FAK Inhibition Induces Glioblastoma Cell Senescence-Like State through p62 and p27


Abstract
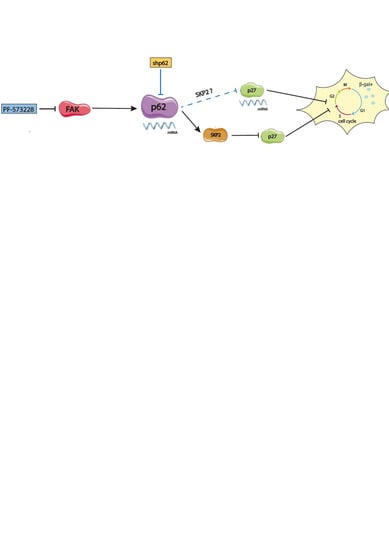
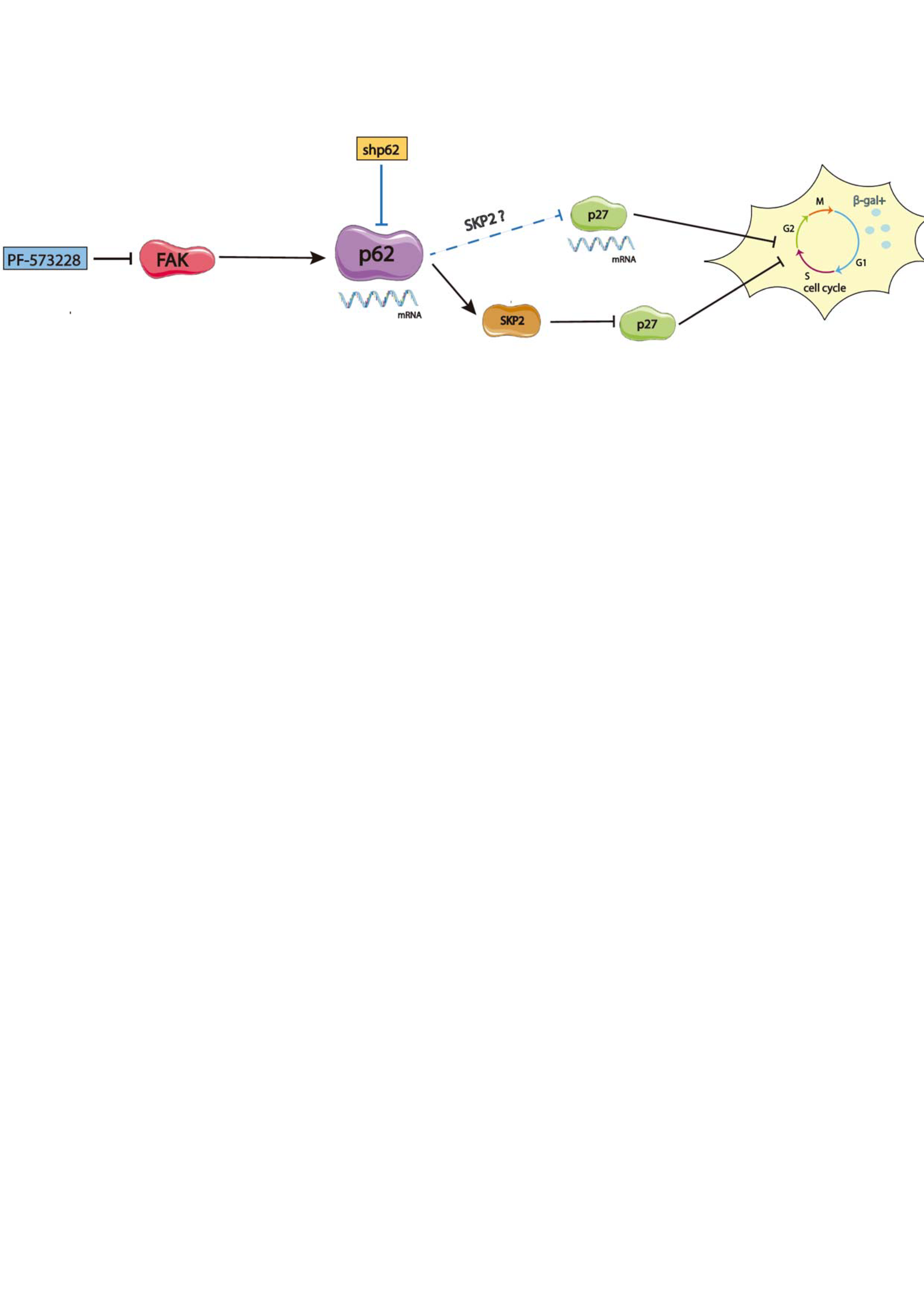
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
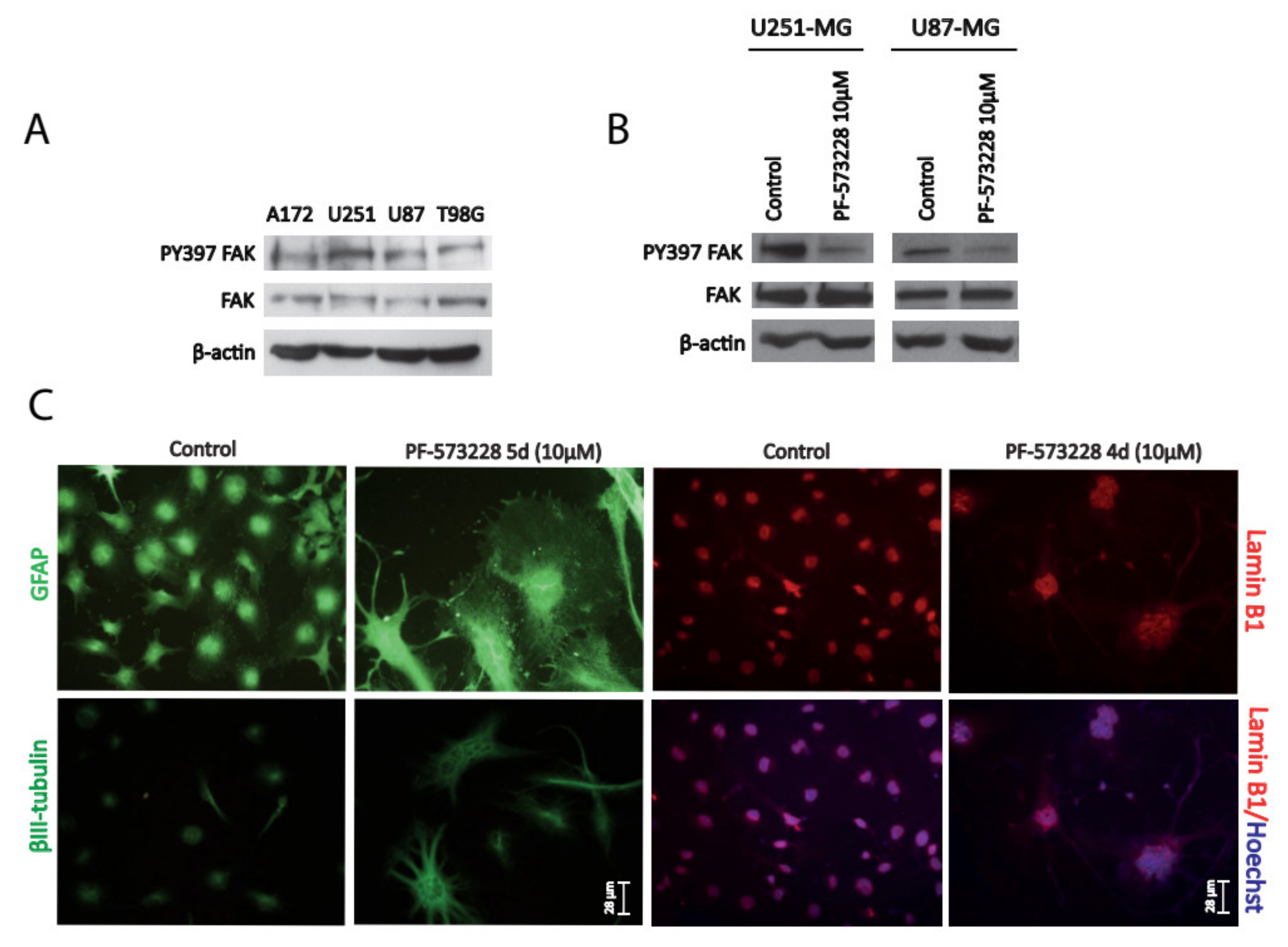
2.1. FAK Inhibition Reshapes GBM Cell Morphology, Increasing Cellular and Nuclear Size
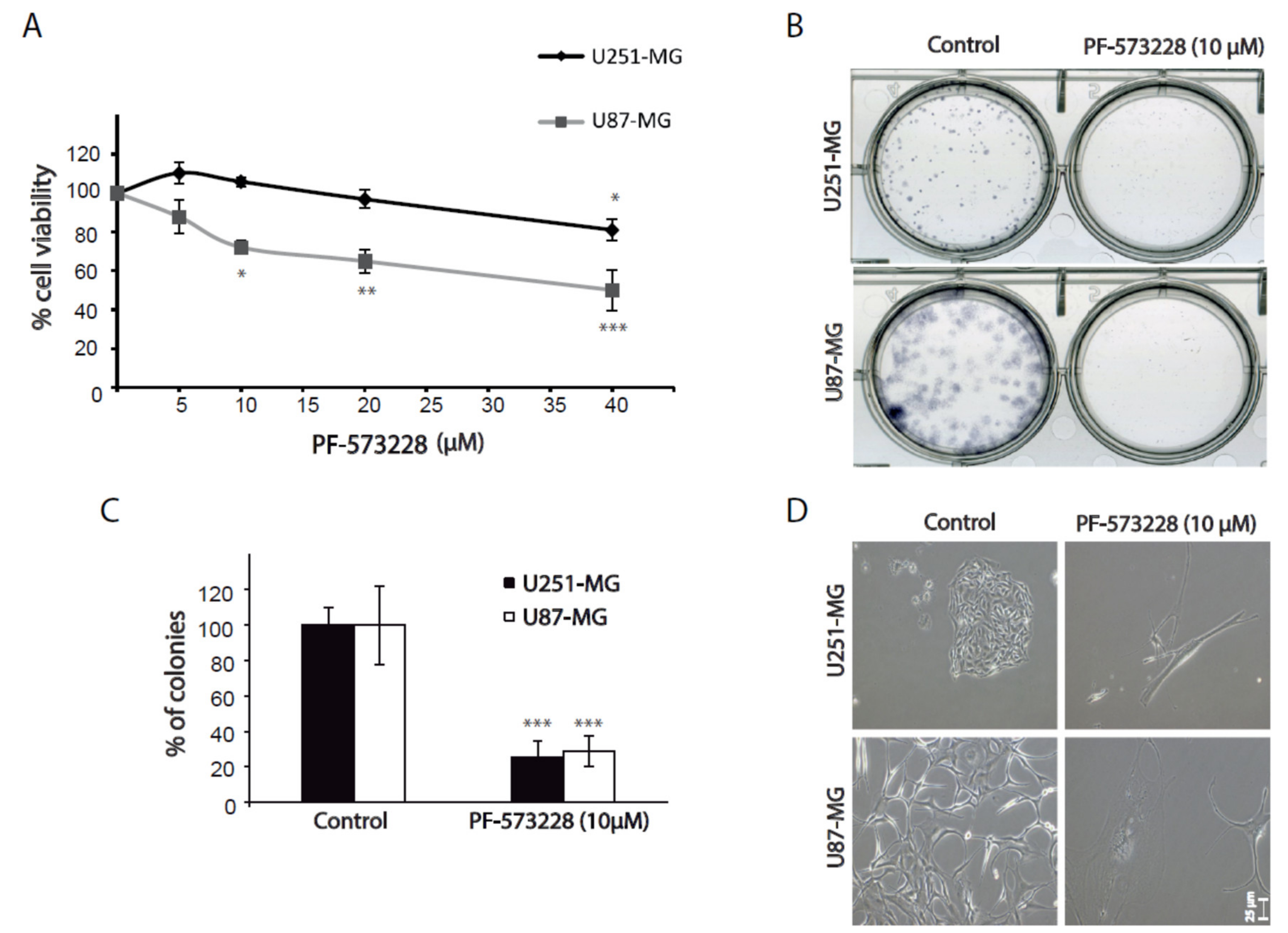
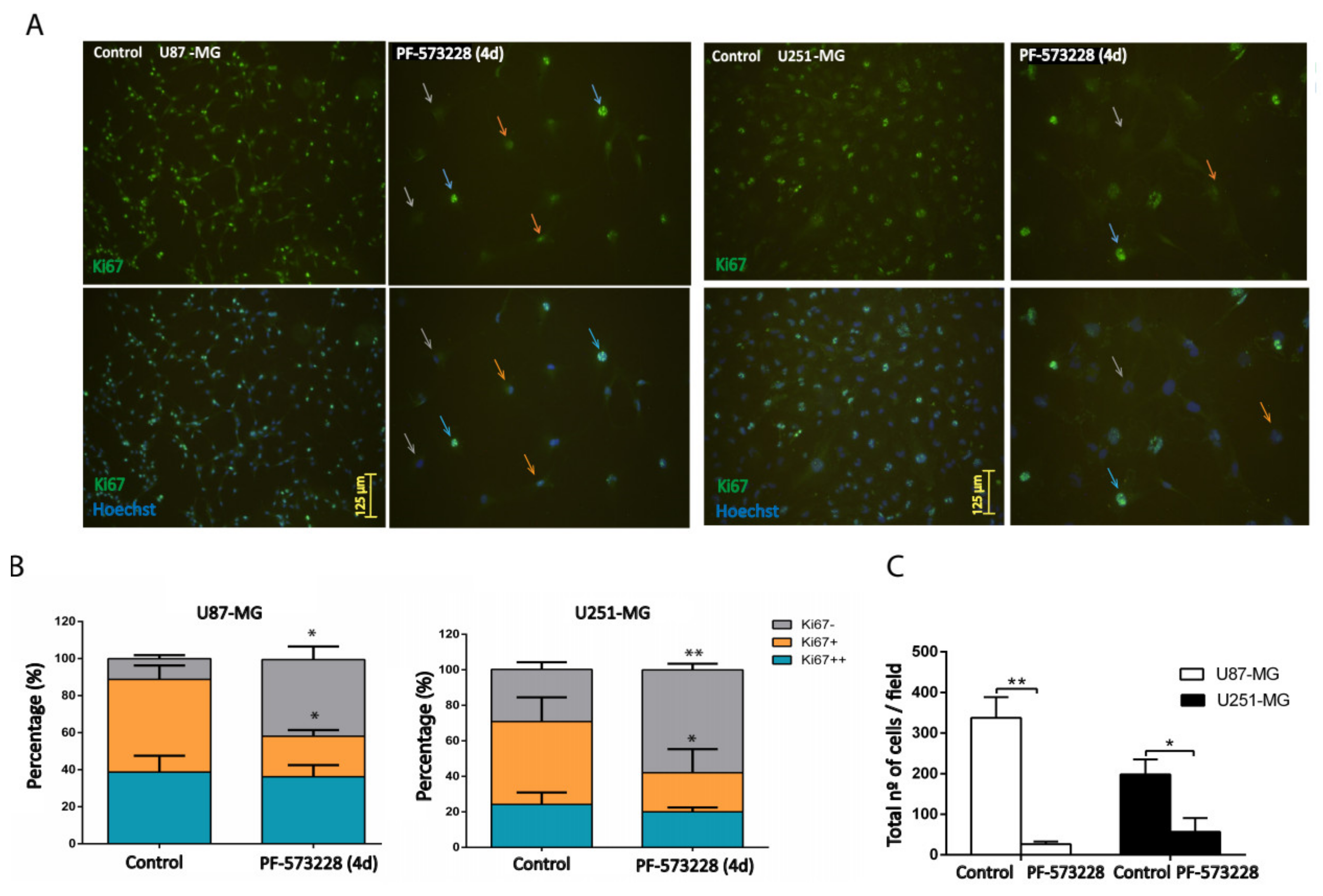
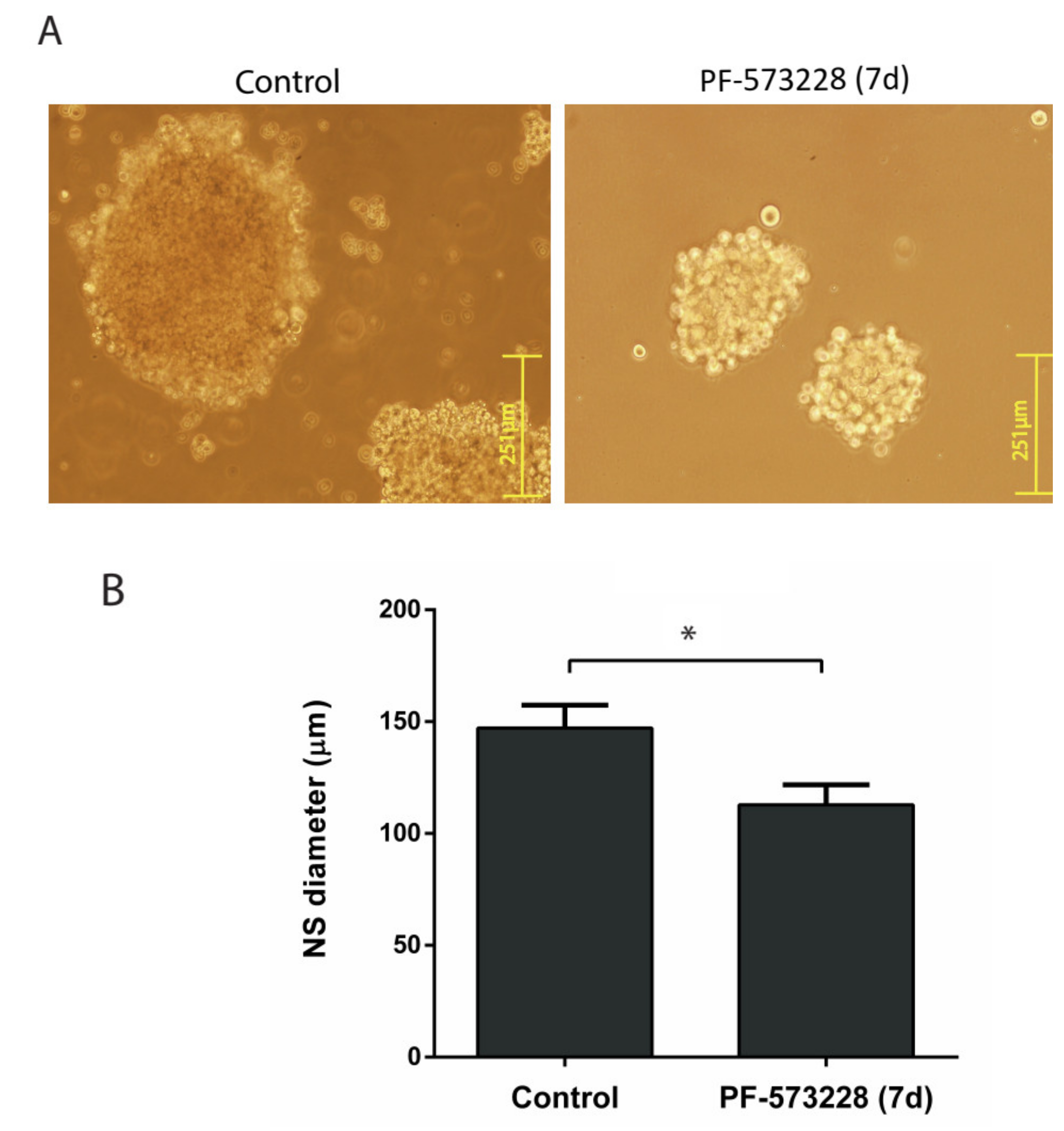
2.2. FAK Inhibition Reduces GBM Cell Proliferation
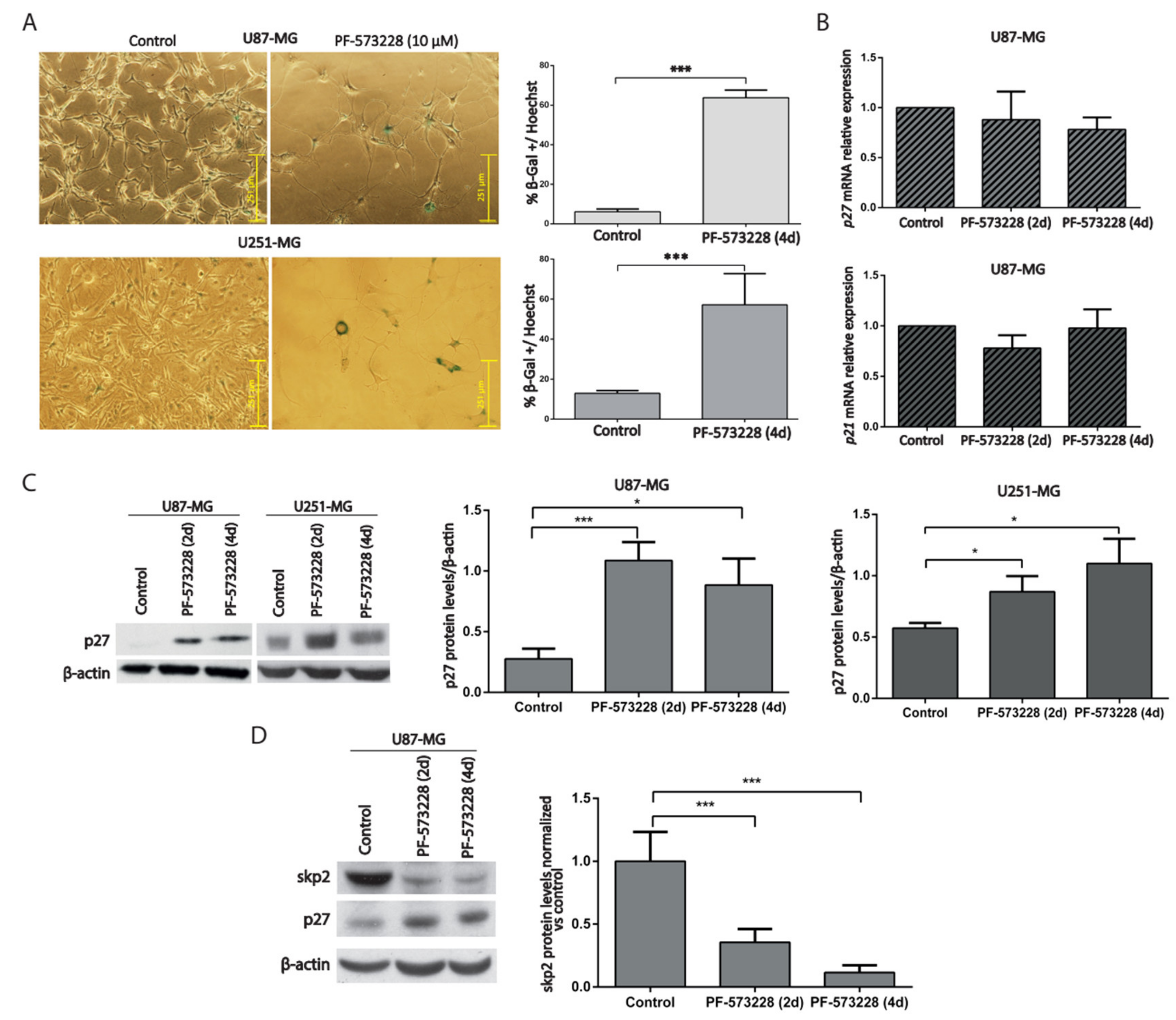
2.3. PF-573228 Increases p27 Protein Levels and β-Galactosidase Activity
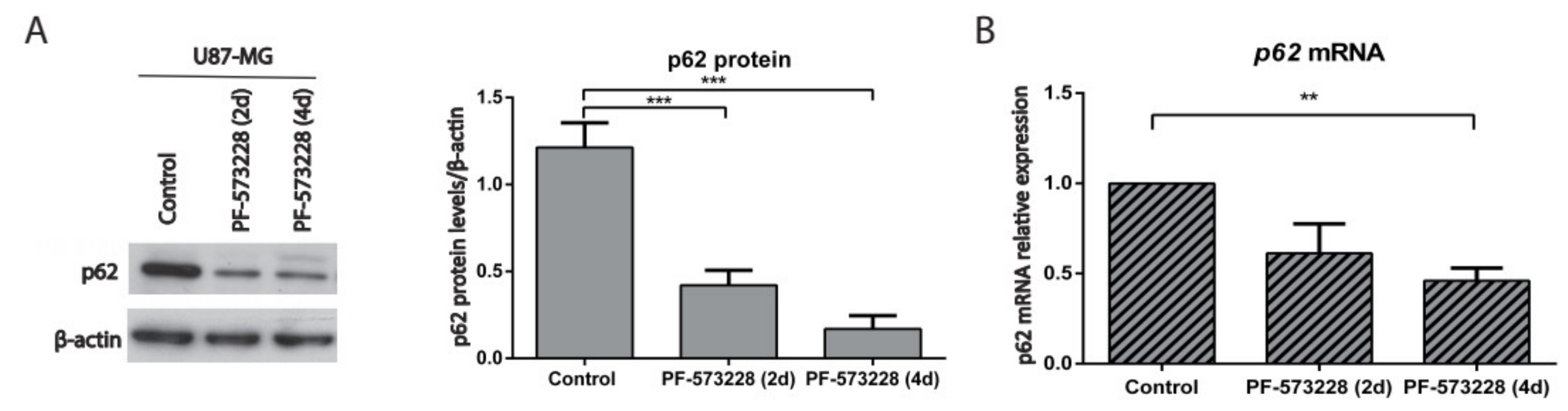
2.4. p62/SQSTM-1 Expression is Reduced upon FAK Inhibition
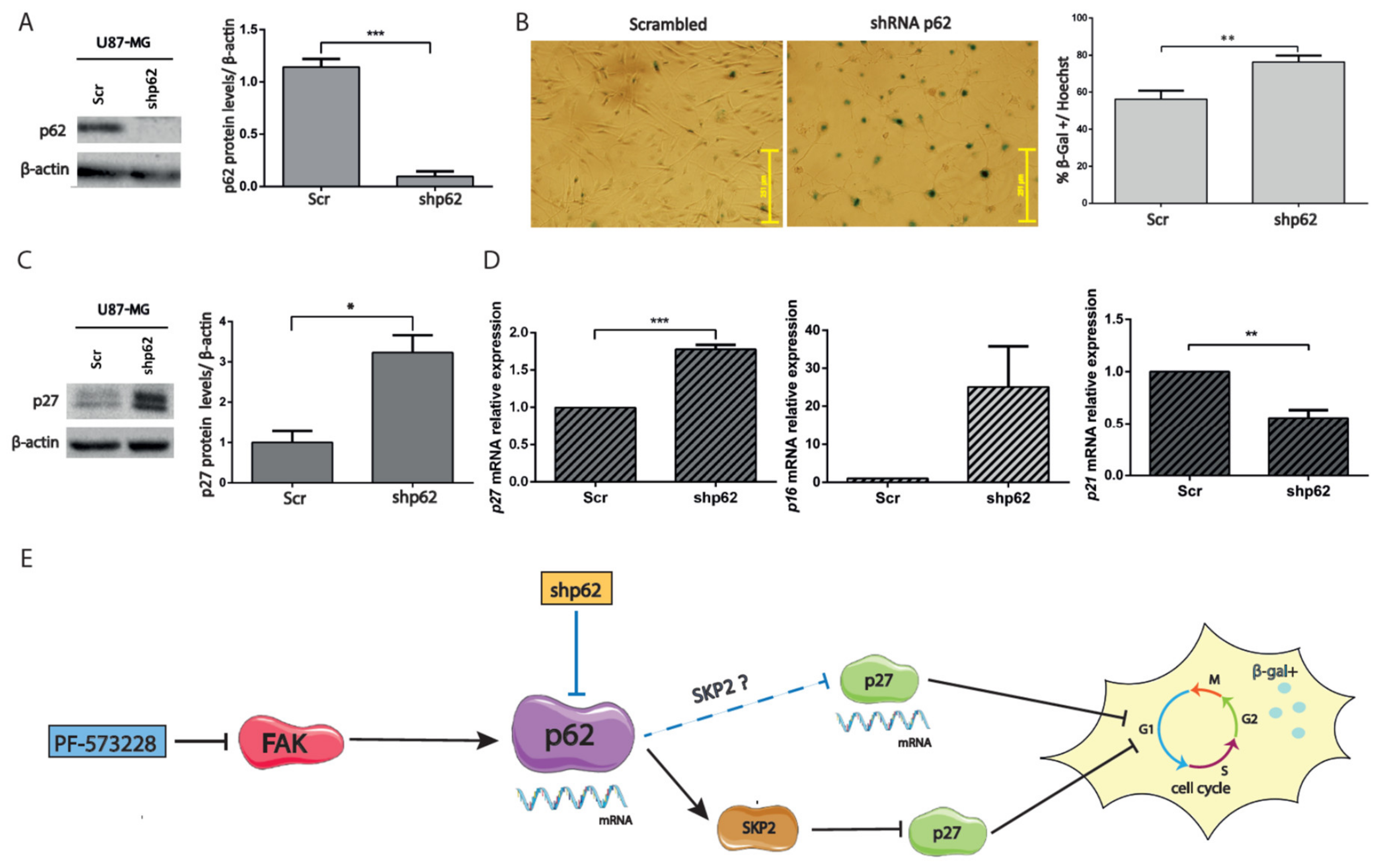
2.5. Depletion of p62 Increases p27 and SA-β-Gal Activity
3. Discussion
4. Material and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Immunoblot Analysis
4.4. shRNA-Induced Gene Silencing by Lentiviral Infection
4.5. Real-Time PCR
4.6. Cell Viability Assay
4.7. Clonogenic Assays
4.8. Immunohistochemistry
4.9. Senescence-Associated β-Galactosidase
4.10. Statistical Analyses and Bioinformatics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tai, Y.L.; Chen, L.C.; Shen, T.L. Emerging roles of focal adhesion kinase in cancer. BioMed Res. Int. 2015, 2015, 690690. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Hu, Y.-L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci. Rep. 2015, 4, 6024. [Google Scholar] [CrossRef] [Green Version]
- Hamadi, A.; Bouali, M.; Dontenwill, M.; Stoeckel, H.; Takeda, K.; Rondé, P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J. Cell Sci. 2005, 118, 4415–4425. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Tamura, M.; Gu, J.; Danen, E.H.J.; Takino, T.; Miyamoto, S.; Yamada, K.M. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-Kinase/Akt Cell survival pathway. J. Biol. Chem. 1999, 274, 20693–20703. [Google Scholar] [CrossRef] [Green Version]
- Storch, K.; Sagerer, A.; Cordes, N. Cytotoxic and radiosensitizing effects of FAK targeting in human glioblastoma cells in vitro. Oncol. Rep. 2015, 33, 2009–2016. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Grammer, R.; Nelson, M.; Guan, J.-L.; Stewart, J.; Gladson, C.L. FAK regulation of the cell cycle p27. J. Biol. Chem. 2004, 27, 1–51. [Google Scholar]
- Gabarra-Niecko, V.; Schaller, M.D.; Dunty, J.M. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003, 22, 359–374. [Google Scholar] [CrossRef]
- Mamillapalli, R.; Gavrilova, N.; Mihaylova, V.T.; Tsvetkov, L.M.; Wu, H.; Zhang, H.; Sun, H. PTEN regulates the ubiquitin-dependent degradation of the CDK inhibitor p27KIP1 through the ubiquitin E3 ligase SCFSKP2. Curr. Biol. 2001, 11, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a graded rather than a binary marker of proliferation versus quiescence. Cell Rep. 2018, 24, 1105–1112.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Evans, M.K. Techniques to induce and quantify cellular senescence. J. Vis. Exp. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Carrano, A.C.; Pagano, M. Role of the F-box protein Skp2 in adhesion-dependent cell cycle progression. J. Cell Biol. 2001, 153, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- Bryant, P.; Zheng, Q.; Pumiglia, K. Focal adhesion kinase controls cellular levels of p27/Kip1 and p21/Cip1 through Skp2-Dependent and -independent mechanisms. Mol. Cell. Biol. 2006, 26, 4201–4213. [Google Scholar] [CrossRef] [Green Version]
- Bond, M.; Sala-Newby, G.B.; Newby, A.C. Focal adhesion kinase (FAK)-dependent regulation of S-phase kinase-associated protein-2 (Skp-2) stability: A novel mechanism regulating smooth muscle cell proliferation. J. Biol. Chem. 2004, 279, 37304–37310. [Google Scholar] [CrossRef] [Green Version]
- Puissant, A.; Fenouille, N.; Auberger, P. When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. 2012, 2, 397–413. [Google Scholar] [PubMed]
- Linares, J.F.; Amanchy, R.; Greis, K.; Diaz-Meco, M.T.; Moscat, J. Phosphorylation of p62 by cdk1 controls the timely transit of cells through mitosis and tumor cell proliferation. Mol. Cell. Biol. 2011, 31, 2171. [Google Scholar] [CrossRef] [Green Version]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed hcc-initiating cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Karras, P.; Riveiro-Falkenbach, E.; Cañón, E.; Tejedo, C.; Calvo, T.G.; Martínez-Herranz, R.; Alonso-Curbelo, D.; Cifdaloz, M.; Perez-Guijarro, E.; Gómez-López, G.; et al. p62/SQSTM1 fuels melanoma progression by opposing mRNA decay of a selective set of pro-metastatic factors. Cancer Cell 2019, 35, 46–63.e10. [Google Scholar] [CrossRef] [Green Version]
- Kumsta, C.; Chang, J.T.; Lee, R.; Tan, E.P.; Yang, Y.; Loureiro, R.; Choy, E.H.; Lim, S.H.Y.; Saez, I.; Springhorn, A.; et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans by inducing autophagy. Nat. Commun. 2019, 10, 5648. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Nishio, K.; Inoue, A. Senescence-associated alterations of cytoskeleton: Extraordinary production of vimentin that anchors cytoplasmic p53 in senescent human fibroblasts. Histochem. Cell Bio. 2005, 123, 263–273. [Google Scholar] [CrossRef]
- Patki, M.; McFall, T.; Rosati, R.; Huang, Y.; Malysa, A.; Polin, L.; Fielder, A.; Wilson, M.R.; Lonardo, F.; Back, J.; et al. Chronic p27Kip1 induction by dexamethasone causes senescence phenotype and permanent cell cycle blockade in lung adenocarcinoma cells over-expressing glucocorticoid receptor. Sci. Rep. 2018, 8, 16006. [Google Scholar] [CrossRef]
- Lee, J.J.; Lee, J.S.; Cui, M.N.; Yun, H.H.; Kim, H.Y.; Lee, S.H.; Lee, J.H. BIS targeting induces cellular senescence through the regulation of 14-3-3 zeta/STAT3/SKP2/p27 in glioblastoma cells. Cell Death Dis. 2014, 5, e1537-12. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Huang, G.; Ho, B.; Yemma, M.; Morrison, C.D.; Lee, J.; Eliceiri, B.P.; Cance, W.G. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol. Cancer Ther. 2013, 12, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.H.; Wang, P.H.; Niu, S.W.; Zhen, Y.Y.; Huang, M.S.; Hsiao, M.; Yang, C.J. Inhibition of FAK signaling elicits lamin A/C-associated nuclear deformity and cellular senescence. Front. Oncol. 2019, 9, 22. [Google Scholar] [CrossRef]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.-N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.-S.; Li, D.-X.; Liu, Y.-H.; Fu, X.-M.; Tang, S.-F.; Li, J. Expression of NANOG in human gliomas and its relationship with undifferentiated glioma cells. Oncol. Rep. 2011, 26, 593–601. [Google Scholar] [CrossRef]
- Ho, B.; Olson, G.; Figel, S.; Gelman, I.; Cance, W.G.; Golubovskaya, V.M. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J. Biol. Chem. 2012, 287, 18656–18673. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.J.; Lui, P.P.W.; Obajdin, J.; Riccio, F.; Stroukov, W.; Willis, T.L.; Spagnoli, F.; Watt, F.M. Mechanisms, hallmarks, and implications of stem cell quiescence. Stem Cell Rep. 2019, 12, 1190–1200. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Sandilands, E.; Serrels, B.; McEwan, D.G.; Morton, J.P.; MacAgno, J.P.; McLeod, K.; Stevens, C.; Brunton, V.G.; Langdon, W.Y.; Vidal, M.; et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat. Cell Biol. 2012, 14, 51–60. [Google Scholar] [CrossRef]
- Nàger, M.; Sallán, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macià, A.; Yeramian, A.; Cantí, C.; Herreros, J. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.G.R.; Harris, J.W.; Wold, B.J.; Lin, F.; Brody, J.P. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene 2003, 22, 2322–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in cancer: Signaling adaptor beyond autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Pan, B.Q.; Shi, F.; Xie, Z.H.; Jiang, Y.Y.; Shang, L.; Zhang, Y.; Xu, X.; Cai, Y.; Hao, J.J.; et al. Sequestosome 1 protects esophageal squamous carcinoma cells from apoptosis via stabilizing SKP2 under serum starvation condition. Oncogene 2018, 37, 3260–3274. [Google Scholar] [CrossRef]
- Jung, D.; Khurana, A.; Roy, D.; Kalogera, E.; Bakkum-Gamez, J.; Chien, J.; Shridhar, V. Quinacrine upregulates p21/p27 independent of p53 through autophagy-mediated downregulation of p62-Skp2 axis in ovarian cancer. Sci. Rep. 2018, 8, 2487. [Google Scholar] [CrossRef] [Green Version]
- Salazar, G.; Cullen, A.; Huang, J.; Zhao, Y.; Serino, A.; Hilenski, L.; Patrushev, N.; Forouzandeh, F.; Hwang, H.S. SQSTM1/p62 and PPARGC1A/PGC-1alpha at the interface of autophagy and vascular senescence. Autophagy 2019, 0, 1–19. [Google Scholar] [CrossRef]
- Pankiv, S.; Lamark, T.; Bruun, J.A.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J. Biol. Chem. 2010, 285, 5941–5953. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in blood-brain barrier penetrance and drug repurposing for improved treatment of glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology 2017, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alza, L.; Nàger, M.; Visa, A.; Cantí, C.; Herreros, J. FAK Inhibition Induces Glioblastoma Cell Senescence-Like State through p62 and p27. Cancers 2020, 12, 1086. https://doi.org/10.3390/cancers12051086
Alza L, Nàger M, Visa A, Cantí C, Herreros J. FAK Inhibition Induces Glioblastoma Cell Senescence-Like State through p62 and p27. Cancers. 2020; 12(5):1086. https://doi.org/10.3390/cancers12051086
Chicago/Turabian StyleAlza, Lía, Mireia Nàger, Anna Visa, Carles Cantí, and Judit Herreros. 2020. "FAK Inhibition Induces Glioblastoma Cell Senescence-Like State through p62 and p27" Cancers 12, no. 5: 1086. https://doi.org/10.3390/cancers12051086
APA StyleAlza, L., Nàger, M., Visa, A., Cantí, C., & Herreros, J. (2020). FAK Inhibition Induces Glioblastoma Cell Senescence-Like State through p62 and p27. Cancers, 12(5), 1086. https://doi.org/10.3390/cancers12051086