Glucocorticoids Promote the Onset of Acute Experimental Colitis and Cancer by Upregulating mTOR Signaling in Intestinal Epithelial Cells


Abstract
:1. Introduction
2. Material and methods
2.1. Mice
2.2. Experimental Colitis and Histological Analyses
2.3. Flow Cytometry
2.4. Quantitative RT-PCR
2.5. Quantification of Tissue Cytokine Levels
2.6. Immunoblot Analysis
2.7. Statistical Analysis
3. Results
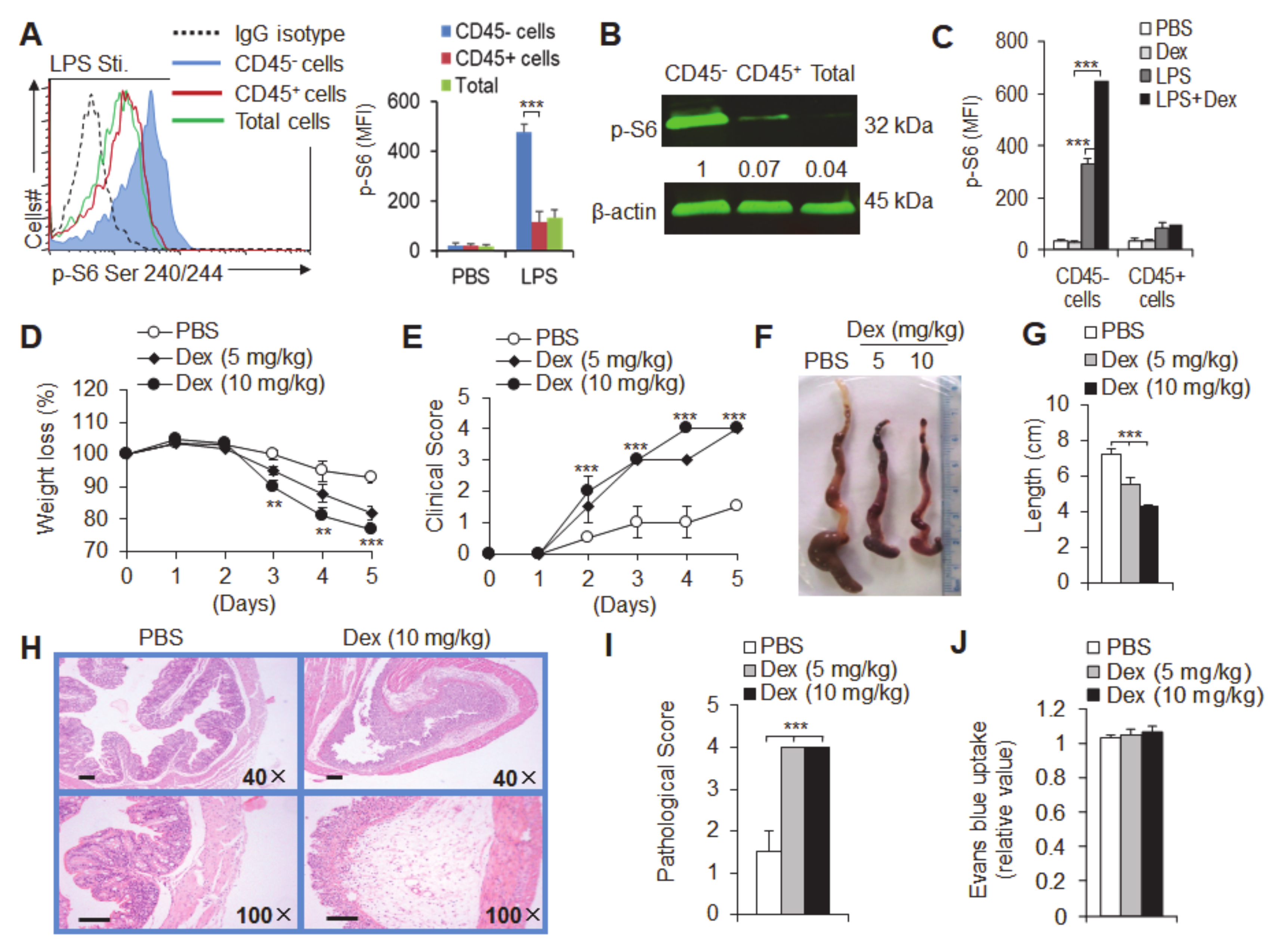
3.1. Dexamethasone (Dex) Treatment Specifically Upregulates mTOR Signaling in Intestinal Epithelial Cells in Acute Experimental Ulcerative Colitis
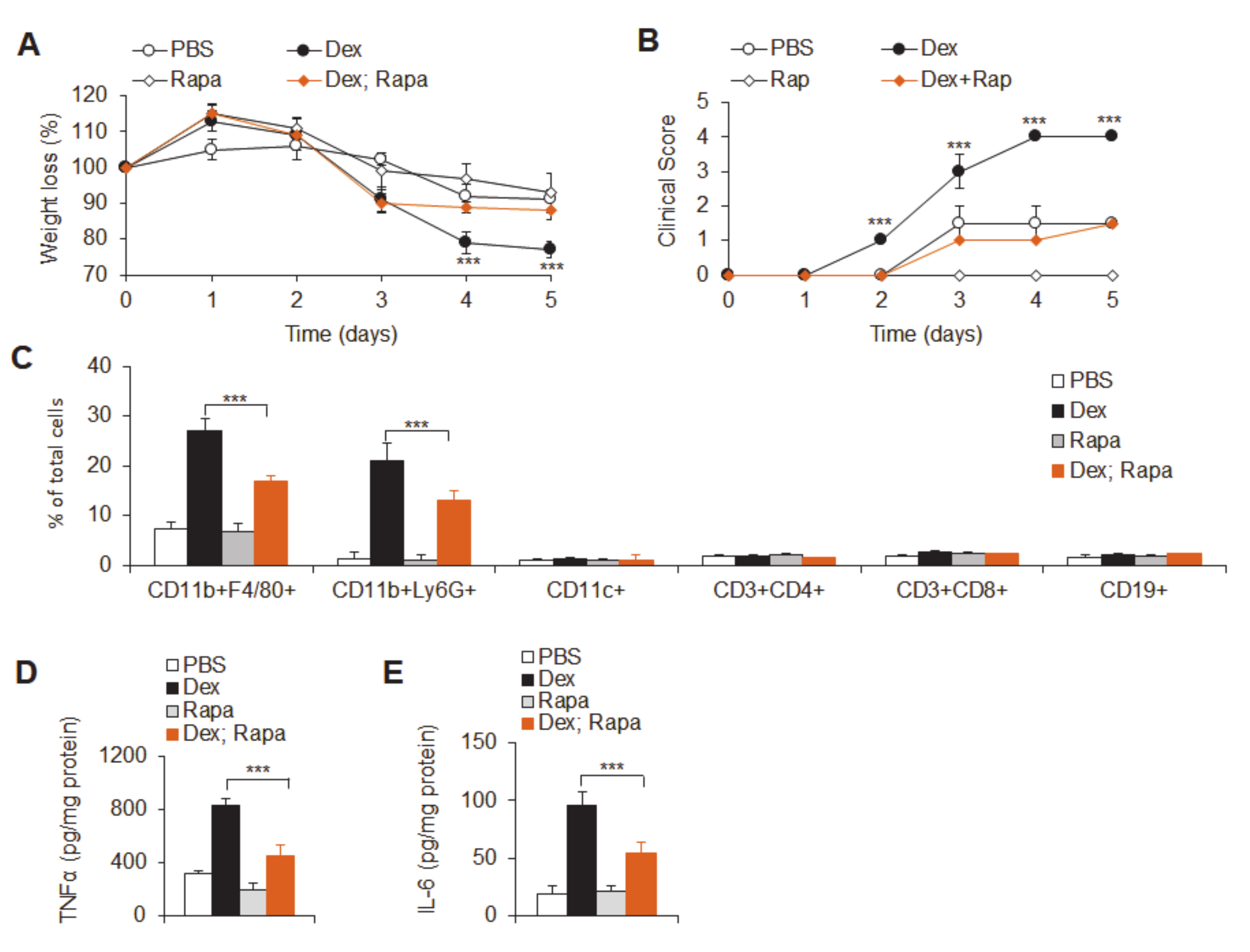
3.2. Dex Treatment Potentiates Acute Experimental Ulcerative Colitis
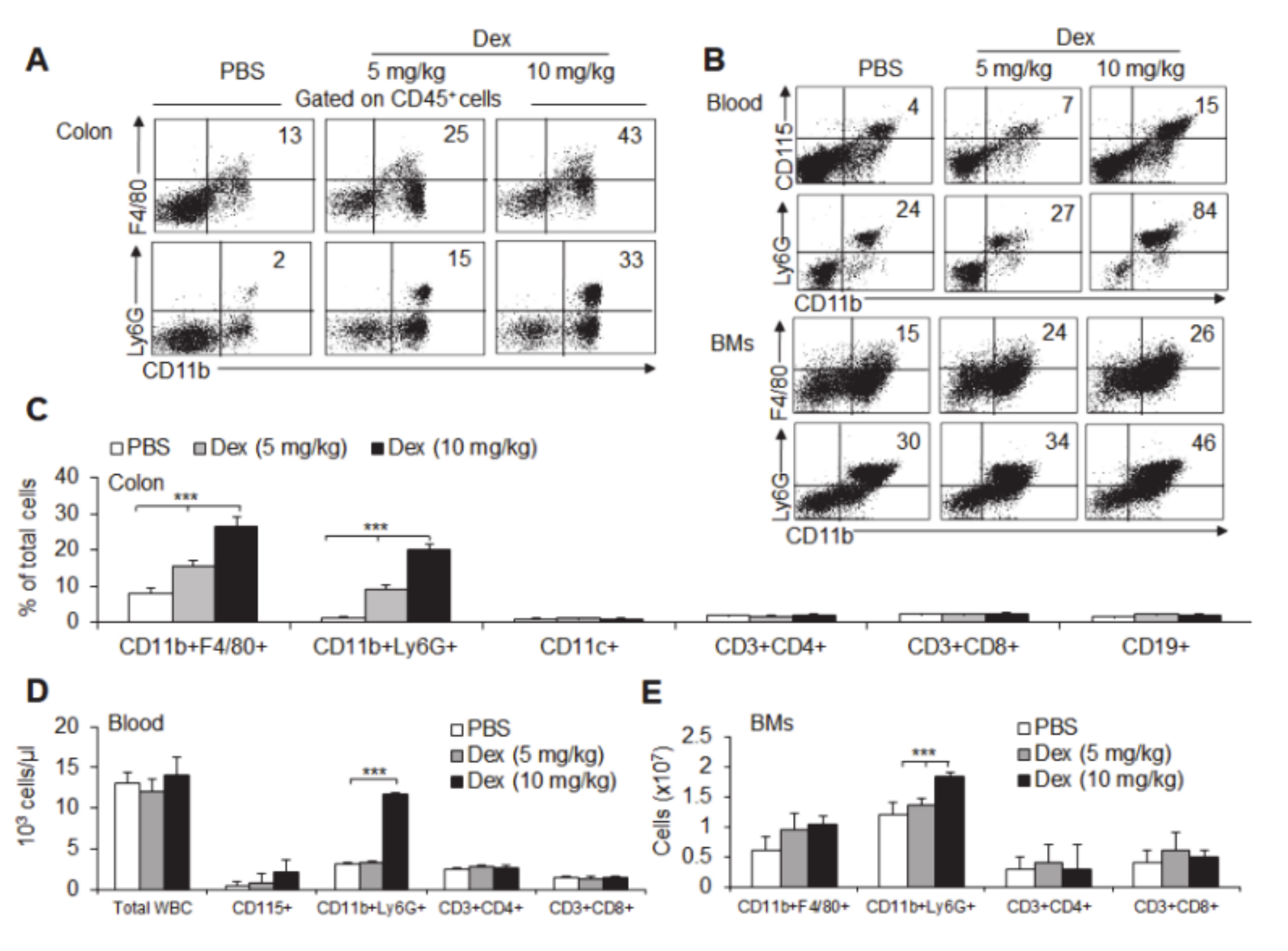
3.3. Dex-Treated Mice Had Increased Mucosal Immune Cell Infiltration
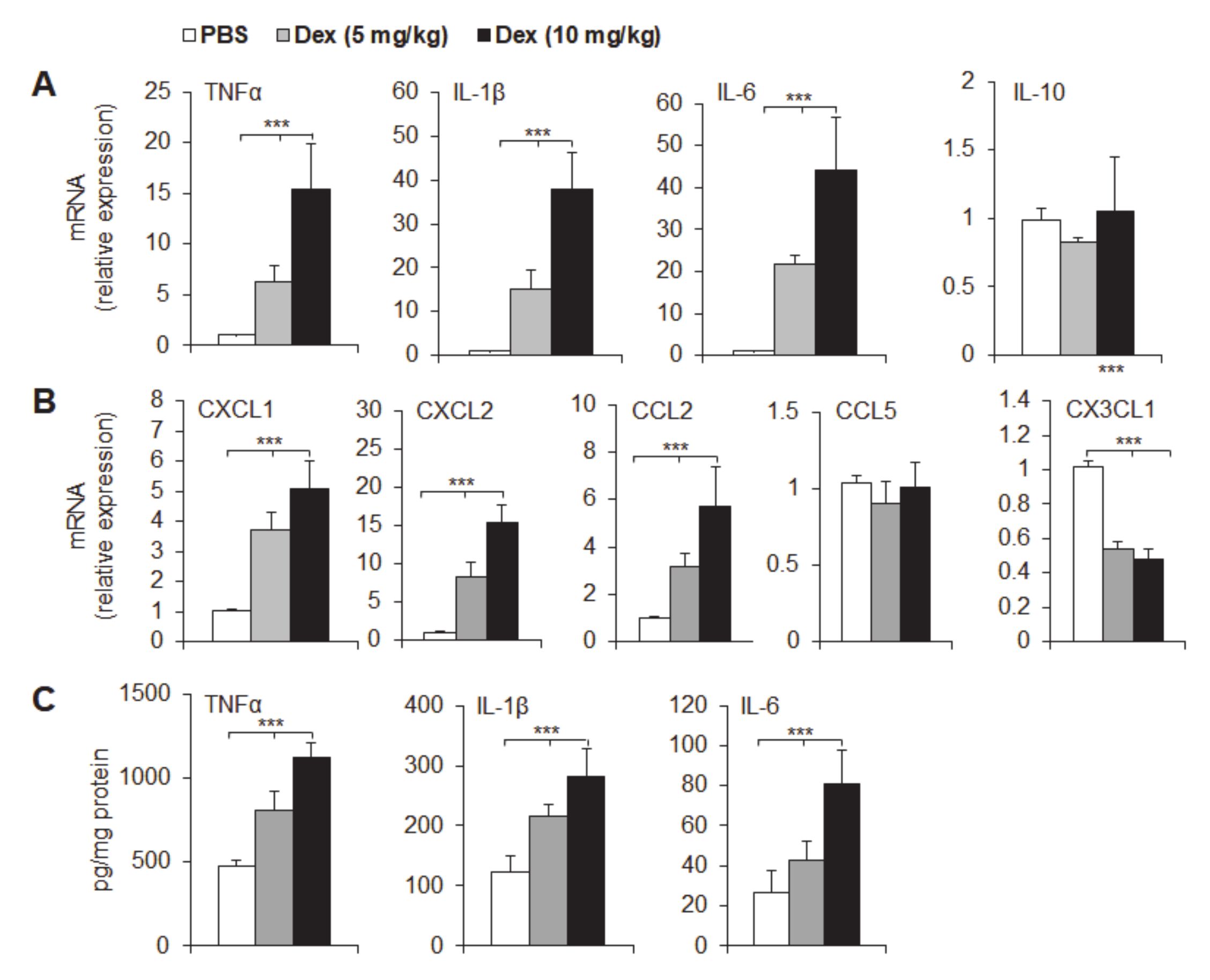
3.4. Dex Treatment Potentiated Mucosal Expression of Pro-Inflammatory Cytokines and Chemokines
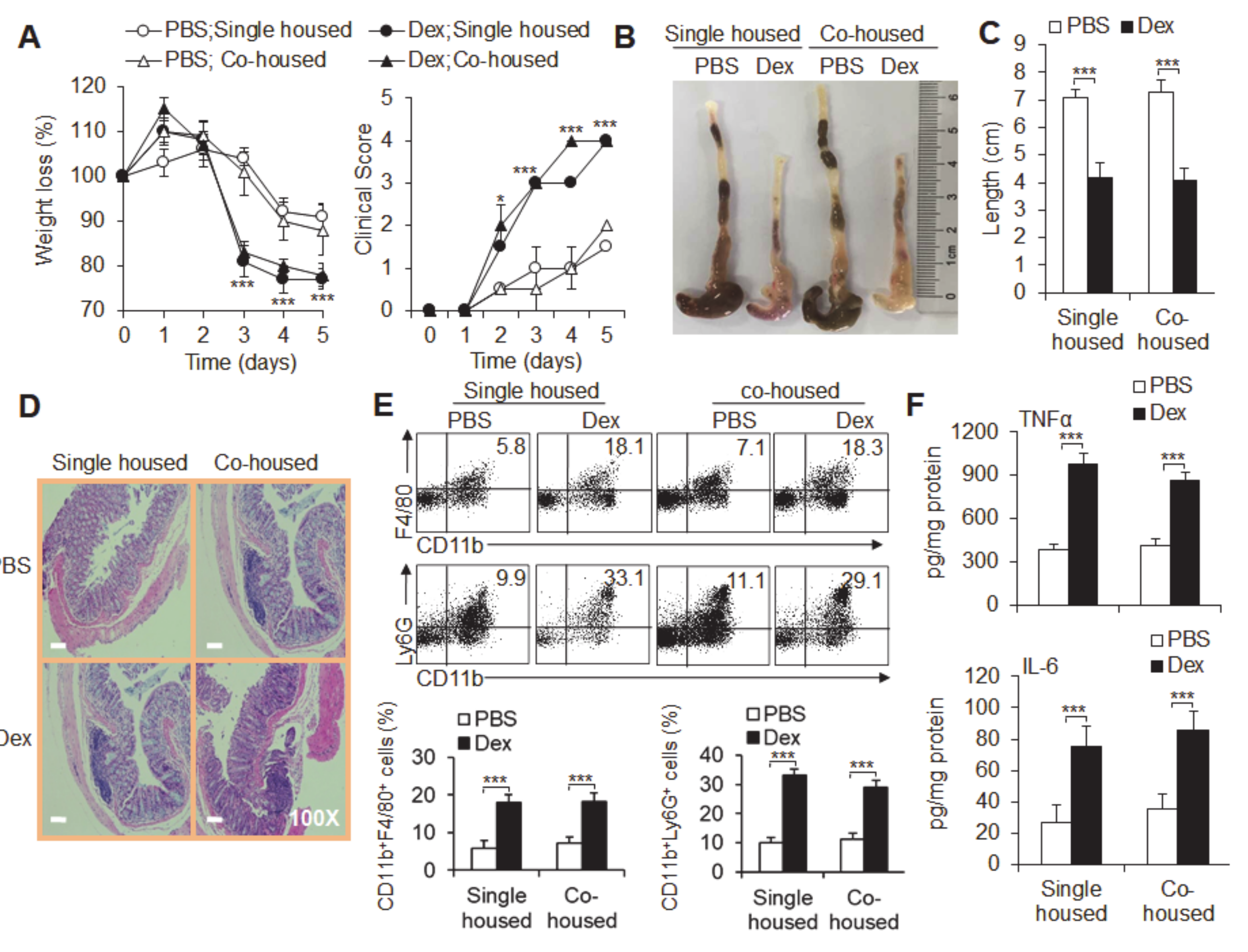
3.5. The Microbiota Is Not Required for Dex-Induced Severe Acute Ulcerative Colitis
3.6. mTOR, but not GR, Is Required for Dex-Induced Severe Ulcerative Colitis
3.7. The Dex-mTOR Signal Axis Potentiated Colitis-Associated Cancer
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Hindryckx, P.; Jairath, V.; D’Haens, G. Acute severe ulcerative colitis: From pathophysiology to clinical management. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, G.; Vermeire, S.; Rutgeerts, P. Management of acute severe ulcerative colitis. Gut 2011, 60, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Leake, I. IBD: Treatment for acute severe ulcerative colitis. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 436. [Google Scholar]
- Lynch, R.W.; Soane, T.; Gibson, R.; Pal, S.; Lees, C.W. Bilateral lower limb weakness in acute severe ulcerative colitis. Lancet 2016, 388, 101–102. [Google Scholar] [CrossRef]
- Halfvarson, J.; Jarnerot, G. Treatment of choice for acute severe steroid-refractory ulcerative colitis is remicade. Inflamm. Bowel Dis. 2009, 15, 143–145. [Google Scholar] [CrossRef]
- Jain, S.; Ahuja, V.; Limdi, J.K. Optimal management of acute severe ulcerative colitis. Postgrad. Med. J. 2019, 95, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Whaley, K.G.; Rosen, M.J. Contemporary medical management of acute severe ulcerative colitis. Inflamm. Bowel Dis. 2019, 25, 56–66. [Google Scholar] [CrossRef]
- Dalton, H.R.; Jewell, D.P. The management of acute severe ulcerative colitis. Ann. Med. 1991, 23, 389–391. [Google Scholar] [CrossRef]
- Cohen, R.D. How should we treat severe acute steroid-refractory ulcerative colitis? Inflamm. Bowel Dis. 2009, 15, 150–151. [Google Scholar] [CrossRef]
- Ventham, N.T.; Kalla, R.; Kennedy, N.A.; Satsangi, J.; Arnott, I.D. Predicting outcomes in acute severe ulcerative colitis. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Sandle, G.I.; Hayslett, J.P.; Binder, H.J. Effect of glucocorticoids on rectal transport in normal subjects and patients with ulcerative-colitis. Gut 1986, 27, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, O.H.; Vainer, B. Effects of glucocorticoids on circulating concentrations of soluble intercellular adhesion molecule-1 (ICAM-1) in ulcerative colitis. Gastroenterology 2001, 120, A186–A186. [Google Scholar] [CrossRef]
- Pacha, J.; Ergang, P.; Vagnerova, K.; Vytackova, K.; Miksik, I. Colitis modulates local metabolism of glucocorticoids not only in colon but also in secondary lymphoid organs. Inflamm. Res. 2011, 60, 240–240. [Google Scholar]
- Cakir, B.; Bozkurt, A.; Ercan, F.; Yegen, B.C. The anti-inflammatory effect of leptin on experimental colitis: Involvement of endogenous glucocorticoids. Gastroenterology 2003, 124, A493–A493. [Google Scholar] [CrossRef]
- Richter, J.M.; Kushkuley, S.; Barrett, J.A.; Oster, G. Treatment of new-onset ulcerative colitis and ulcerative proctitis: a retrospective study. Aliment Pharmacol Ther. 2012, 36, 248–256. [Google Scholar]
- Zimmerman, M.J.; Jewell, D.P. Cytokines and mechanisms of action of glucocorticoids and aminosalicylates in the treatment of ulcerative colitis and Crohn’s disease. Aliment. Pharmacol. Ther. 1996, 10, 93–98. [Google Scholar] [CrossRef]
- Liu, L.; Yuan, S.; Long, Y.; Guo, Z.; Sun, Y.; Li, Y.; Niu, Y.; Li, C.; Mei, Q. Immunomodulation of Rheum tanguticum polysaccharide (RTP) on the immunosuppressive effects of dexamethasone (DEX) on the treatment of colitis in rats induced by 2,4,6-trinitrobenzene sulfonic acid. Int. Immunopharmacol. 2009, 9, 1568–1577. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. MTOR interacts with Raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, J.T.; Ray, C.; Fox, A.L.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. Mammalian EAK-7 activates alternative mTOR signaling to regulate cell proliferation and migration. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, J.T.; Haidar, F.S.; Fox, A.L.; Ray, C.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. mEAK-7 forms an alternative mTOR complex with DNA-PKcs in human cancer. Iscience 2019, 17, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.W.; Bi, Y.; Shen, B.; Yang, H.; Zhang, Y.; Wang, X.; Liu, H.; Lu, Y.; Liao, J.; Chen, X.; et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1 alpha-Dependent glycolysis. Cancer Res. 2014, 74, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Yang, T.; Li, L.; Sun, L.; Hou, Y.; Hu, X.; Zhang, L.; Tian, H.; Zhao, Q.; Peng, J.; et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat. Commun. 2014, 5, 4696. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Z.; Bi, Y.; Fu, Z.; Gong, P.; Li, Y.; Yu, Q.; Jia, A.; Wang, J.; Xue, L.; et al. mTOR signaling disruption from myeloid-derived suppressive cells protects against immune-mediated hepatic injury through the HIF1 alpha-dependent glycolytic pathway. J. Leukoc. Biol. 2016, 100, 1349–1362. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Li, D.; Ren, J.; Li, X.; Wang, T.; Dou, H.; Hou, Y. mTOR inhibitor INK128 attenuates dextran sodium sulfate-induced colitis by promotion of MDSCs on Treg cell expansion. J. Cell. Physiol. 2019, 234, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhao, Y.; Shi, L.; Li, W.; Chen, K.; Li, M.; Chen, X.; Zhang, H.; Li, T.; Matsuzawa-Ishimoto, Y.; et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J. Clin. Investig. 2020, 130, 2111–2128. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Wang, X.; Bi, Y.; Shen, B.; Shao, K.; Yang, H.; Lu, Y.; Zhang, Z.; Chen, X.; Liu, H.; et al. Dexamethasone potentiates myeloid-derived suppressor cell function in prolonging allograft survival through nitric oxide. J. Leukoc. Biol. 2014, 96, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T.; Haidinger, M.; Katholnig, K.; Kopecky, C.; Poglitsch, M.; Lassnig, C.; Rosner, M.; Zlabinger, G.J.; Hengstschläger, M.; Müller, M.; et al. Inhibition of mTOR blocks the anti-inflammatory effects of glucocorticoids in myeloid immune cells. Blood 2011, 117, 4273–4283. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, R.; Sagmeister, M.; Offner, F. Acute ulcerative colitis during successful interferon/ribavirin treatment for chronic hepatitis. Gut 2005, 54, 438–439. [Google Scholar]
- Zhao, J.; Cao, J.; Yu, L.; Ma, H. Dehydroepiandrosterone resisted E. Coli O157:H7-induced inflammation via blocking the activation of p38 MAPK and NF-kappaB pathways in mice. Cytokine 2020, 127, 154955. [Google Scholar] [CrossRef]
- Sander, L.E.; Obermeier, F.; Dierssen, U.; Kroy, D.C.; Singh, A.K.; Seidler, U.; Streetz, K.L.; Lutz, H.H.; Müller, W.; Tacke, F.; et al. Gp130 signaling promotes development of acute experimental colitis by facilitating early neutrophil/macrophage recruitment and activation. J. Immunol. 2008, 181, 3586–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieleman, L.A.; Ridwan, B.U.; Tennyson, G.S.; Beagley, K.W.; Bucy, R.P.; Elson, C.O. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 1994, 107, 1643–1652. [Google Scholar] [CrossRef]
- Zabana, Y.; Domènech, E.; Latorre, N.; Ojanguren, I.; Mañosa, M.; Gassull, M.A. Acute appendicitis in inactive extensive ulcerative colitis. Gastroenterol. Hepatol. 2007, 30, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bi, Y.; Chen, X.; Li, C.; Li, Y.; Zhang, Z.; Wang, J.; Lu, Y.; Yu, Q.; Su, H.; et al. Histone Deacetylase SIRT1 negatively regulates the differentiation of Interleukin-9-Producing CD4(+) T cells. Immunity 2016, 44, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. J. Tech. Methods Pathol. 1993, 69, 238–249. [Google Scholar]
- Liu, G.; Bi, Y.; Wang, R.; Shen, B.; Zhang, Y.; Yang, H.; Wang, X.; Liu, H.; Lu, Y.; Han, F. Kinase AKT1 negatively controls neutrophil recruitment and function in mice. J. Immunol. 2013, 191, 2680–2690. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Bi, Y.; Xue, L.; Zhang, Y.; Yang, H.; Chen, X.; Lu, Y.; Zhang, Z.; Liu, H.; Wang, X.; et al. Dendritic cell SIRT1-HIF1alpha axis programs the differentiation of CD4+ T cells through IL-12 and TGF-beta1. Proc. Natl. Acad. Sci. USA 2015, 112, E957–E965. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; He, Y.; Zhou, S.; Cao, Y.; Li, Y.; Bi, Y.; Liu, G. HIF1alpha-Dependent metabolic signals control the differentiation of follicular helper T cells. Cells 2019, 8, E1450. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Bi, Y.; Li, Y.; Yang, H.; Yu, Q.; Wang, J.; Wang, Y.; Su, H.; Jia, A.; Hu, Y.; et al. Dendritic cell MST1 inhibits Th17 differentiation. Nat. Commun. 2017, 8, 14275. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, L.; Zhang, H.; Xiao, Y.; Shao, L.; Li, H.; Yin, H.; Wang, R.; Liu, G.; Corley, D.; et al. Disruption of TSC1/2 signaling complex reveals a checkpoint governing thymic CD4+ CD25+ Foxp3+ regulatory T-cell development in mice. FASEB J. 2013, 27, 3979–3990. [Google Scholar] [CrossRef]
- Liu, L.; Lu, Y.; Martinez, J.; Bi, Y.; Lian, G.; Wang, T.; Milasta, S.; Wang, J.; Yang, M.; Liu, G.; et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1alpha-dependent. Proc. Natl. Acad. Sci. USA 2016, 113, 1564–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Burns, S.; Huang, G.; Boyd, K.; Proia, R.L.; Flavell, R.A.; Chi, H. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat. Immunol. 2009, 10, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani, M.; Hoshii, T.; Fujii, H.; Koyasu, S.; Hirao, A.; Matsuda, S. Cutting edge: mTORC1 in intestinal CD11c+ CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J. Immunol. 2012, 188, 4736–4740. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Chapman, J.R.; Wang, L.; Harris, T.E.; Shabanowitz, J.; Hunt, D.F.; Fu, Z. Intestinal cell kinase (ICK) promotes activation of mTOR complex 1 (mTORC1) through phosphorylation of Raptor Thr-908. J. Biol. Chem. 2012, 287, 12510–12519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujishita, T.; Aoki, M.; Taketo, M.M. The role of mTORC1 pathway in intestinal tumorigenesis. Cell Cycle 2009, 8, 3684–3687. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Zhang, L.; Li, X.; Zhang, X.; Liu, S.; Gao, N.; Li, L.; Gao, G.; Wei, G.; Chen, Z.; et al. Repression of mammalian target of rapamycin complex 1 inhibits intestinal regeneration in acute inflammatory bowel disease models. J. Immunol. 2015, 195, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.M.; Lawrence, T.; Kleiman, A.; Warden, P.; Medghalchi, M.; Tuckermann, J.; Saklatvala, J.; Clark, A.R. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med. 2006, 203, 1883–1889. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Zhang, W.; Tang, K.; Zhang, H.; Zhang, Y.; Li, D.; Li, Y.; Xu, P.; Luo, S.; Cai, W.; et al. Switch of glycolysis to gluconeogenesis by dexamethasone for treatment of hepatocarcinoma. Nat. Commun. 2013, 4, 2508. [Google Scholar] [CrossRef] [Green Version]
- Pandit, S.; Geissler, W.; Harris, G.; Sitlani, A. Allosteric effects of dexamethasone and RU486 on glucocorticoid receptor-DNA interactions. J. Biol. Chem. 2002, 277, 1538–1543. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Brown, D.E.; Brewer, J.A.; Vogt, S.K.; Muglia, L.J. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 2007, 109, 4313–4319. [Google Scholar] [CrossRef] [Green Version]
- Hackstein, H.; Thomson, A.W. Dendritic cells: Emerging pharmacological targets of immunosuppressive drugs. Nat. Rev. Immunol. 2004, 4, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Zhong, S.; Geng, Y.; Munirathinam, G.; Cha, I.; Reardon, C.; Getz, G.S.; van Rooijen, N.; Kang, Y.; Wang, B.; et al. Dexamethasone promotes tolerance in vivo by enriching CD11clo CD40lo tolerogenic macrophages. Eur. J. Immunol. 2013, 43, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, A.L.; Ng, S.C. Review article: The optimal medical management of acute severe ulcerative colitis. Aliment. Pharmacol. Ther. 2010, 32, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Gulliford, S.R.; Limdi, J.K. Acute severe ulcerative colitis: Timing is everything. Postgrad. Med. J. 2011, 87, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeds, I.L.; Truta, B.; Parian, A.M.; Chen, S.Y.; Efron, J.E.; Gearhart, S.L.; Safar, B.; Fang, S.H. Early surgical intervention for acute ulcerative colitis is associated with improved postoperative outcomes. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2017, 21, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Turnquist, H.R.; Cardinal, J.; Macedo, C.; Rosborough, B.R.; Sumpter, T.L.; Geller, D.A.; Metes, D.; Thomson, A.W. mTOR and GSK-3 shape the CD4+ T-cell stimulatory and differentiation capacity of myeloid DCs after exposure to LPS. Blood 2010, 115, 4758–4769. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Readinger, J.A.; DuBois, W.; Janka-Junttila, M.; Robinson, R.; Pruitt, M.; Bliskovsky, V.; Wu, J.Z.; Sakakibara, K.; Patel, J.; et al. Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production. Blood 2011, 117, 1228–1238. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; Pearce, L.R.; Garcia-Martinez, J.M. New insights into mTOR signaling: mTORC2 and beyond. Sci. Signal. 2009, 2, pe27. [Google Scholar] [CrossRef]
- Yang, K.; Chi, H. Tuning mTOR activity for immune balance. J. Clin. Investig. 2013, 123, 5001–5004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalle Pezze, P.; Sonntag, A.G.; Thien, A.; Prentzell, M.T.; Gödel, M.; Fischer, S.; Neumann-Haefelin, E.; Huber, T.B.; Baumeister, R.; Shanley, D.P.; et al. A dynamic network model of mTOR signaling reveals TSC-independent mTORC2 regulation. Sci. Signal. 2012, 5, ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limon, J.J.; So, L.; Jellbauer, S.; Chiu, H.; Corado, J.; Sykes, S.M.; Raffatellu, M.; Fruman, D.A. mTOR kinase inhibitors promote antibody class switching via mTORC2 inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, E5076–E5085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T.; Säemann, M.D. The multiple facets of mTOR in immunity. Trends Immunol. 2009, 30, 218–226. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Names | Primer Sequences (5ʹ-3ʹ) |
|---|---|
| HPRT | Forward: AGTACAGCCCCAAAATGGTTAAG |
| Reverse: CTTAGGCTTTGTATTTGGCTTTTC | |
| IL-1β | Forward: TGGGAAACAACAGTGGTCAGG |
| Reverse: CCATCAGAGGCAAGGAGGAA | |
| IL-6 | Forward: GCAATGGCAATTCTGATTGTATG |
| Reverse: CCAGTGCCTCTTTGCTGCTTTC | |
| TNFα | Forward: GAGTGACAAGCCTGTAGCC |
| Reverse: CTCCTGGTATGAGATAGCAAA | |
| IL-10 | Forward: GCTCTTACTGACTGGCATGAG |
| Reverse: CAA TACCATTGACCTGCCGAT | |
| CXCL1 | Forward: GCACCCAAACCGAAGTCATAG |
| Reverse: AGAAGCCAGCGTTCACCAGA | |
| CXCL2 | Forward: GCCCAGACAGAAGTCATAGCC |
| Reverse: CTCCTCCTTTCCAGGTCAGTTA | |
| CX3CL1 | Forward: CGCAATCATCTTGGAGACGA |
| Reverse: GTGCCGCCATTTCGAGTTA | |
| CCL1 | Forward: TCAGCCAGATGCAGTTAACGC |
| Reverse: TGATCCTCTTGTAGCTCTCCAGC | |
| CCL5 | Forward: GATGGACATAGAGGACACAACT |
| Reverse: TGGGACGGCAGATCTGAGGG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Dong, L.; Jia, A.; Chen, X.; Yang, Q.; Wang, Y.; Wang, Y.; Liu, R.; Cao, Y.; He, Y.; et al. Glucocorticoids Promote the Onset of Acute Experimental Colitis and Cancer by Upregulating mTOR Signaling in Intestinal Epithelial Cells. Cancers 2020, 12, 945. https://doi.org/10.3390/cancers12040945
Zhang Z, Dong L, Jia A, Chen X, Yang Q, Wang Y, Wang Y, Liu R, Cao Y, He Y, et al. Glucocorticoids Promote the Onset of Acute Experimental Colitis and Cancer by Upregulating mTOR Signaling in Intestinal Epithelial Cells. Cancers. 2020; 12(4):945. https://doi.org/10.3390/cancers12040945
Chicago/Turabian StyleZhang, Zhengguo, Lin Dong, Anna Jia, Xi Chen, Qiuli Yang, Yufei Wang, Yuexin Wang, Ruichen Liu, Yejin Cao, Ying He, and et al. 2020. "Glucocorticoids Promote the Onset of Acute Experimental Colitis and Cancer by Upregulating mTOR Signaling in Intestinal Epithelial Cells" Cancers 12, no. 4: 945. https://doi.org/10.3390/cancers12040945
APA StyleZhang, Z., Dong, L., Jia, A., Chen, X., Yang, Q., Wang, Y., Wang, Y., Liu, R., Cao, Y., He, Y., Bi, Y., & Liu, G. (2020). Glucocorticoids Promote the Onset of Acute Experimental Colitis and Cancer by Upregulating mTOR Signaling in Intestinal Epithelial Cells. Cancers, 12(4), 945. https://doi.org/10.3390/cancers12040945