer activity on lung cancer cell lines. Exp. Biol. Med. 2014, 239, 638–645. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Locus (Human) | Name | Full Name |
|---|---|---|
| sp|P10809|CH60 | HSPD1 | 60 kDa heat shock protein, mitochondrial precursor |
| sp|P05141|ADT2 | SLC25A5 | ADP/ATP translocase 2 |
| sp|Q99832|TCPH | CCT7 | T-complex protein 1 subunit eta |
| sp|P15311|EZRI | EZR | Ezrin |
| sp|P15328|FOLR1 | FOLR1 | Folate receptor alpha |
| sp|P08107|HSP71 | HSPA1A | Heat shock 70 kDa protein 1A/1B |
| sp|P11142|HSP7C | HSPA8 | Heat shock cognate 71 kDa protein |
| sp|P07900|HS90A | HSP90AA1 | Heat shock protein HSP 90-alpha |
| sp|P08238|HS90B | HSP90AB1 | Heat shock protein HSP 90-beta |
| tr|B7Z904|B7Z904 | TXNRD1 | Thioredoxin reductase 1 |
| sp|P07195|LDHB | LDHB | L-lactate dehydrogenase B chain |
| sp|P60660|MYL6 | MYL6 | Smooth muscle of Myosin light polypeptide 6 |
| sp|O75475|PSIP1 | PSIP1 | PC4 and SFRS1-interacting protein |
| sp|P02545|LMNA | LMNA | Prelamin-A/C |
| sp|Q92841|DDX17 | DDX17 | Probable ATP-dependent RNA helicase DDX17 |
| sp|P17844|DDX5 | DDX5 | Probable ATP-dependent RNA helicase DDX5 |
| sp|Q9UQ80|PA2G4 | PA2G4 | Proliferation-associated protein 2G4 |
| sp|P30101|PDIA3 | PDIA3 | Protein disulfide-isomerase A3 |
| sp|P30153|2AAA | PPP2R1A | Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha |
| sp|P26639|SYTC | TARS | Threonyl-tRNA synthetase |
| sp|P02786|TFR1 | TFRC | Transferrin receptor protein 1 |
| sp|Q71U36|TBA1A | TUBA1A | Tubulin alpha-1A chain |
| sp|P68363|TBA1B | TUBA1B | Tubulin alpha-1B chain |
| sp|Q9BQE3|TBA1C | TUBA1C | Tubulin alpha-1C chain |
| sp|P68366|TBA4A | TUBA4A | Tubulin alpha-4A chain |
| sp|Q9NY65|TBA8 | TUBA8 | Tubulin alpha-8 chain |
| sp|P78371|TCPB | CCT2 | T-complex protein 1 subunit beta |
| sp|O00571|DDX3X | DDX3X | ATP-dependent RNA helicase DDX3X |
| sp|Q02543|RL18A | RPL18 | 60S ribosomal protein L18 |
| sp|Q02878|RL6 | RPL6 | 60S ribosomal protein L6 |
| sp|P01891|1A68 | HLA-A | HLA class I histocompatibility antigen, A-68 alpha chain |
| sp|P49368|TCPG | CCT3 | T-complex protein 1 subunit gamma |
| sp|P05187|PPB1 | ALPP | Alkaline phosphatase, placental type |
| sp|P23526|SAHH | AHCY | Adenosylhomocysteinase |
| sp|P55072|TERA | VCP | Transitional endoplasmic reticulum ATPase |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, H.-Y.; Cho, Y.S.; Kim, J.Y.; Yun, K.N.; Yoo, J.S.; Lee, E.; Kim, I.; Kwon, H.J. Autophagic Inhibition via Lysosomal Integrity Dysfunction Leads to Antitumor Activity in Glioma Treatment. Cancers 2020, 12, 543. https://doi.org/10.3390/cancers12030543
Hwang H-Y, Cho YS, Kim JY, Yun KN, Yoo JS, Lee E, Kim I, Kwon HJ. Autophagic Inhibition via Lysosomal Integrity Dysfunction Leads to Antitumor Activity in Glioma Treatment. Cancers. 2020; 12(3):543. https://doi.org/10.3390/cancers12030543
Chicago/Turabian StyleHwang, Hui-Yun, Yoon Sun Cho, Jin Young Kim, Ki Na Yun, Jong Shin Yoo, Eunhyeong Lee, Injune Kim, and Ho Jeong Kwon. 2020. "Autophagic Inhibition via Lysosomal Integrity Dysfunction Leads to Antitumor Activity in Glioma Treatment" Cancers 12, no. 3: 543. https://doi.org/10.3390/cancers12030543
APA StyleHwang, H. -Y., Cho, Y. S., Kim, J. Y., Yun, K. N., Yoo, J. S., Lee, E., Kim, I., & Kwon, H. J. (2020). Autophagic Inhibition via Lysosomal Integrity Dysfunction Leads to Antitumor Activity in Glioma Treatment. Cancers, 12(3), 543. https://doi.org/10.3390/cancers12030543