The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles

 ,
, 
Abstract
:1. Introduction
2. Results
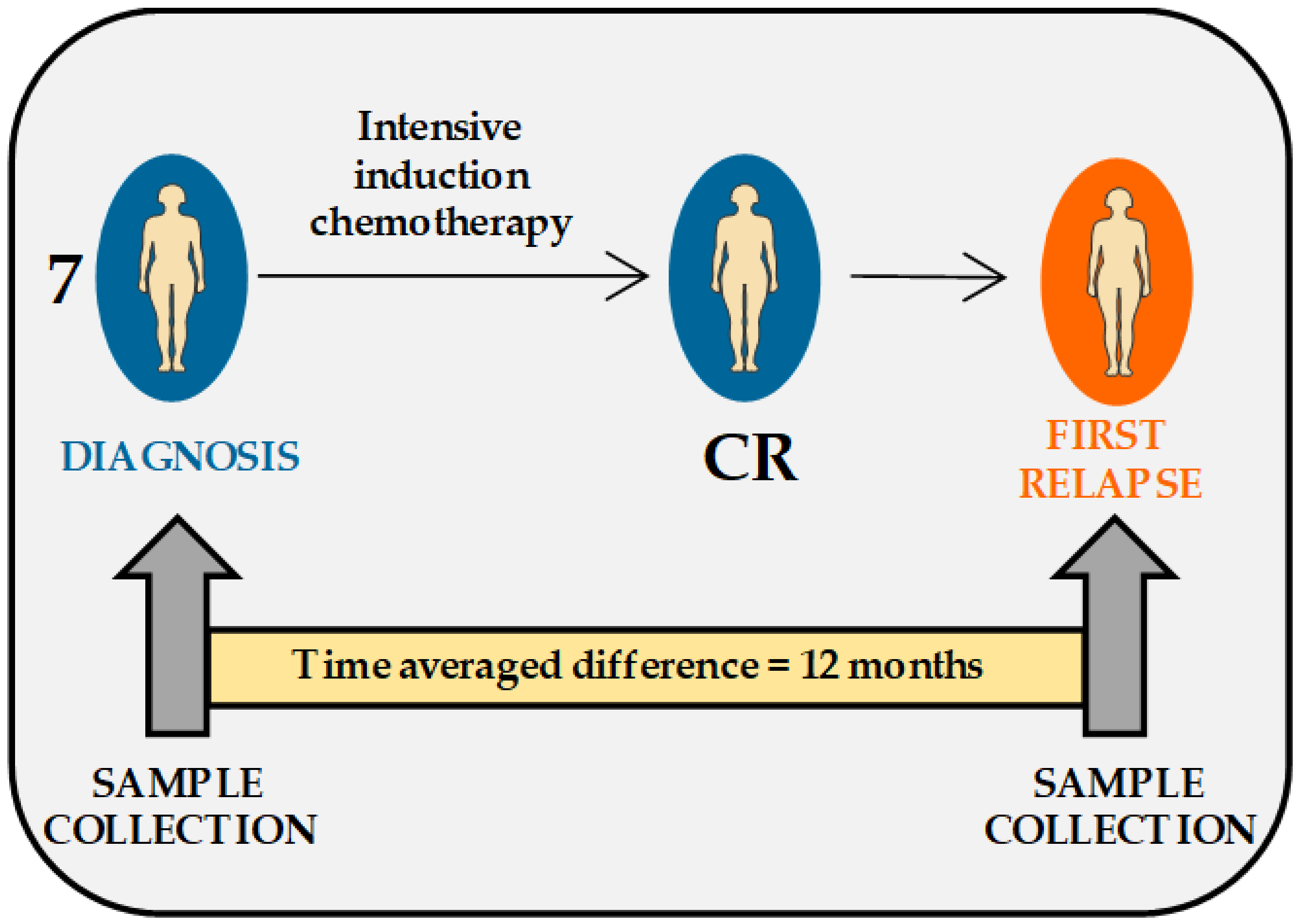
2.1. Description of AML Patients and Patients’ Cells Included in the Study
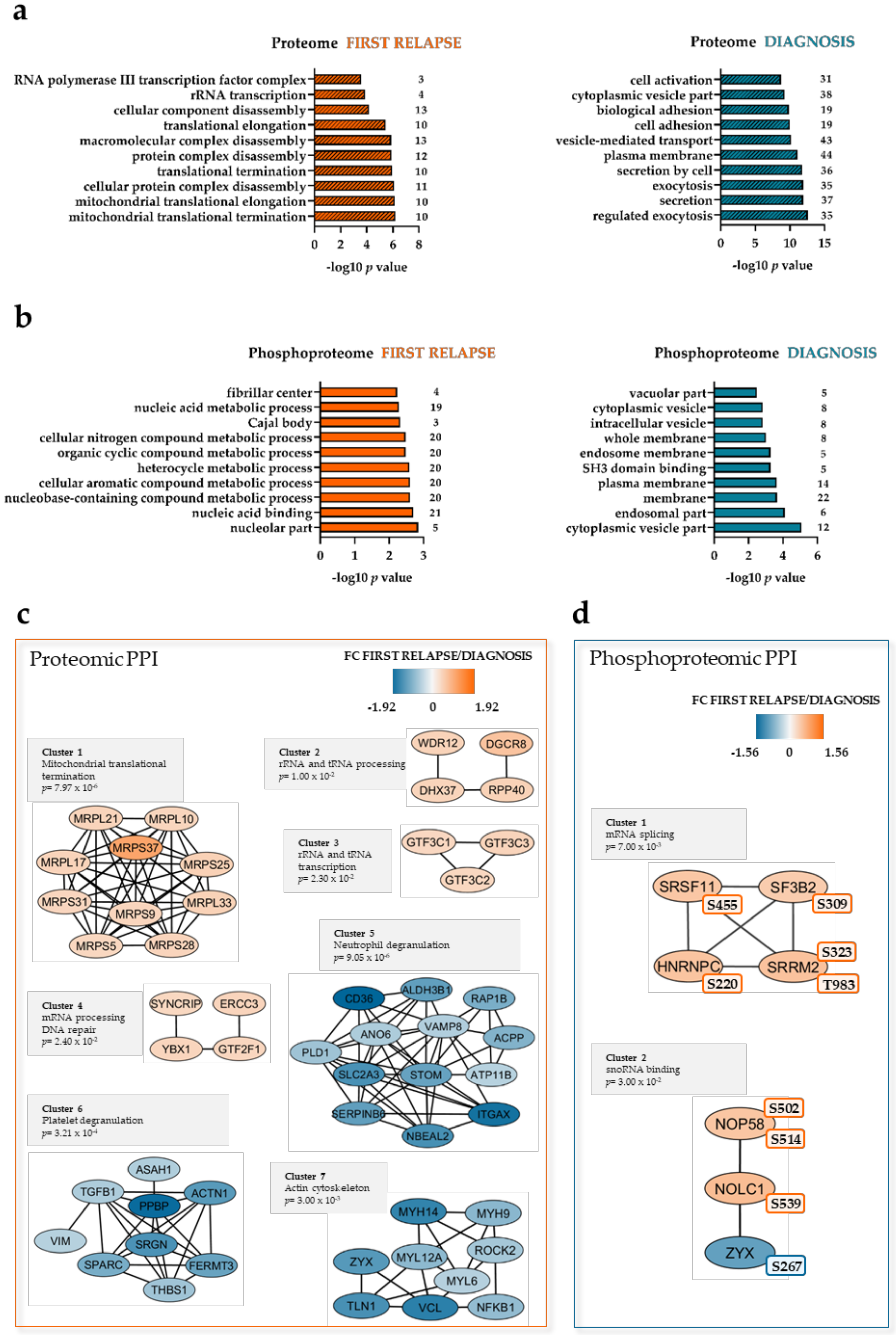
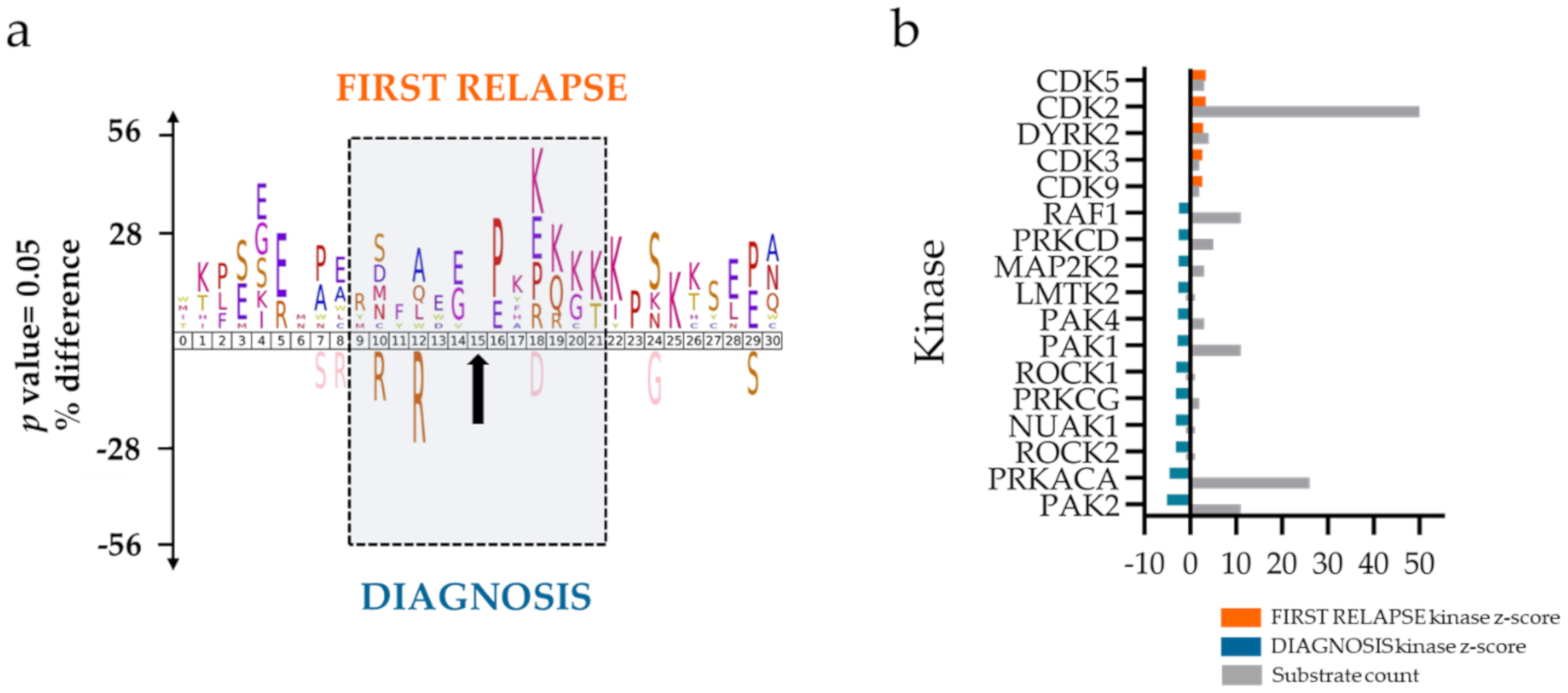
2.2. Mitochondrial Processing and Immune Responses at Relapse
3. Discussion
4. Materials and Methods
4.1. AML Patients and Sample Collection
4.2. Sample Preparation for LC–MS/MS
4.3. Nanoflow LC–MS/MS and Data Analysis
4.4. Western Blots
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrara, F.; Schiffer, C.A. Acute myeloid leukaemia in adults. Lancet 2013, 381, 484–495. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Versluis, J.; Cornelissen, J.J.; Craddock, C.; Sanz, M.A.; Canaani, J.; Nagler, A. Acute Myeloid Leukemia in Adults. In The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies; Carreras, E., Dufour, C., Mohty, M., Kroger, N., Eds.; Springer: Cham, Switzerland, 2019; pp. 507–521. [Google Scholar] [CrossRef]
- Mariani, S.; Trisolini, S.M.; Minotti, C.; Breccia, M.; Cartoni, C.; De Propris, M.S.; Loglisci, G.; Latagliata, R.; Limongi, M.Z.; Testi, A.M.; et al. Very late acute myeloid leukemia relapse: Clinical features, treatment and outcome. Leuk. Lymphoma 2020, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Bene, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [Green Version]
- Paietta, E. Consensus on MRD in AML? Blood 2018, 131, 1265–1266. [Google Scholar] [CrossRef]
- Rowe, J.M. Progress and predictions: AML in 2018. Best Pract. Res. Clin. Haematol. 2018, 31, 337–340. [Google Scholar] [CrossRef]
- Corces, M.R.; Chang, H.Y.; Majeti, R. Preleukemic Hematopoietic Stem Cells in Human Acute Myeloid Leukemia. Front. Oncol. 2017, 7, 263. [Google Scholar] [CrossRef] [Green Version]
- Ediriwickrema, A.; Aleshin, A.; Reiter, J.G.; Corces, M.R.; Kohnke, T.; Stafford, M.; Liedtke, M.; Medeiros, B.C.; Majeti, R. Single-cell mutational profiling enhances the clinical evaluation of AML MRD. Blood Adv. 2020, 4, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Horibata, S.; Alyateem, G.; DeStefano, C.B.; Gottesman, M.M. The Evolving AML Genomic Landscape: Therapeutic Implications. Curr. Cancer Drug Targets 2020. [Google Scholar] [CrossRef]
- Ramos, N.R.; Mo, C.C.; Karp, J.E.; Hourigan, C.S. Current Approaches in the Treatment of Relapsed and Refractory Acute Myeloid Leukemia. J. Clin. Med. 2015, 4, 665–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, D.J.; Wilkes, E.; Casado, P.; Rajeeve, V.; Fitzgibbon, J.; Gribben, J.; Cutillas, P.R. Proteomic Analysis Directs Effective Drug Selection in Relapsed AML By Quantifying Drug Targets. Blood 2016, 128. [Google Scholar] [CrossRef]
- Ball, B.; Stein, E.M. Which are the most promising targets for minimal residual disease-directed therapy in acute myeloid leukemia prior to allogeneic stem cell transplant? Haematologica 2019, 104, 1521–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schraw, J.M.; Junco, J.J.; Brown, A.L.; Scheurer, M.E.; Rabin, K.R.; Lupo, P.J. Metabolomic profiling identifies pathways associated with minimal residual disease in childhood acute lymphoblastic leukaemia. Ebiomedicine 2019, 48, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, M.; Wang, F.; Loghavi, S.; Bueso-Ramos, C.; Gumbs, C.; Little, L.; Song, X.Z.; Zhang, J.H.; Kadia, T.; Borthakur, G.; et al. Late relapse in acute myeloid leukemia (AML): Clonal evolution or therapy-related leukemia? Blood Cancer J. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Cocciardi, S.; Dolnik, A.; Kapp-Schwoerer, S.; Rucker, F.G.; Lux, S.; Blatte, T.J.; Skambraks, S.; Kronke, J.; Heidel, F.H.; Schnoder, T.M.; et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hollein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 mutated AML can relapse with wild-type NPM1: Persistent clonal hematopoiesis can drive relapse. Blood Adv. 2018, 2, 3118–3125. [Google Scholar] [CrossRef]
- Cutillas, P.R.; (Queen Mary University of London, London, UK). Personal communication, 2020.
- Aasebo, E.; Berven, F.S.; Bartaula-Brevik, S.; Stokowy, T.; Hovland, R.; Vaudel, M.; Doskeland, S.O.; McCormack, E.; Batth, T.S.; Olsen, J.V.; et al. Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers 2020, 12, 709. [Google Scholar] [CrossRef] [Green Version]
- Ramadass, M.; Catz, S.D. Molecular mechanisms regulating secretory organelles and endosomes in neutrophils and their implications for inflammation. Immunol. Rev. 2016, 273, 249–265. [Google Scholar] [CrossRef]
- Kadry, Y.A.; Calderwood, D.A. Chapter 22: Structural and signaling functions of integrins. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183206. [Google Scholar] [CrossRef]
- Michael, M.; Parsons, M. New perspectives on integrin-dependent adhesions. Curr. Opin. Cell Biol. 2020, 63, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.; Rodriguez-Prados, J.C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-Substrate Enrichment Analysis Provides Insights into the Heterogeneity of Signaling Pathway Activation in Leukemia Cells. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Wiredja, D.D.; Koyuturk, M.; Chance, M.R. The KSEA App: A web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.W.; Tsai, C.H.; Lin, C.C.; Tien, F.M.; Chen, Y.W.; Lin, H.Y.; Yao, M.; Lin, Y.C.; Lin, C.T.; Cheng, C.L.; et al. Cytogenetics and mutations could predict outcome in relapsed and refractory acute myeloid leukemia patients receiving BCL-2 inhibitor venetoclax. Ann. Hematol. 2020, 99, 501–511. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Elson, P.; Kalaycio, M.E.; Advani, A.S.; Copelan, E.A.; Faderl, S.; Kantarjian, H.M.; Estey, E. Time from diagnosis to treatment initiation predicts survival in younger, but not older, acute myeloid leukemia patients. Blood 2009, 113, 28–36. [Google Scholar] [CrossRef]
- Bruserud, O.; Gjertsen, B.T.; Foss, B.; Huang, T.S. New strategies in the treatment of acute myelogenous leukemia (AML): In Vitro culture of aml cells—The present use in experimental studies and the possible importance for future therapeutic approaches. Stem Cells 2001, 19, 1–11. [Google Scholar] [CrossRef]
- Skrtic, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Schimmer, A.D.; Skrtic, M. Therapeutic potential of mitochondrial translation inhibition for treatment of acute myeloid leukemia. Expert Rev. Hematol. 2012, 5, 117–119. [Google Scholar] [CrossRef] [Green Version]
- Sharon, D.; Cathelin, S.; Subedi, A.; Williams, R.; Benicio, M.; Ketela, T.; Chan, S.M. Targeting Mitochondrial Translation Overcomes Venetoclax Resistance in Acute Myeloid Leukemia (AML) through Activation of the Integrated Stress Response. Blood 2017, 130, 297. [Google Scholar]
- Sharon, D.; Cathelin, S.; Mirali, S.; Di Trani, J.M.; Yanofsky, D.J.; Keon, K.A.; Rubinstein, J.L.; Schimmer, A.D.; Ketela, T.; Chan, S.M. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Gausdal, G.; Gjertsen, B.T.; McCormack, E.; Damme, P.; Hovland, R.; Krakstad, C.; Bruserud, O.; Gevaert, K.; Vandekerckhove, J.; Doskeland, S.O. Abolition of stress-induced protein synthesis sensitizes leukemia cells to anthracycline-induced death. Blood 2008, 111, 2866–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruserud, O.; Ryningen, A.; Olsnes, A.M.; Stordrange, L.; Oyan, A.M.; Kalland, K.H.; Gjertsen, B.T. Subclassification of patients with acute myelogenous leukemia based on chemokine responsiveness and constitutive chemokine release by their leukemic cells. Haematologica 2007, 92, 332–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reikvam, H.; Hatfield, K.J.; Lassalle, P.; Kittang, A.O.; Ersvaer, E.; Bruserud, O. Targeting the angiopoietin (Ang)/Tie-2 pathway in the crosstalk between acute myeloid leukaemia and endothelial cells: Studies of Tie-2 blocking antibodies, exogenous Ang-2 and inhibition of constitutive agonistic Ang-1 release. Expert Opin. Investig. Drugs 2010, 19, 169–183. [Google Scholar] [CrossRef]
- Reikvam, H.; Hatfield, K.J.; Oyan, A.M.; Kalland, K.H.; Kittang, A.O.; Bruserud, O. Primary human acute myelogenous leukemia cells release matrix metalloproteases and their inhibitors: Release profile and pharmacological modulation. Eur. J. Haematol. 2010, 84, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Bademosi, A.T.; Luo, J.; Meunier, F.A. Actin Remodeling in Regulated Exocytosis: Toward a Mesoscopic View. Trends Cell Biol. 2018, 28, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Brenner, A.K.; Reikvam, H.; Bruserud, O. A Subset of Patients with Acute Myeloid Leukemia Has Leukemia Cells Characterized by Chemokine Responsiveness and Altered Expression of Transcriptional as well as Angiogenic Regulators. Front. Immunol. 2016, 7, 205. [Google Scholar] [CrossRef] [Green Version]
- Honnemyr, M.; Bruserud, O.; Brenner, A.K. The constitutive protease release by primary human acute myeloid leukemia cells. J. Cancer Res. Clin. Oncol. 2017, 143, 1985–1998. [Google Scholar] [CrossRef]
- Lee, D.J.; Zeidner, J.F. Cyclin-dependent kinase (CDK) 9 and 4/6 inhibitors in acute myeloid leukemia (AML): A promising therapeutic approach. Expert Opin. Investig. Drugs 2019, 28, 989–1001. [Google Scholar] [CrossRef]
- Xie, S.; Jiang, H.; Zhai, X.W.; Wei, F.; Wang, S.D.; Ding, J.; Chen, Y. Antitumor action of CDK inhibitor LS-007 as a single agent and in combination with ABT-199 against human acute leukemia cells. Acta Pharmacol. Sin. 2016, 37, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Li, K.L.; Bray, S.C.; Iarossi, D.; Adams, J.; Zhong, L.J.; Noll, B.; Rahaman, M.H.; Richmond, J.; To, L.B.; Lewis, I.D.; et al. Investigation of a Novel Cyclin-Dependent-Kinase (CDK) Inhibitor Cdki-73 As an Effective Treatment Option for MLL-AML. Blood 2015, 126. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, G.; Schuurhuis, G.J. MRD in AML: Does it already guide therapy decision-making? Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, K.J.; Hovland, R.; Øyan, A.M.; Kalland, K.H.; Ryningen, A.; Gjertsen, B.T.; Bruserud, Ø. Release of angiopoietin-1 by primary human acute myelogenous leukemia cells is associated with mutations of nucleophosmin, increased by bone marrow stromal cells and possibly antagonized by high systemic angiopoietin-2 levels. Leukemia 2008, 22, 287–293. [Google Scholar] [CrossRef]
- Reikvam, H.; Hovland, R.; Forthun, R.B.; Erdal, S.; Gjertsen, B.T.; Fredly, H.; Bruserud, O. Disease-stabilizing treatment based on all-trans retinoic acid and valproic acid in acute myeloid leukemia-identification of responders by gene expression profiling of pretreatment leukemic cells. BMC Cancer 2017, 17, 630. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Hernandez-Valladares, M.; Aasebø, E.; Mjaavatten, O.; Vaudel, M.; Bruserud, Ø.; Berven, F.; Selheim, F. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biol. Proced. Online 2016, 18, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–D1106. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Arntzen, M.Ø.; Koehler, C.J.; Barsnes, H.; Berven, F.S.; Treumann, A.; Thiede, B. IsobariQ: Software for isobaric quantitative proteomics using IPTL, iTRAQ, and TMT. J. Proteome Res. 2011, 10, 913–920. [Google Scholar] [CrossRef]
- Scholz, C.; Lyon, D.; Refsgaard, J.C.; Jensen, L.J.; Choudhary, C.; Weinert, B.T. Avoiding abundance bias in the functional annotation of post-translationally modified proteins. Nat. Methods 2015, 12, 1003–1004. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patient Code | Treatment at Diagnosis | Chemotherapy Cycles before CR | Time from CR1 to Relapse (Months) | Chemotherapy after Relapse | Response to Intensive Chemotherapy | Survival after Diagnosed First Relapse (Months) |
|---|---|---|---|---|---|---|
| PX1 | ICT/CCT | 2 | 8 | ICT | No CR2, death from chemoresistant disease | 3 |
| PX2 | ICT/CCT | 1 | 16 | ST | No CR2 | 1 |
| PX3 | ICT/CCT | 1 | 27 | ICT | Allogeneic retransplantation in CR2, death due to chemoresistant second relapse | 10 |
| PX4 | ICT/CCT | 1 | 13 | ST | No CR2 | 2 |
| PX5 | ICT/CCT | 1 | 5 | ICT | No CR2, death due to infection during neutropenia | 1 |
| PX6 | ICT/CCT | 1 | 3 | ICT | No CR2, death from chemoresistant disease | 2 |
| PX7 | ICT/CCT | 2 | 2 | ICT | No CR2 | 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aasebø, E.; Berven, F.S.; Hovland, R.; Døskeland, S.O.; Bruserud, Ø.; Selheim, F.; Hernandez-Valladares, M. The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles. Cancers 2020, 12, 1466. https://doi.org/10.3390/cancers12061466
Aasebø E, Berven FS, Hovland R, Døskeland SO, Bruserud Ø, Selheim F, Hernandez-Valladares M. The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles. Cancers. 2020; 12(6):1466. https://doi.org/10.3390/cancers12061466
Chicago/Turabian StyleAasebø, Elise, Frode S. Berven, Randi Hovland, Stein Ove Døskeland, Øystein Bruserud, Frode Selheim, and Maria Hernandez-Valladares. 2020. "The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles" Cancers 12, no. 6: 1466. https://doi.org/10.3390/cancers12061466
APA StyleAasebø, E., Berven, F. S., Hovland, R., Døskeland, S. O., Bruserud, Ø., Selheim, F., & Hernandez-Valladares, M. (2020). The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles. Cancers, 12(6), 1466. https://doi.org/10.3390/cancers12061466