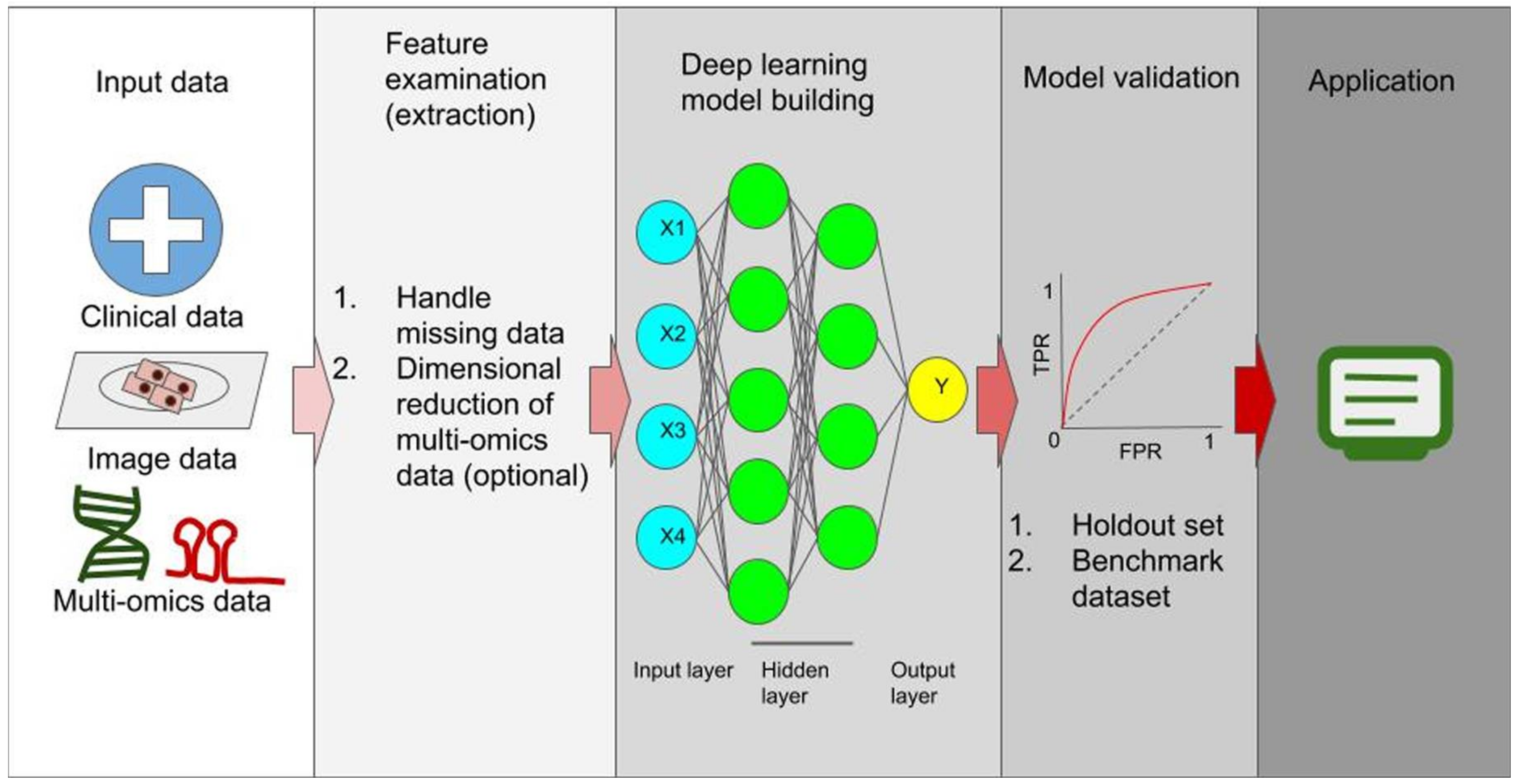
The Application of Deep Learning in Cancer Prognosis Prediction


Abstract
:1. Current Development in Cancer Prognosis Prediction
2. Overview of Deep Learning
3. Current Application of Deep Learning in Cancer Prognosis
3.1. NN Models with no Feature Extraction
3.2. Feature Extraction from Gene Expression Data to Build Fully Connected NNs
3.3. CNN-Based Models
4. Challenges in the Application of Deep Learning in Cancer Prognosis
5. Conclusions and Summary
6. Key Points
- Deep learning (DNN) models accept lots of data in different formats. It is a great tool to be used in cancer prognosis prediction since patient’s health data contain multi-source data.
- Using feature extraction could be one way to efficiently extract data from multi-omics data to train neural networks and possibly improve cancer prognosis prediction.
- Fully connected NN and CNN models have been tested in a number of studies to predict cancer prognosis and showed good performance.
- Current deep learning models in cancer prognosis studies still require further testing and validation in larger datasets.
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, F.E.; Vos, P.W.; Holbert, D. Modeling survival in colon cancer: A methodological review. Mol. Cancer 2007, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, K.Y.; Ma, J.; Fisher, J.; Kreisberg, J.F.; Raphael, B.J.; Ideker, T. Visible Machine Learning for Biomedicine. Cell 2018, 173, 1562–1565. [Google Scholar]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation From Incomplete Observations. Publ. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- NW, M. Evaluation of Survival Data and Two New Rank Order Statistics Arising In Its Consideration. Cancer Chemother. Rep. 1966, 50, 163–170. [Google Scholar]
- Peto, R.; Peto, J. Asymptotically efficient rank invariant test procedures. J. R. Stat. Soc. Ser. A 1972, 135, 185–198. [Google Scholar] [CrossRef]
- Harrington, D. Linear Rank Tests in Survival Analysis. In Encyclopedia of Biostatistics, 2nd ed.; Armitage, P., Colton, T., Eds.; Wiley: New York, NY, USA, 2005. [Google Scholar]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Alexe, G.; Dalgin, G.; Ganesan, S.; Delisi, C.; Bhanot, G. Analysis of breast cancer progression using principal component analysis and clustering. J. Biosci. 2007, 32, 1027–1039. [Google Scholar] [CrossRef] [Green Version]
- Hemsley, P.A. An outlook on protein S-acylation in plants: What are the next steps? J. Exp. Bot. 2017. [Google Scholar] [CrossRef] [Green Version]
- Kretowska, M. Computational Intelligence in Survival Analysis. In Encyclopedia of Business Analytics and Optimization; IGI Global: Warsaw, Poland, 2014; pp. 491–501. [Google Scholar]
- Petalidis, L.P.; Oulas, A.; Backlund, M.; Wayland, M.T.; Liu, L.; Plant, K.; Happerfield, L.; Freeman, T.C.; Poirazi, P.; Collins, V.P. Improved grading and survival prediction of human astrocytic brain tumors by artificial neural network analysis of gene expression microarray data. Mol. Cancer Ther. 2008, 7, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, C.L.; Street, W.N.; Wolberg, W.H. Application of Artificial Neural Network-Based Survival Analysis on Two Breast Cancer Datasets. Amia. Annu. Symp. Proc. 2007, 11, 130–134. [Google Scholar]
- Van IJzendoorn, D.G.; Szuhai, K.; Briaire-de Bruijn, I.H.; Kostine, M.; Kuijjer, M.L.; Bovée, J.V. Machine learning analysis of gene expression data reveals novel diagnostic and prognostic biomarkers and identifies therapeutic targets for soft tissue sarcomas. PLoS Comput. Biol. 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N. The Cancer Genome Atlas Pan-Cancer analysis project. Chin. J. Lung Cancer 2013, 45, 1113–1120. [Google Scholar]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Chandran, U.R.; Medvedeva, O.P.; Michael, B.M.; Blood, P.D.; Anish, C.; Soumya, L.; Antonio, F.; Wong, K.F.; Lee, A.V.; Zhihui, Z. TCGA Expedition: A Data Acquisition and Management System for TCGA Data. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Haeussler, M.; Zweig, A.S.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Hinrichs, A.S.; Gonzalez, J.N. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 2018, 47, D853–D858. [Google Scholar] [CrossRef] [Green Version]
- Deng, M.; Brägelmann, J.; Kryukov, I.; Saraiva-Agostinho, N.; Perner, S. FirebrowseR: An R client to the Broad Institute’s Firehose Pipeline. Database J. Biol. Databases Curation 2017. [Google Scholar] [CrossRef] [Green Version]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [PubMed] [Green Version]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 13, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.G. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteva, A.; Robicquet, A.; Ramsundar, B.; Kuleshov, V.; DePristo, M.; Chou, K.; Cui, C.; Corrado, G.; Thrun, S.; Dean, J. A guide to deep learning in healthcare. Nat. Med. 2019, 25, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.S. The Origins of Logistic Regression. Soc. Sci. Electron. Publ. 2003. [Google Scholar] [CrossRef] [Green Version]
- Boser, B.E.; Guyon, I.M.; Vapnik, V.N. A Training Algorithm for Optimal Margin Classifiers. Proc. Fifth Annu. Workshop Comput. Learn. Theory 2008, 5, 144–152. [Google Scholar]
- Maron, M.E. Automatic Indexing: An Experimental Inquiry. J. ACM 1961, 8, 404–417. [Google Scholar] [CrossRef]
- Breiman, L.; Friedman, J.H.; Olshen, R.A. Classification and Regression Trees; Routledge: New York, NY, USA, 2017. [Google Scholar]
- Friedman, J.H. Stochastic gradient boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Breiman, L. Arcing the Edge; Technical Report; Statistics Department, University of California: Berkeley, CA, USA, 1997. [Google Scholar]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef] [Green Version]
- Sibson, R. SLINK: An optimally efficient algorithm for the single-link cluster method. Comput. J. 1973, 16, 30–34. [Google Scholar] [CrossRef]
- Defays, D. An efficient algorithm for a complete link method. Comput. J. 1977, 20, 364–366. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, S. Least squares quantization in PCM. IEEE Trans. Inf. Theory 1982, 28, 129–137. [Google Scholar] [CrossRef]
- MacQueen, J. Some methods for classification and analysis of multivariate observations. In Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability, Los Angeles, CA, USA, 21 June–18 July 1967; pp. 281–297. [Google Scholar]
- Blei, D.M.; Ng, A.Y.; Jordan, M.I. Latent dirichlet allocation. J. Mach. Learn. Res. 2003, 3, 993–1022. [Google Scholar]
- Pearson, K. Principal components analysis. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901, 6, 559. [Google Scholar] [CrossRef] [Green Version]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.S.; Dean, J. Distributed representations of words and phrases and their compositionality. Adv. Neural Inf. Process. Syst. 2013, 26, 3111–3119. [Google Scholar]
- Sutton, R.S.; Barto, A.G. Reinforcement Learning: An Introduction; MIT Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Hinton, G.E. Learning distributed representations of concepts. In ; , 1991. In Proceedings of the Eighth Annual Conference of the Cognitive Science Society, Hillsdale, NJ, USA, 7–10 August 1991; p. 12. [Google Scholar]
- Bengio, Y. Learning deep architectures for AI. Found. Trends® Mach. Learn. 2009, 2, 1–127. [Google Scholar] [CrossRef]
- Szegedy, C.; Liu, W.; Jia, Y.; Sermanet, P.; Reed, S.; Anguelov, D.; Erhan, D.; Vanhoucke, V.; Rabinovich, A. Going deeper with convolutions. In Proceedings of the 2015 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Boston, MA, USA, 7–12 June 2015. [Google Scholar]
- Simonyan, K.; Zisserman, A. Very deep convolutional networks for large-scale image recognition. arXiv 2014, arXiv:1409.1556. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Hilton Head Island, SC, USA, 15 June 2000; pp. 770–778. [Google Scholar]
- Chollet, F. Xception: Deep learning with depthwise separable convolutions. In Proceedings of the 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Honolulu, HI, USA, 21–26 July 2017; pp. 1251–1258. [Google Scholar]
- Jordan, M. Serial Order: A Parallel Distributed Processing Approach; Technical Report; California University: San Diego, CA, USA, 1986. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Farahbakhsh-Farsi, P.; Djalali, M.; Koohdani, F.; Saboor-Yaraghi, A.A.; Eshraghian, M.R.; Javanbakht, M.H.; Chamari, M.; Djazayery, A. Effect of omega-3 supplementation versus placebo on acylation stimulating protein receptor gene expression in type 2 diabetics. J. Diabetes. Metab. Disord. 2014, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poplin, R.; Varadarajan, A.V.; Blumer, K.; Liu, Y.; McConnell, M.V.; Corrado, G.S.; Peng, L.; Webster, D.R. Prediction of cardiovascular risk factors from retinal fundus photographs via deep learning. Nat. Biomed. Eng. 2018, 2, 158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yao, H.; Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing 2016, 184, 232–242. [Google Scholar] [CrossRef]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Biganzoli, E.; Boracchi, P.; Mariani, L.; Marubini, E. Feed forward neural networks for the analysis of censored survival data: A partial logistic regression approach. Stat. Med. 1998, 17, 1169–1186. [Google Scholar] [CrossRef]
- Cox, D.R. Regression models and life-tables. J. R. Stat. Soc. Ser. B 1972, 34, 187–202. [Google Scholar] [CrossRef]
- Joshi, R.; Reeves, C. Beyond the Cox model: Artificial neural networks for survival analysis part II. In Proceedings of the Eighteenth International Conference on Systems Engineering, Coventry, UK, 3–10 May 2003; pp. 179–184. [Google Scholar]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox-nnet: An artificial neural network method for prognosis prediction of high-throughput omics data. PLoS Comput. Biol. 2018, 14. [Google Scholar] [CrossRef]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar] [CrossRef]
- Jing, B.; Zhang, T.; Wang, Z.; Jin, Y.; Liu, K.; Qiu, W.; Ke, L.; Sun, Y.; He, C.; Hou, D. A deep survival analysis method based on ranking. Artif. Intell. Med. 2019, 98, 1–9. [Google Scholar] [CrossRef]
- Hao, J.; Kim, Y.; Kim, T.-K.; Kang, M. PASNet: Pathway-associated sparse deep neural network for prognosis prediction from high-throughput data. BMC Bioinform. 2018, 19, 510. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, A. Multi-view Factorization AutoEncoder with Network Constraints for Multi-omic Integrative Analysis. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Madrid, Spain, 3–6 December 2018. [Google Scholar]
- Meng, C.; Zeleznik, O.A.; Thallinger, G.G.; Kuster, B.; Gholami, A.M.; Culhane, A.C. Dimension reduction techniques for the integrative analysis of multi-omics data. Brief. Bioinform. 2016, 17, 628–641. [Google Scholar] [CrossRef]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef]
- Sun, D.; Wang, M.; Li, A. A multimodal deep neural network for human breast cancer prognosis prediction by integrating multi-dimensional data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhan, X.; Xiang, S.; Johnson, T.S.; Helm, B.; Yu, C.Y.; Zhang, J.; Salama, P.; Rizkalla, M.; Han, Z. SALMON: Survival Analysis Learning With Multi-Omics Neural Networks on Breast Cancer. Front. Genet. 2019, 10, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep Learning-Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. 2017, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, H.; Nakayama, K.I. A 23 gene–based molecular prognostic score precisely predicts overall survival of breast cancer patients. EBioMedicine 2019, 46, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, K. Normalized imqcm: An algorithm for detecting weak quasi-cliques in weighted graph with applications in gene co-expression module discovery in cancers. Cancer Inform. 2014, 13, CIN-S14021. [Google Scholar] [CrossRef] [Green Version]
- Steck, H.; Krishnapuram, B.; Dehing-oberije, C.; Lambin, P.; Raykar, V.C. On ranking in survival analysis: Bounds on the concordance index. In Proceedings of the Advances in Neural Information Processing Systems, Malvern, PA, USA, 8–10 December 2008; pp. 1209–1216. [Google Scholar]
- Esteva, A.; Kuprel, B.; Novoa, R.A.; Ko, J.; Swetter, S.M.; Blau, H.M.; Thrun, S. Dermatologist-level classification of skin cancer with deep neural networks. Nature 2017, 542, 115–118. [Google Scholar] [CrossRef]
- Levine, A.B.; Schlosser, C.; Grewal, J.; Coope, R.; Jones, S.J.M.; Yip, S. Rise of the Machines: Advances in Deep Learning for Cancer Diagnosis. Trends Cancer 2019, 5, 157–169. [Google Scholar] [CrossRef]
- Kather, J.N.; Krisam, J.; Charoentong, P.; Luedde, T.; Herpel, E.; Weis, C.-A.; Gaiser, T.; Marx, A.; Valous, N.A.; Ferber, D. Predicting survival from colorectal cancer histology slides using deep learning: A retrospective multicenter study. PLoS Med. 2019, 16. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Damodaran, K.; Soylemezoglu, A.C.; Uhler, C.; Shivashankar, G.V. Machine Learning for Nuclear Mechano-Morphometric Biomarkers in Cancer Diagnosis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Rajkomar, A.; Oren, E.; Chen, K.; Dai, A.M.; Hajaj, N.; Hardt, M.; Liu, P.J.; Liu, X.; Marcus, J.; Sun, M. Scalable and accurate deep learning with electronic health records. NPJ Digit. Med. 2018, 1, 18. [Google Scholar] [CrossRef]
- Shameer, K.; Johnson, K.W.; Yahi, A.; Miotto, R.; Li, L.; Ricks, D.; Jebakaran, J.; Kovatch, P.; Sengupta, P.P.; GELIJNS, S. Predictive modeling of hospital readmission rates using electronic medical record-wide machine learning: A case-study using Mount Sinai heart failure cohort. Pac. Symp. Biocomput. 2017, 22, 276–287. [Google Scholar]
- Elfiky, A.A.; Pany, M.J.; Parikh, R.B.; Obermeyer, Z. Development and application of a machine learning approach to assess short-term mortality risk among patients with cancer starting chemotherapy. JAMA Netw. Open 2018, 1. [Google Scholar] [CrossRef] [Green Version]
- Mathotaarachchi, S.; Pascoal, T.A.; Shin, M.; Benedet, A.L.; Rosa-Neto, P. Identifying incipient dementia individuals using machine learning and amyloid imaging. Neurobiol. Aging 2017, 59, 80. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Delving deep into rectifiers: Surpassing human-level performance on imagenet classification. In Proceedings of the 2015 IEEE International Conference on Computer Vision (ICCV), Santiago, Chile, 7–13 December 2015; pp. 1026–1034. [Google Scholar]
- Korfiatis, P.; Kline, T.L.; Lachance, D.H.; Parney, I.F.; Buckner, J.C.; Erickson, B.J. Residual Deep Convolutional Neural Network Predicts MGMT Methylation Status. J. Digit. Imaging 2017, 30, 622–628. [Google Scholar] [CrossRef]
- Han, L.; Kamdar, M. MRI to MGMT: Predicting Drug Efficacy for Glioblastoma Patients. Pac. Symp. Biocomput. Pac. Symp. Biocomput. 2018, 23, 331–338. [Google Scholar]
- Mobadersany, P.; Yousefi, S.; Amgad, M.; Gutman, D.A.; Barnholtz-Sloan, J.S.; Velázquez Vega, J.E.; Brat, D.J.; Cooper, L.A.D. Predicting cancer outcomes from histology and genomics using convolutional networks. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [Green Version]
- Bychkov, D.; Linder, N.; Turkki, R.; Nordling, S.; Kovanen, P.E.; Verrill, C.; Walliander, M.; Lundin, M.; Haglund, C.; Lundin, J. Deep learning based tissue analysis predicts outcome in colorectal cancer. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Courtiol, P.; Maussion, C.; Moarii, M.; Pronier, E.; Pilcer, S.; Sefta, M.; Manceron, P.; Toldo, S.; Zaslavskiy, M.; Le Stang, N. Deep learning-based classification of mesothelioma improves prediction of patient outcome. Nat. Med. 2019, 25, 1519–1525. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Z.; Rong, Y.; Zhou, B.; Bai, Y.; Wei, W.; Wang, M.; Guo, Y.; Tian, J. Deep learning provides a new computed tomography-based prognostic biomarker for recurrence prediction in high-grade serous ovarian cancer. Radiother. Oncol. 2019, 132, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Christopher, M.; Belghith, A.; Bowd, C.; Proudfoot, J.A.; Goldbaum, M.H.; Weinreb, R.N.; Girkin, C.A.; Liebmann, J.M.; Zangwill, L.M. Performance of Deep Learning Architectures and Transfer Learning for Detecting Glaucomatous Optic Neuropathy in Fundus Photographs. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Sohn, J.H.; Kawczynski, M.G.; Trivedi, H.; Harnish, R.; Jenkins, N.W.; Lituiev, D.; Copeland, T.P.; Aboian, M.S.; Mari Aparici, C. A Deep learning model to predict a diagnosis of alzheimer disease by using 18F-FDG PET of the brain. Radiology 2018, 290, 456–464. [Google Scholar] [CrossRef]
- Raghu, M.; Zhang, C.; Kleinberg, J.; Bengio, S. Transfusion: Understanding transfer learning for medical imaging. Adv. Neural Inf. Process. Syst. 2019, 3342–3352. [Google Scholar]
- Vinyals, O.; Blundell, C.; Lillicrap, T.; Wierstra, D. Matching networks for one shot learning. Adv. Neural Inf. Process. Syst. 2016, 3630–3638. [Google Scholar]
- Triantafillou, E.; Zemel, R.; Urtasun, R. Few-shot learning through an information retrieval lens. Adv. Neural Inf. Process. Syst. 2017, 2255–2265. [Google Scholar]
- Buuren, S.V.; Groothuis-Oudshoorn, K. mice: Multivariate imputation by chained equations in R. J. Stat. Softw. 2010, 1–68. [Google Scholar] [CrossRef] [Green Version]
- Rendleman, M.C.; Buatti, J.M.; Braun, T.A.; Smith, B.J.; Nwakama, C.; Beichel, R.R.; Brown, B.; Casavant, T.L. Machine learning with the TCGA-HNSC dataset: Improving usability by addressing inconsistency, sparsity, and high-dimensionality. BMC Bioinform. 2019, 20, 339. [Google Scholar] [CrossRef]
- Raghunathan, T.E.; Lepkowski, J.M.; Van Hoewyk, J.; Solenberger, P. A multivariate technique for multiply imputing missing values using a sequence of regression models. Surv. Methodol. 2001, 27, 85–96. [Google Scholar]
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef]
- Ren, K.; Qin, J.; Zheng, L.; Yang, Z.; Zhang, W.; Qiu, L.; Yu, Y. Deep recurrent survival analysis. In Proceedings of the AAAI Conference on Artificial Intelligence, New York, NY, USA, 7–12 February 2020; pp. 4798–4805. [Google Scholar]
- Deng, J.; Russakovsky, O.; Krause, J.; Bernstein, M.S.; Berg, A.; Fei-Fei, L. Scalable multi-label annotation. In Proceedings of the SIGCHI Conference on Human Factors in Computing Systems, Toronto, ON, Canada, 26–27 April 2014; pp. 3099–3102. [Google Scholar]
- Deng, J.; Dong, W.; Socher, R.; Li, L.-J.; Li, K.; Fei-Fei, L. Imagenet: A large-scale hierarchical image database. In Proceedings of the 2009 IEEE Conference on Computer Vision and Pattern Recognition, Miami, FL, USA, 20–25 June 2009; pp. 248–255. [Google Scholar]
- Russakovsky, O.; Deng, J.; Su, H.; Krause, J.; Satheesh, S.; Ma, S.; Huang, Z.; Karpathy, A.; Khosla, A.; Bernstein, M. Imagenet large scale visual recognition challenge. Int. J. Comput. Vis. 2015, 115, 211–252. [Google Scholar] [CrossRef] [Green Version]
- Kermany, D.S.; Goldbaum, M.; Cai, W.; Valentim, C.C.; Liang, H.; Baxter, S.L.; McKeown, A.; Yang, G.; Wu, X.; Yan, F. Identifying medical diagnoses and treatable diseases by image-based deep learning. Cell 2018, 172, 1122–1131. [Google Scholar] [CrossRef]
- Goswami, C.P.; Nakshatri, H. PROGgene: Gene expression based survival analysis web application for multiple cancers. J. Clin. Bioinform. 2013, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Elfilali, A.; Lair, S.; Verbeke, C.; La, R.P.; Radvanyi, F.; Barillot, E. ITTACA: A new database for integrated tumor transcriptome array and clinical data analysis. Nucleic Acids Res. 2006, 34, D613–D616. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xie, L.; Dang, Y.; Sun, X.; Xie, T.; Guo, J.; Han, Y.; Yan, Z.; Zhu, W.; Wang, Y. OSlms: A Web Server to Evaluate the Prognostic Value of Genes in Leiomyosarcoma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, F.; Lv, J.; Xin, J.; Xie, L.; Zhu, W.; Tang, Y.; Li, Y.; Zhao, X.; Wang, Y. Interactive online consensus survival tool for esophageal squamous cell carcinoma prognosis analysis. Oncol. Lett. 2019, 18, 1199–1206. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Q.; Yang, M.; Yuan, Q.; Dang, Y.; Sun, X.; An, Y.; Dong, H.; Xie, L.; Zhu, W. OSblca: A Web Server for Investigating Prognostic Biomarkers of Bladder Cancer Patients. Front. Oncol. 2019, 9, 466. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Wang, Q.; Sun, X.; Ban, B.; Lu, Z.; Dang, Y.; Xie, L.; Zhang, L.; Li, Y.; Guo, X. OSbrca: A Web Server for Breast Cancer Prognostic Biomarker Investigation with Massive Data from tens of Cohorts. Front. Oncol. 2019, 9, 1349. [Google Scholar] [CrossRef]
- Xie, L.; Wang, Q.; Dang, Y.; Ge, L.; Sun, X.; Li, N.; Han, Y.; Yan, Z.; Zhang, L.; Li, Y. OSkirc: A web tool for identifying prognostic biomarkers in kidney renal clear cell carcinoma. Future Oncol. 2019, 15, 3103–3110. [Google Scholar] [CrossRef]
- Xie, L.; Wang, Q.; Nan, F.; Ge, L.; Dang, Y.; Sun, X.; Li, N.; Dong, H.; Han, Y.; Zhang, G. OSacc: Gene Expression-Based Survival Analysis Web Tool For Adrenocortical Carcinoma. Cancer Manag. Res. 2019, 11, 9145–9152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, Q.; Li, N.; Ge, L.; Yang, M.; An, Y.; Zhang, G.; Dong, H.; Ji, S.; Zhu, W. OSuvm: An interactive online consensus survival tool for uveal melanoma prognosis analysis. Mol. Carcinog. 2020, 59, 56–61. [Google Scholar] [CrossRef] [PubMed]
- McMahan, H.B.; Moore, E.; Ramage, D.; Hampson, S. Communication-efficient learning of deep networks from decentralized data. arXiv 2016, arXiv:1602.05629. [Google Scholar]
- Su, H.; Deng, J.; Fei-Fei, L. Crowdsourcing annotations for visual object detection. In Proceedings of the Workshops at the Twenty-Sixth AAAI Conference on Artificial Intelligence, Toronto, ON, Canada, 22–26 July 2012. [Google Scholar]
- Avati, A.; Jung, K.; Harman, S.; Downing, L.; Ng, A.; Shah, N.H. Improving palliative care with deep learning. BMC Med. Inform. Decis. Mak. 2018, 18, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfiky, A.A.; Elshemey, W.M. Molecular dynamics simulation revealed binding of nucleotide inhibitors to ZIKV polymerase over 444 nanoseconds. J. Med. Virol. 2018, 90, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Horng, S.; Sontag, D.A.; Halpern, Y.; Jernite, Y.; Shapiro, N.I.; Nathanson, L.A. Creating an automated trigger for sepsis clinical decision support at emergency department triage using machine learning. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Henry, K.E.; Hager, D.N.; Pronovost, P.J.; Saria, S. A targeted real-time early warning score (TREWScore) for septic shock. Sci. Transl. Med. 2015, 7, ra122–ra299. [Google Scholar] [CrossRef] [Green Version]
- Culliton, P.; Levinson, M.; Ehresman, A.; Wherry, J.; Steingrub, J.S.; Gallant, S.I. Predicting severe sepsis using text from the electronic health record. arXiv 2017, arXiv:1711.11536. [Google Scholar]
- Oh, J.; Makar, M.; Fusco, C.; McCaffrey, R.; Rao, K.; Ryan, E.E.; Washer, L.; West, L.R.; Young, V.B.; Guttag, J. A generalizable, data-driven approach to predict daily risk of Clostridium difficile infection at two large academic health centers. Infect. Control Hosp. Epidemiol. 2018, 39, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Miotto, R.; Li, L.; Kidd, B.A.; Dudley, J.T. Deep patient: An unsupervised representation to predict the future of patients from the electronic health records. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, Y.; Jiang, Y.; Sun, Y.; Zhang, Y.-J.; Luo, P. Clinical assistant diagnosis for electronic medical record based on convolutional neural network. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- De Langavant, L.C.; Bayen, E.; Yaffe, K. Unsupervised machine learning to identify high likelihood of dementia in population-based surveys: Development and validation study. J. Med. Internet Res. 2018, 20. [Google Scholar] [CrossRef]


{kind=link}
| Publication a | Type of Cancer | Type of Data | Sample Size | Methods | Architecture | Outputs | Hyperparameters | Validation | NN Model Performance |
|---|---|---|---|---|---|---|---|---|---|
| Joshi et al., 2006 [58] | Melanoma | Clinical data of tumors | 1946 (1160 females and 786 males) | 3 layers NN | Normalized input | Survival time | Sigmoid activation | Not reported | Achieved similar performance as Cox and Kaplan Meier statistical methods |
| Chi et al., 2007 [14] | Breast cancer | Cell images to measure 30 nuclear features | Dataset 1: 198 cases; Dataset 2: 462 cases | 3 layers NN | 30 input nodes, 20 hidden nods | Survival time | Epoch = 1000, Sigmoid activation | 10 fold cross validation | As good as conventional methods |
| Petalidis et al., 2008 [13] | Astrocytic brain tumor | A list of genes expression from microarray data | 65 | A single layer perceptron and an output (multiple binary models) | Number of inputs equals to the number of classifier genes in different models | Tumor grades | Lr 1 = 0.05, Epoch = 100 | Leave-one-out cross validation | 44, 9 and 7 probe sets have achieved 93.3%, 84.6%, and 95.6% validation success rates, respectively. |
| Ching et al., 2018 [59] | 10 types of cancer | TCGA gene expression data, clinical data and survival data | 5031 | NN | Input normalization and log-transformed, 0–2 hidden layers (143 nodes) | Survival time | L1, L2, or MCP 2 regularization, tanh activation for hidden layer(s), dropout, Cox regression as output layer | 5-fold cross validation | Similar or in some cases better performance than Cox-PH, Cox-boosting or RF |
| Katzman et al. 2018 [60] | Breast cancer | METABRIC 3, 4 genes data and clinical information, GBSG 4: clinical data | METABRIC: 1980, GBSG: 1546 training, 686 testing | NN | METABRIC: 1 dense layer, 41 nodes GBSG: patients clinical information, 1 dense layer, 8 nodes | Survival | SELU 5 activation, Adam optimizer, dropout, LR decay, momentum | 20% of METABRIC patients used as test set GBSG has split test dataset | C-index: 0.654 for METABRIC and 0.676 for GBSG, both are better than CoxPH |
| Jing et al. 2019 [61] | Breast cancer, nasopharyngeal carcinoma | METABRIC, GBSG, NPC 7: 8–9 clinical features | METABRIC: 1980 NPC: 4630 | DNN | METABRIC: 4 hidden layers, 45 nodes of each; GBSG: 3 hidden layers, 84, 84 and 70 nodes, respectively NPC, 3 layers, 120 nodes of each layer in model 1, 108, 108 and 90 nodes respectively in model 2. | Survival | ELU 6, dropout, L1 and L2, momentum, LR decay, batch size. Loss function equals to mean square error and a pairwise ranking loss | After removed patients with missing data, 20% used as test set | C-index: 0.661 for METABRIC and 0.688 for GBSG, both are better than CoxPH and DeepSurv. c-index ranges 0.681–0.704 depends on input data for NPC, better than CoxPH. |
| Publication a | Type of Cancer | Type of Data | Sample Size | Methods Used in Feature Extraction | Architecture | Outputs | Hyperparameters | Validation | NN Model Performance |
|---|---|---|---|---|---|---|---|---|---|
| Sun et al., 2018 [66] | Breast cancer | Gene expression profile, CAN 1 profile and clinical data | 1980 (1489 LTS 2, 491 non-LTS) | mRMR (extracted 400 features from gene expression and 200 features from CNA) | 4 hidden layers (1000, 500, 500, and 100 nodes, respectively) | Survival time | Lr 3 = 1e–3, Tanh activation, epoch 10–100, batch size = 64 | 10-fold cross validation | ROC4: 0.845 (better than SVM, RF5, and LR 6), Sp 7: 0.794–0.826, Pre 8: 0.749–0.875, Sn 9: 0.2–0.25, Mcc10: 0.356–0.486 |
| Huang et al., 2019 [67] | Breast cancer | mRNA, miRNA, CNB 11, TMB 12, clinical data | 583 (80% for training, 20% for testing in each fold of cross validation) | lmQCM 13, Epigengene matrix to extract 57 dimensions from mRNA data and 12 dimensions from miRNA data | Hybrid network, mRNA and miRNA dimension reduction inputs have 1 hidden layer (8 and 4 nodes, respectively), CNB, TMB and clinical data have no hidden layer | Survival time | Adam optimizer, LASSO 14 regularization, Epoch = 100, sigmoid activation, Cox regression as output, batch size = 64 | 5-fold cross validation | Multi-omics data network reached a median c-index15 of 0.7285 |
| Hao et al., 2018 [62] | Glioblastoma multiforme | Gene expression (TCGA), pathway (MsigDB 16) | 475 (376 non-LTS, 99 LTS) | Pathway based analysis (12,024 genes from mRNA data to 574 pathways and 4359 genes) | 4 layers NN: gene layer—pathway layer—hidden layer—output | Survival time | Lr = 1e−4, L2 = 3e−4, dropout, softmax output | 5-fold validation | AUC17 = 0. 66 ± 0.013, F1 score = 0.3978 ± 0.016 |
| Chaudhary et al., 2018 [68] | Liver cancer | mRNA, miRNA, methylation data, and clinical data (TCGA) | 360 samples training, (5 additional cohorts, 230, 221, 166, 40 and 27 samples for validation) | Autoencoder unsupervised NN to extract 100 features from mRNA, miRNA and methylation data | 3 hidden layers NN (500, 100, 500 nodes, respectively) and a bottleneck layer | Feature reduction | Epoch = 10, Dropout = 0.5, SGD 18 | Not reported | NN outputs were used for K means clustering. |
| Shimizu and Nakayama, 2019, [69] | Breast cancer | METABRIC 19 | 1903 (METABRIC, 952 samples for training) | Select 23 genes by statistical methods | 3 layers NN | Survival time | Lr = 0.001, Epoch = 1000, Cross entropy for loss function Relu activation for hidden nodes, softmax function for output layer | 951 samples from METABRIC | NN node weights were used to calculate a mPS20 |
| Publication a | Type of Cancer | Type of Data | Sample Size | Architecture | Outputs | Hyperparameters | Validation | NN Model Performance |
|---|---|---|---|---|---|---|---|---|
| Korfiatis et al., 2017 [81] | Glioblastoma multiforme | MRI images | 155 (66 methylated and 89 unmethylated tumors) Training: 7856 images (934 methylated, 1621 unmethylated, 5301 no tumor) Testing: 2612 images (250 methylated, 335 unmethylated, 2027 no tumor) | Base model: ResNet18 ResNet34 ResNet50 | 3 classes, methylated, unmethylated, or no tumor | Lr1 = 0.01, mini Batch = 32, momentum = 0.5, weight decay = 0.1, Relu activation, Epoch = 50, SGD2 as optimizer, batch normalization | Stratified cross-validation | ResNet50 based model validation dataset performance: Accuracy = 94.9%, Precision = 96%, Recall = 95% ResNet34 Accuracy = 80.72%, Precision = 93%, Recall = 81%, ResNet18 Accuracy = 76.75%, Precision:80%, Recall = 77% |
| Han et al., 2018 [81] | Glioblastoma multiforme | MRI images | 458,951 image frames from 5235 MRI scans of 262 patients (TCIA3) | 3 convolutional layers, 2 fully connected layers, 1 bi-directional GRU4 layer (RNN), 1 fully connected layer, softmax output | 2 classes (positive and negative methylation status) | Data augmentation, (rotation and flipping, 90-fold increase of the dataset), Lr = 5e−6 – 5e−1, dropout (0–0.5), Adam optimizer, Epoch = 10, L2 regularization, batch norm, relu activation | Validation set reached a precision of 0.67, an AUC of 0.56. | Training data set obtained 0.97 accuracy. 0.67 and 0.62 accuracies on the validation and test set, respectively |
| Mobadersany et al., 2018 [83] | Low grade glioma and glioblastoma | H&E images, genomics data, clinical data | 769 gliomas from TCGA, containing genomics data (IDH mutation and 1 p/19 q codeletion), clinical data and 1061 slides. | VGG19 is the base model and cox regression used as output, Built 2 models with or without genomics data | Survival | Data augmentation, Lr = 0.001, epoch = 100, exponential learning decay | Monte Carlo cross-validation | SCNN median c-index is 0.754, GSCNN (adding IDH mutation and 1 p/19 q codeletion as features) improved the median c-index to 0.801 |
| Kather et al., 2019 [74] | Colorectal cancer | H&E tissue slides | Training set (tissue): 86 H&E slides to create 100,000 image patches Testing set (tissue): 25 H&E slides of 7180 image patches Training set (OS5): 862 H&E slides from 500 TCGA patients Validation set (OS): 409 H&E slides from 409 DACHS patients | Base models: VGG19, AlexNet, GoogLeNet, SqueezeNet and Resnet50, add an output softmax layer | 9 tissue type classification | Lr = 3e −4, Iteration = 8, Batch size = 360, softmax function | An independent cohort of 409 samples | VGG19 gets the best results, 94–99% accuracy in tissue class prediction |
| Bychkov et al., 2018 [84] | Colorectal cancer | H&E images of tumor tissue microarray | 420 patients (equal number of survived or died within five years after diagnosis) | VGG16 to generate a 16 × 16 feature from input data, followed with 3 layers LSTM 6 (264, 128 and 64 LSTM cells, respectively) | Survival | Default hyperparameters in VGG16, LSTM used hyperbolic tangent as activation, binary cross entropy loss function, Adadelta optimizer | 60 samples for validation, 140 samples for testing | CNN + LSTM model reached an AUC 7 of 0.69, better than CNN + SVM, CNN + LR 8, or CNN + NB 9 |
| Courtiol et al., 2019 [85] | Mesothelioma | H&E slides | 2981 patient slides (MESOPATH/MESOBANK, 2300 training, 681 testing) Validation: 56 patients (TCGA) | Divided each slide to up to 10,000 tiles as input data 3 classes of each tile: epithelioid, sarcomatoid or biphasic. ResNet50 for feature extraction | Survival | Multi-layer perceptron with sigmoid activation, Autoencoder | 56 patient slides | MesoNet outperformed histology-based classification but no better than a linear regression based model (Meanpool) |
| Wang et al., 2019 [86] | High grade serous ovarian cancer | CT scanning venous phase images | Feature learning cohort: 8917 CT images from 102 patients | Five convolutional layers (24, 16, 16, 16, 16 filters, respectively) | 16 dimensional feature vector | Batch normalization, average pooling between adjacent convolutional layers | Not reported | CNN outputs were used to build Cox-PH model |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Xie, L.; Han, J.; Guo, X. The Application of Deep Learning in Cancer Prognosis Prediction. Cancers 2020, 12, 603. https://doi.org/10.3390/cancers12030603
Zhu W, Xie L, Han J, Guo X. The Application of Deep Learning in Cancer Prognosis Prediction. Cancers. 2020; 12(3):603. https://doi.org/10.3390/cancers12030603
Chicago/Turabian StyleZhu, Wan, Longxiang Xie, Jianye Han, and Xiangqian Guo. 2020. "The Application of Deep Learning in Cancer Prognosis Prediction" Cancers 12, no. 3: 603. https://doi.org/10.3390/cancers12030603
APA StyleZhu, W., Xie, L., Han, J., & Guo, X. (2020). The Application of Deep Learning in Cancer Prognosis Prediction. Cancers, 12(3), 603. https://doi.org/10.3390/cancers12030603