DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome


Abstract
:1. Introduction
2. Main Molecular Hallmarks of Ovarian Cancer and Association with DNA Repair System
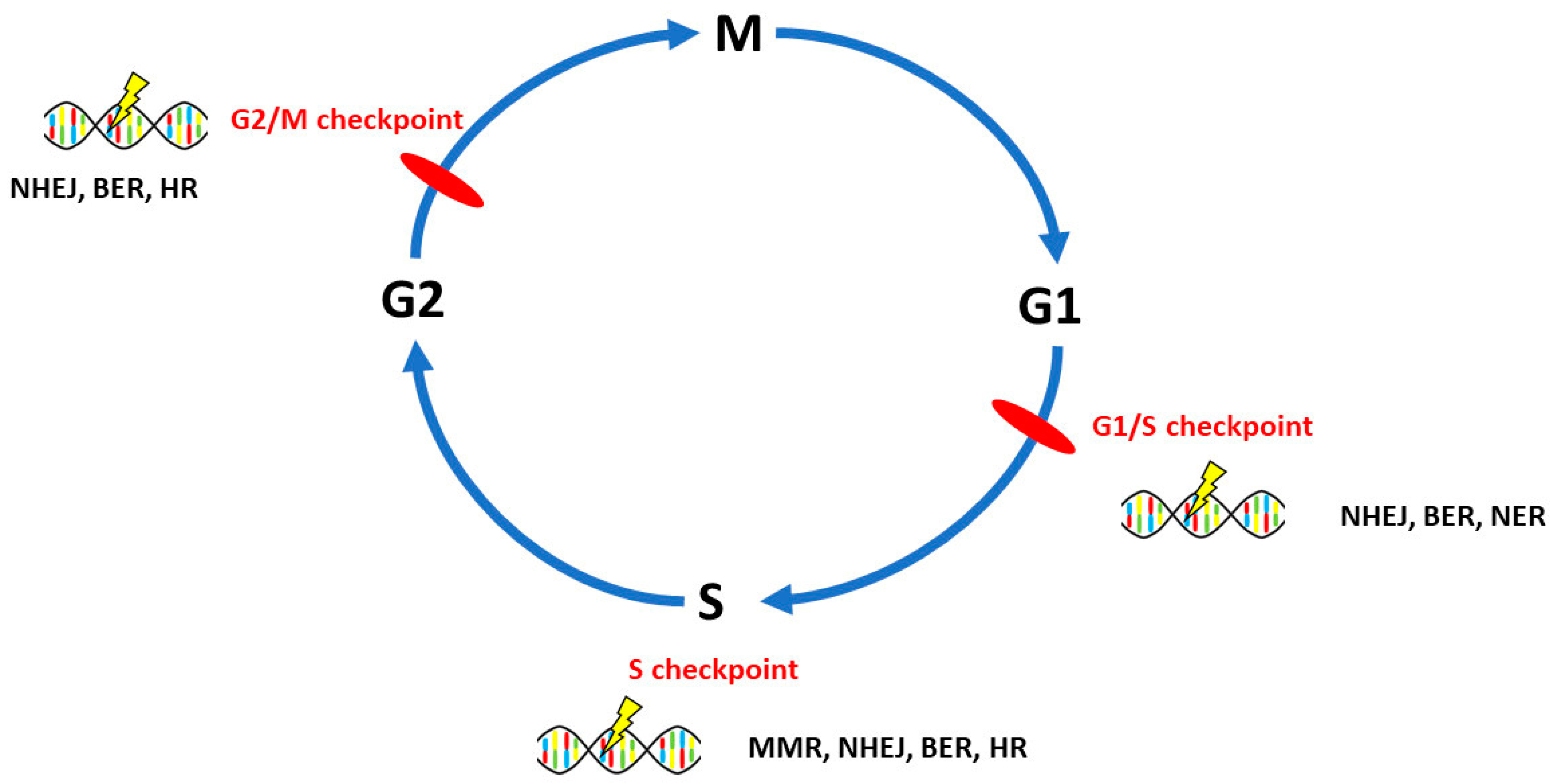
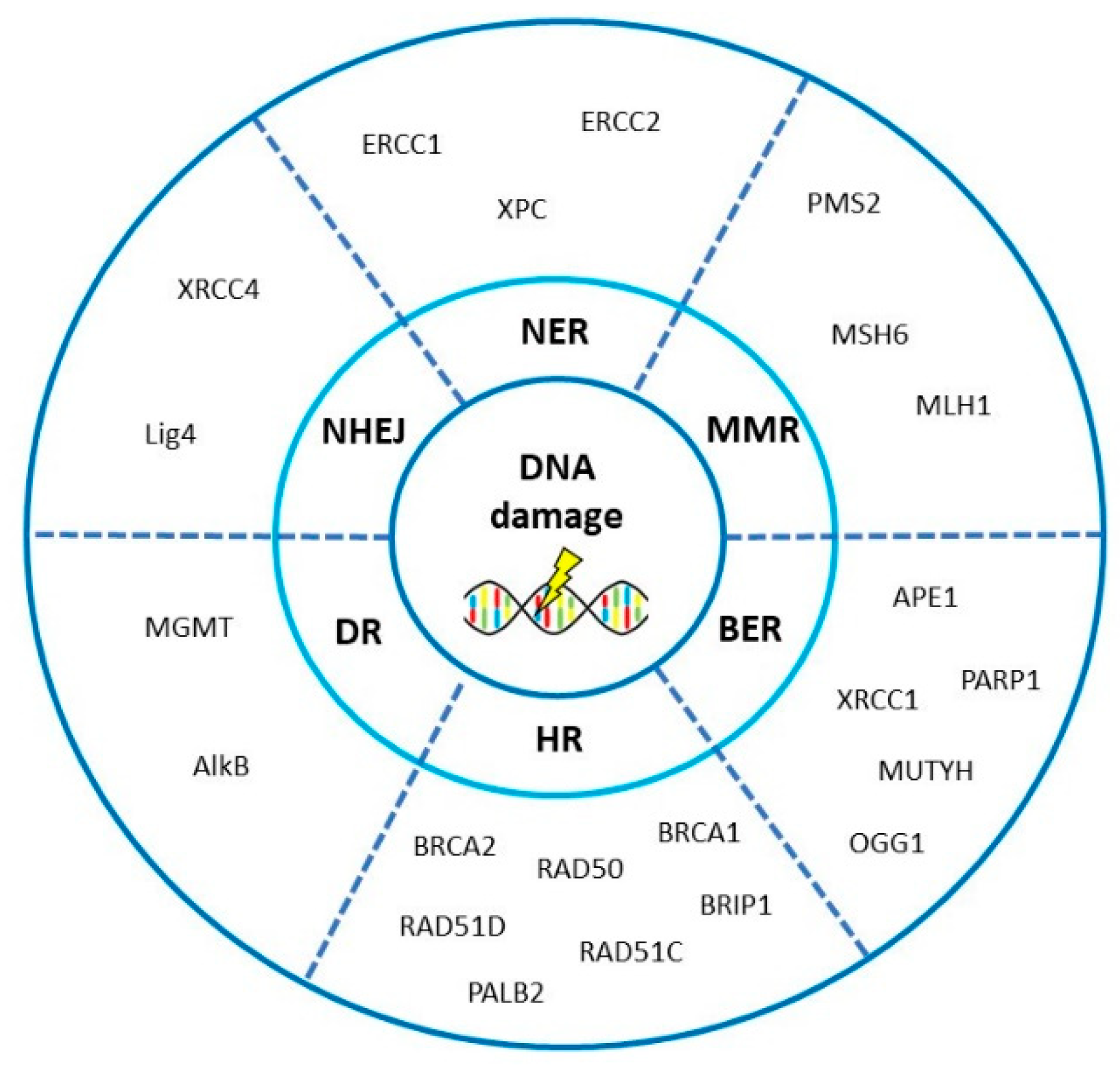
3. DNA Repair Pathways Involved in the Onset, Progression and Prognosis of Ovarian Cancer
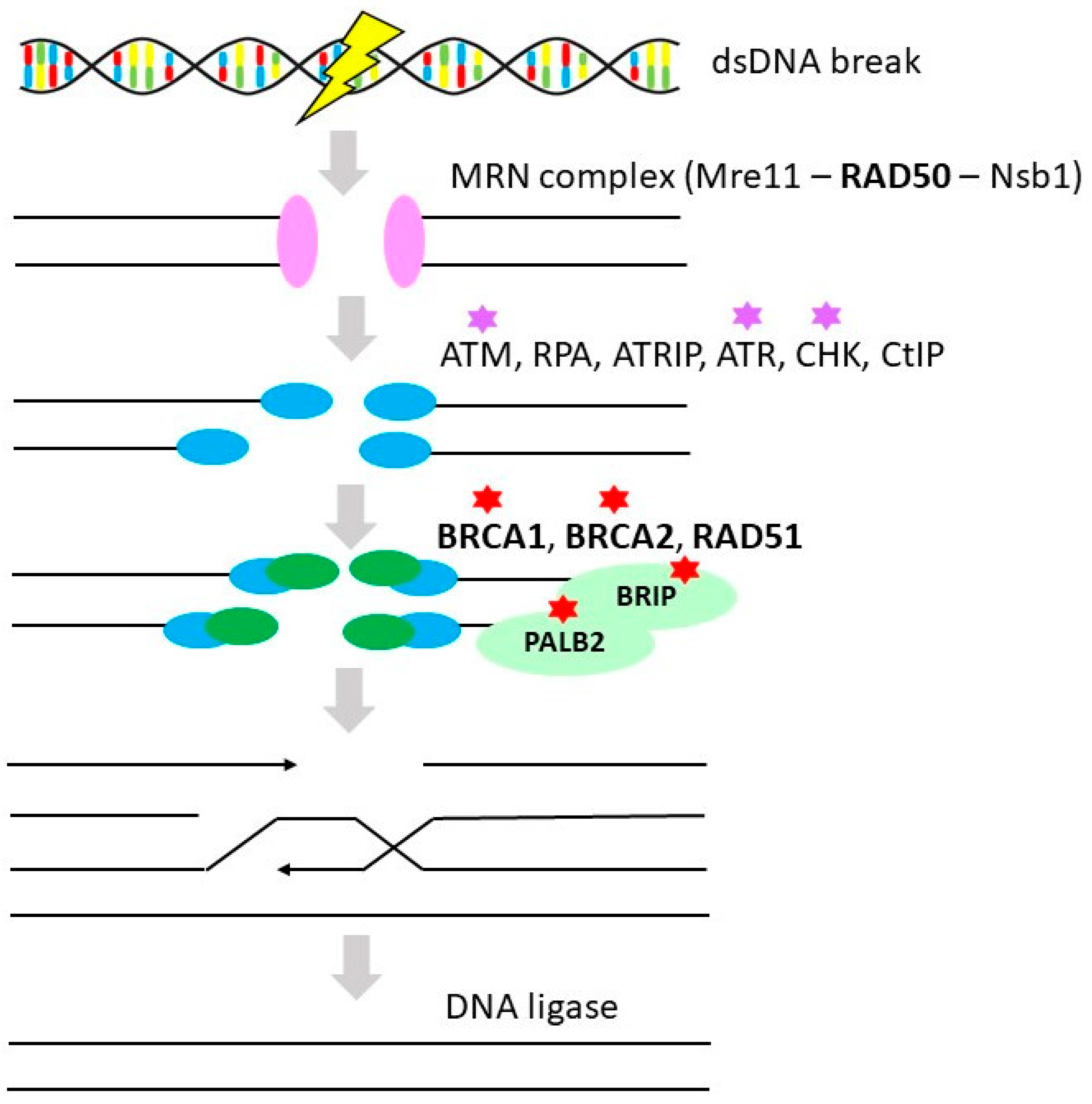
3.1. Homologous Recombination Repair
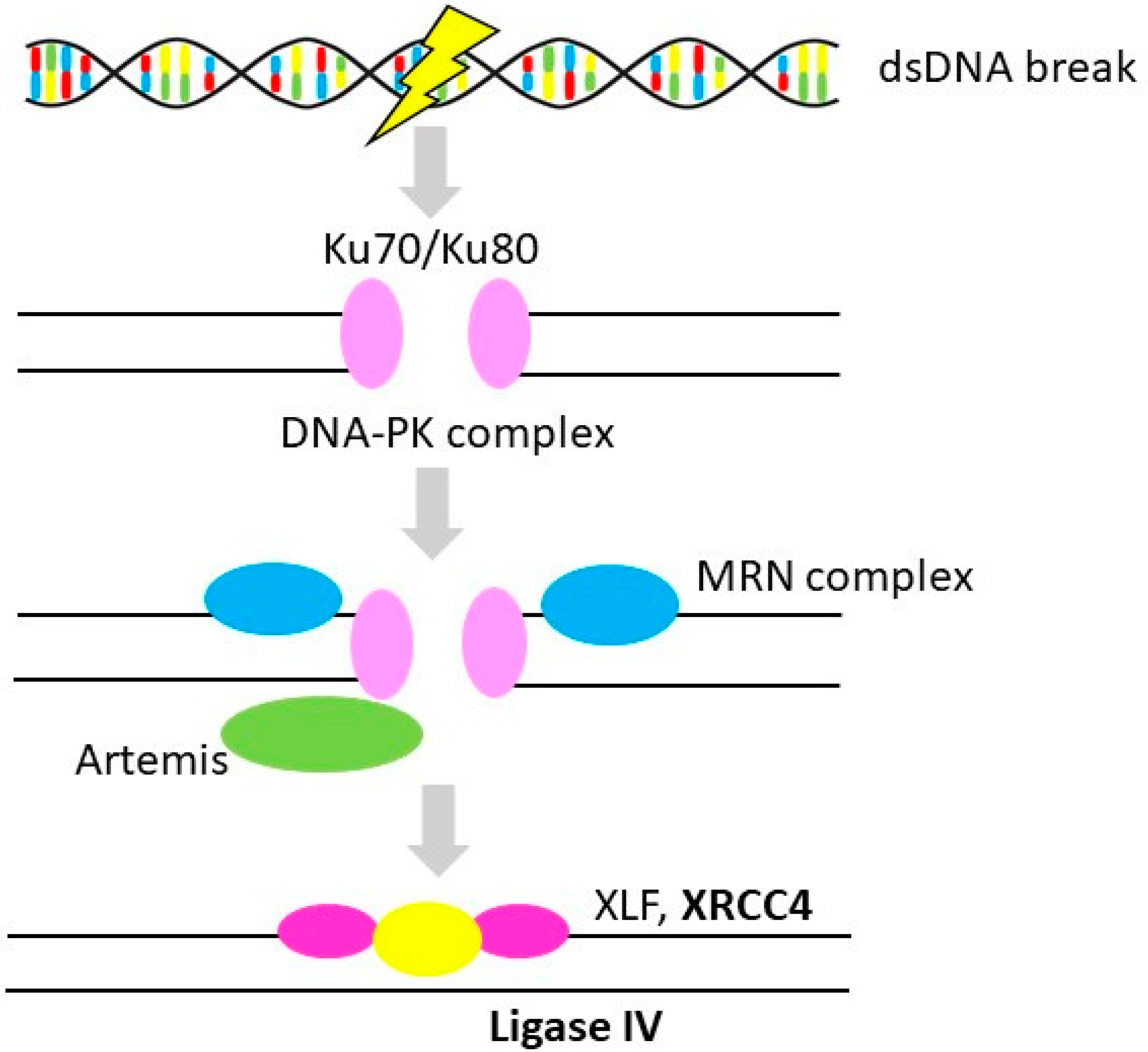
3.2. Non-Homologous End-Joining
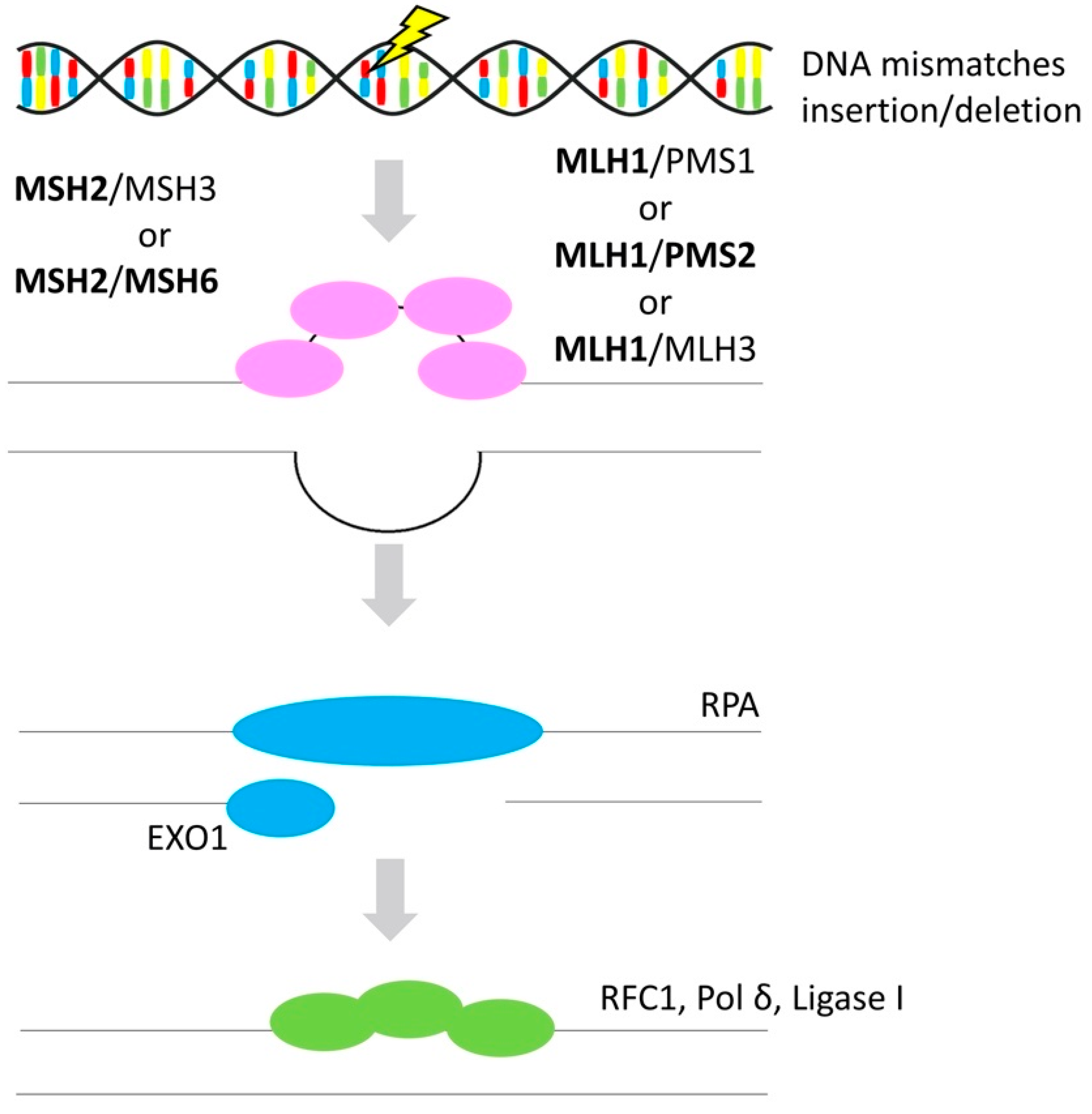
3.3. Mismatch Repair
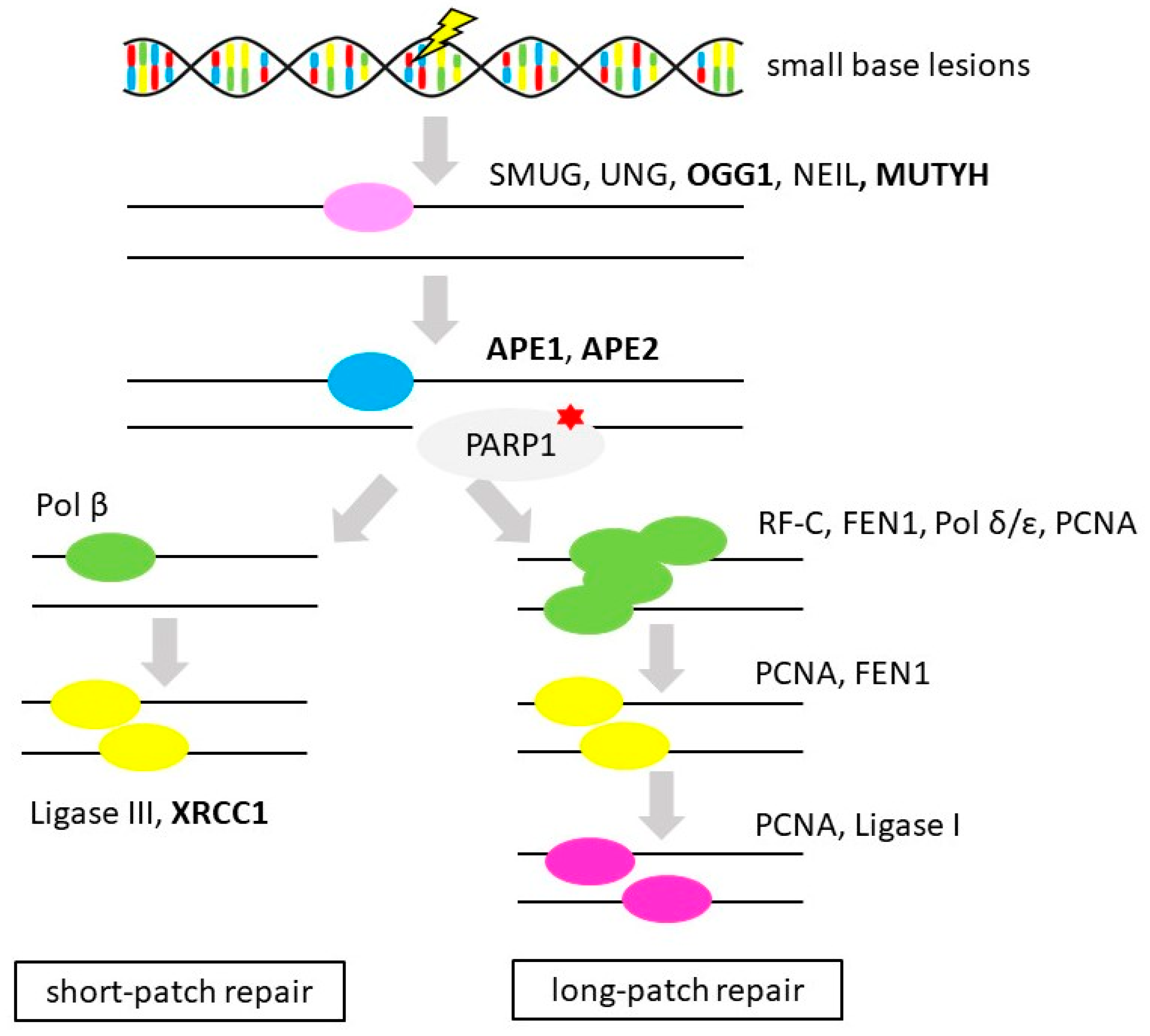
3.4. Base Excision Repair
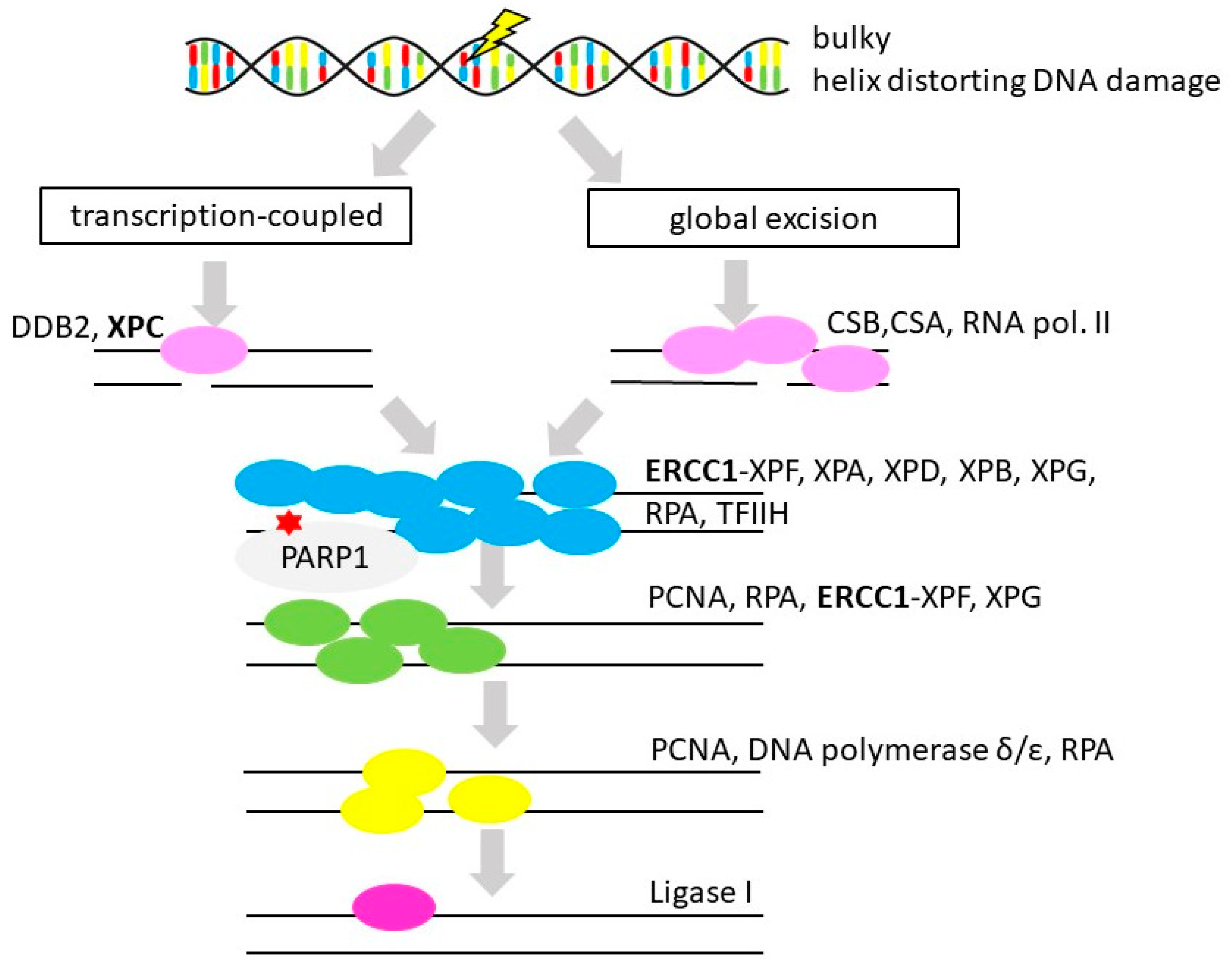
3.5. Nucleotide Excision Repair
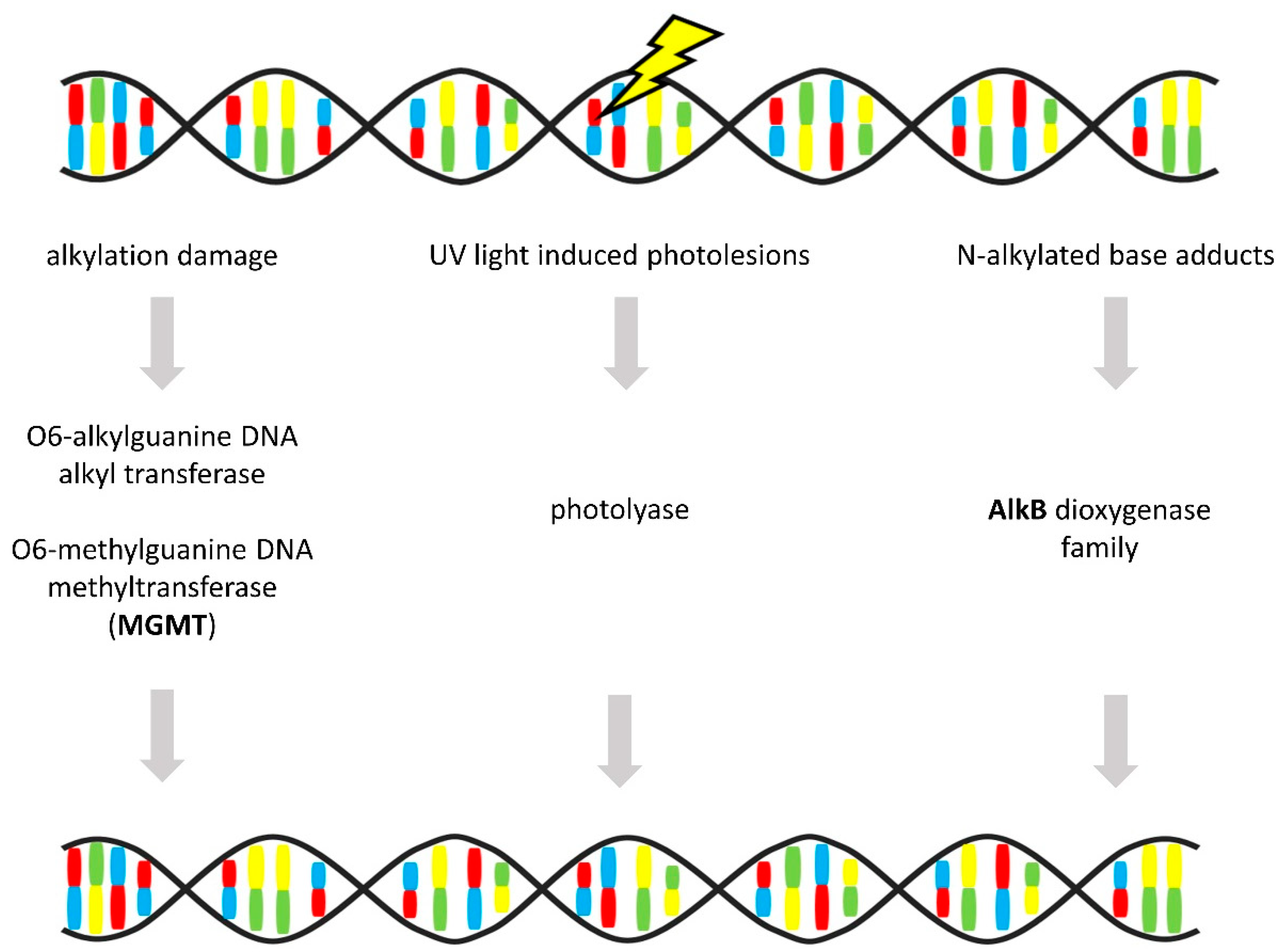
3.6. Direct Repair
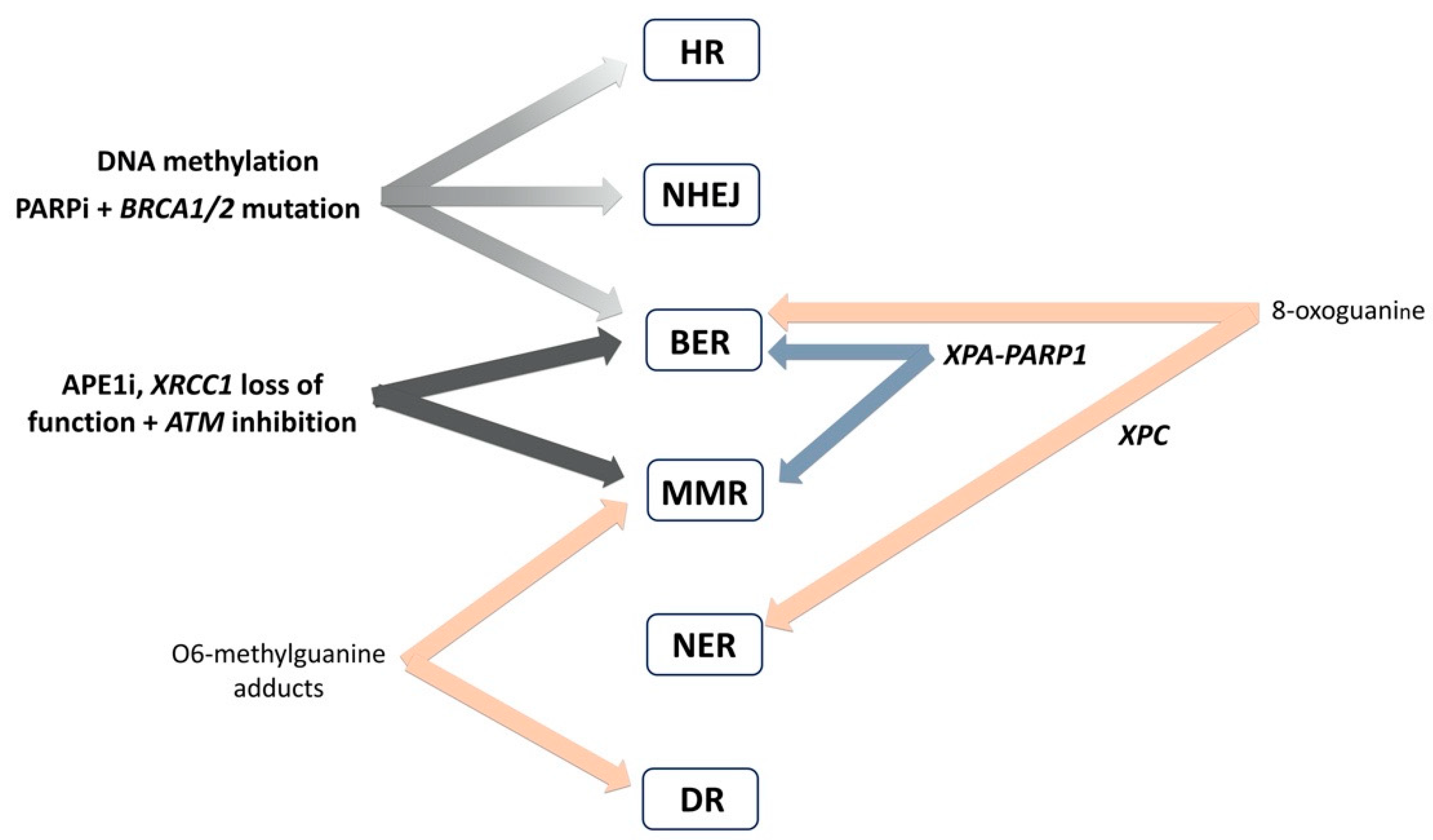
4. Interplay of DNA Repair Pathways
5. Therapeutic Perspectives–Targeting of DNA Repair System in Ovarian Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Repair Pathway | Gene Targets | In Vitro/In Vivo Efficiency | Pre-Clinical/Clinical Studies |
|---|---|---|---|
| Base Excision Repair | PARPi | Talazoparib and veliparib are in advanced clinical trials at the moment. Clinically available PARPi olaparib, rucaparib and niraparib are currently approved for the therapy of OvC on the basis of their BRCA1/2 status (summarized in [210]) | Olaparib-approved by FDA and EMA for use in OvC therapy [144] Rucaparib-approved by FDA and EMA for use in OvC therapy [144] Niraparib-approved by FDA and EMA for use in OvC therapy [144] Veliparib–advanced clinical trials in combination with carboplatin and paclitaxel. Veliparib induction therapy followed by veliparib maintenance therapy led to significantly longer PFS than carboplatin plus paclitaxel induction therapy alone [193,194] Talazoparib–ongoing advanced clinical trials [194,195] |
| Cell cycle checkpoints | CHEK1i | The CHEK1i V158411, PF-477736 and AZD7762 inhibited the proliferation of OvC cells [202] AZD7762 in combination with cisplatin suggested synergistic effects in ovarian clear cell carcinoma cell lines in vitro and suppressed growth of tumors in vivo [203] Prexasertib–effective in monotherapy in PARPi-resistant HGSOC cell lines and mouse xenografts [204] Combination of prexasertib mesylate monohydrate (LY2606368), a CHEK1 and CHEK2 inhibitor, and a PARPi, olaparib synergistically decreased cell viability in HGSOC cell lines (OVCAR3, OV90, PEO1 and PEO4) cell lines and induced greater DNA damage and apoptosis than the control and/or monotherapies [204,211] | Prexasertib–effective in clinical phase II study in recurrent HGSOC [201] |
| ATRi | ATRi (VE-821, VE-822, AZ20) resensitized PARPi-resistant BRCA1-mutated human OvC cell line to PARPi [206] AZD6738 efficient in in ATM-deficient cells and in vivo in PDX mouse models with complete ATM loss [208] Combination PARPi with ATRi (AZD6738) and CHEK1i (MK8776) is more effective than PARPi alone in reducing tumor burden in BRCA1/2 mutated HGSOC cells and PDX models [209] | Ongoing clinical PhaseII CAPRI Study of ATRi AZD6738 (ceralasertib) in combination with PARPi olaparib in HGSOC patients [212] | |
| ATMi | ATMi KU55933 enhanced the response to ionizing radiation in A2780 and OVCAR3 OvC cells [213] | ||
| WEE1i | Adavosertib (AZD 1775 alias MK1775)–efficient in vitro in SKOV-3 and ID8 OvC cell lines, efficient in vivo in ID8 ovarian tumors in monotherapy independent on TP53 or BRCA1 status [214] | AZD1775–active in phase I clinical study of monotherapy in OvC patients carrying BRCA mutations [215] AZD1775–combination therapy with AZD1775 enhanced carboplatin efficacy in TP53-mutated ovarian tumors in phase II clinical study [216] |
6. Conclusions and Future Perspectives
- (i)
- We are facing the lack of systematic knowledge of DNA repair at various levels (i.e., genetic, epigenetic, protein, and functional) and their dynamic in the course of the disease. No available complex functional studies are characterizing any of the DNA repair pathways, as they do exist for other malignancies [226,227,228].
- (ii)
- Although genetic alterations in HR repair pathway and their role in OvC are characterized decently, very little is known about the main pathway restoring DSBs, NHEJ. What is its importance in OvC onset, prognosis, and prediction? In the context of the previous point, further studies are needed on mechanisms (involvement of DSB repair?) underlying chromosomal instability in OvC (such as amplifications, deletions, translocations).
- (iii)
- There is limited knowledge on the interaction of MMR (substantial in OvC etiology) with other DNA repair pathways. In this context, generally, more effort should be dedicated to the links between MMR (and other DNA repair pathways?) with immune response and with the microenvironment. These aspects may impact the patient’s prognosis, as they do in colon cancer.
- (iv)
- In general, there is a poor understanding of interactions among individual DDR players.
- (v)
- Contemporary studies illuminated interesting links between DNA damage, DNA repair, and DNA methylation/demethylation. This important aspect may exert future implications and consequences (epigenetic regulations).
- (vi)
- Epigenetic regulation of DNA repair/DDR via non-coding RNAs should further be addressed in relation to the disease onset, prognosis, and therapy outcome.
- (vii)
- There is a need to characterize OvC patients with a good and poor response with respect to the DNA repair system and its changes. Disclosure of critical determinants in DNA repair/DDR machinery could significantly contribute to the improvement of therapy success in OvC patients with multidrug-resistant tumors.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APE1i | APE1 inhibitors |
| ATMi | ATM inhibitors |
| ATRi | ATR inhibitors |
| BER | base excision repair |
| CHEK1i | CHEK1 inhibitors |
| DDR | DNA damage response |
| DSB | double-strand break |
| EMA | European Medicine Agency |
| EOC | epithelial ovarian carcinoma |
| FDA | Food and Drug Administration U.S. agency |
| GWAS | genome wide association study |
| HGSOC | high-grade serous ovarian carcinoma |
| HR | homologous recombination repair |
| MMR | mismatch repair |
| MSI | microsatellite instability |
| NER | nucleotide excision repair |
| NHEJ | non-homologous end joining |
| OvC | ovarian cancer |
| OS | overall survival |
| PARPi | poly(ADP-ribose) polymerase inhibitors |
| PFS | progression-free survival |
| SNP | single-nucleotide polymorphism |
| SSB | single-strand break |
| WEE1i | WEE1 inhibitors |
| wt | wild-type |
References
- Reilly, N.M.; Novara, L.; Di Nicolantonio, F.; Bardelli, A. Exploiting DNA repair defects in colorectal cancer. Mol. Oncol. 2019, 13, 681–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodicka, P.; Vodenkova, S.; Buchler, T.; Vodickova, L. DNA repair capacity and response to treatment of colon cancer. Pharmacogenomics 2019, 20, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Vodenkova, S.; Opattova, A.; Vodickova, L. DNA damage and repair measured by comet assay in cancer patients. Mutat. Res. 2019, 843, 95–110. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, G.; Fan, T.; Silberstein, P.T. Hereditary cancer syndromes: Utilizing DNA repair deficiency as therapeutic target. Fam. Cancer 2016, 15, 359–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grady, W.M.; Markowitz, S.D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig. Dis. Sci. 2015, 60, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Niskakoski, A.; Pasanen, A.; Porkka, N.; Eldfors, S.; Lassus, H.; Renkonen-Sinisalo, L.; Kaur, S.; Mecklin, J.P.; Butzow, R.; Peltomaki, P. Converging endometrial and ovarian tumorigenesis in Lynch syndrome: Shared origin of synchronous carcinomas. Gynecol. Oncol. 2018, 150, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, F.C.; van Overeem Hansen, T.; Sorensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medcines 2018, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurman, R.J.; Shih Ie, M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Markman, M. Optimizing primary chemotherapy in ovarian cancer. Hematol. Oncol. Clin. N. Am. 2003, 17, 957–968. [Google Scholar] [CrossRef]
- Davis, A.; Tinker, A.V.; Friedlander, M. “Platinum resistant” ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol. Oncol. 2014, 133, 624–631. [Google Scholar] [CrossRef]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef] [Green Version]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [Green Version]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global surveillance of cancer survival 1995-2009: Analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef] [Green Version]
- The World Ovarian Cancer Coalition Atlas, Global Trends in Incidence, Mortality and Survival. Available online: https://worldovariancancercoalition.org/wp-content/uploads/2018/10/THE-WORLD-OVARIAN-CANCER-COALITION-ATLAS-2018.pdf (accessed on 18 March 2020).
- DNA Repair Genes Pertinent Cancer Susceptibility (Version 1.1). Available online: https://panelapp.genomicsengland.co.uk/panels/256/ (accessed on 27 May 2020).
- Mirza-Aghazadeh-Attari, M.; Ostadian, C.; Saei, A.A.; Mihanfar, A.; Darband, S.G.; Sadighparvar, S.; Kaviani, M.; Samadi Kafil, H.; Yousefi, B.; Majidinia, M. DNA damage response and repair in ovarian cancer: Potential targets for therapeutic strategies. DNA Repair 2019, 80, 59–84. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.E.; Faraahi, Z.; McCormick, A.; Edmondson, R.J. DNA damage repair in ovarian cancer: Unlocking the heterogeneity. J. Ovarian Res. 2018, 11, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, B.; Zhang, Z.; Li, Y. Association of MGMT promoter methylation with tumorigenesis features in patients with ovarian cancer: A systematic meta-analysis. Mol. Genet. Genom. Med. 2018, 6, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Brandsma, I.; Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Silwal-Pandit, L.; Langerod, A.; Borresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect Med. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Hernandez, G.; Ramirez, M.J.; Minguillon, J.; Quiles, P.; Ruiz de Garibay, G.; Aza-Carmona, M.; Bogliolo, M.; Pujol, R.; Prados-Carvajal, R.; Fernandez, J.; et al. Decapping protein EDC4 regulates DNA repair and phenocopies BRCA1. Nat. Commun. 2018, 9, 967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan Coyne, G.; Chen, A.P.; Meehan, R.; Doroshow, J.H. PARP Inhibitors in Reproductive System Cancers: Current Use and Developments. Drugs 2017, 77, 113–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rejhova, A.; Opattova, A.; Cumova, A.; Sliva, D.; Vodicka, P. Natural compounds and combination therapy in colorectal cancer treatment. Eur. J. Med. Chem. 2018, 144, 582–594. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Geuting, V.; Lobrich, M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother. Oncol. 2011, 101, 7–12. [Google Scholar] [CrossRef]
- Covo, S.; Ma, W.; Westmoreland, J.W.; Gordenin, D.A.; Resnick, M.A. Understanding the origins of UV-induced recombination through manipulation of sister chromatid cohesion. Cell Cycle 2012, 11, 3937–3944. [Google Scholar] [CrossRef] [Green Version]
- Reliene, R.; Bishop, A.J.; Schiestl, R.H. Involvement of homologous recombination in carcinogenesis. Adv. Genet. 2007, 58, 67–87. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, C.; He, C. DNA repair by reversal of DNA damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012575. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsopoulos, J.; Gronwald, J.; Karlan, B.; Rosen, B.; Huzarski, T.; Moller, P.; Lynch, H.T.; Singer, C.F.; Senter, L.; Neuhausen, S.L.; et al. Age-specific ovarian cancer risks among women with a BRCA1 or BRCA2 mutation. Gynecol. Oncol. 2018, 150, 85–91. [Google Scholar] [CrossRef]
- Doufekas, K.; Olaitan, A. Clinical epidemiology of epithelial ovarian cancer in the UK. Int. J. Womens Health 2014, 6, 537–545. [Google Scholar] [CrossRef] [Green Version]
- SEER Cancer Statistics Review (CSR) 1975–2015. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 18 March 2020).
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [Green Version]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012, 307, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Ruscito, I.; Dimitrova, D.; Vasconcelos, I.; Gellhaus, K.; Schwachula, T.; Bellati, F.; Zeillinger, R.; Benedetti-Panici, P.; Vergote, I.; Mahner, S.; et al. BRCA1 gene promoter methylation status in high-grade serous ovarian cancer patients—A study of the tumour Bank ovarian cancer (TOC) and ovarian cancer diagnosis consortium (OVCAD). Eur. J. Cancer 2014, 50, 2090–2098. [Google Scholar] [CrossRef]
- Gourley, C.; Michie, C.O.; Roxburgh, P.; Yap, T.A.; Harden, S.; Paul, J.; Ragupathy, K.; Todd, R.; Petty, R.; Reed, N.; et al. Increased incidence of visceral metastases in scottish patients with BRCA1/2-defective ovarian cancer: An extension of the ovarian BRCAness phenotype. J. Clin. Oncol. 2010, 28, 2505–2511. [Google Scholar] [CrossRef]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Vaz, F.; Hanenberg, H.; Schuster, B.; Barker, K.; Wiek, C.; Erven, V.; Neveling, K.; Endt, D.; Kesterton, I.; Autore, F.; et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat. Genet. 2010, 42, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.M.; Cicek, M.S.; Larson, N.B.; Davila, J.; Wang, C.; Larson, M.C.; Song, H.; Dicks, E.M.; Harrington, P.; Wick, M.; et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci. Rep. 2014, 4, 4026. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ruark, E.; Xicola, R.M.; Ramsay, E.; Hughes, D.; Warren-Perry, M.; Snape, K.; Breast Cancer Susceptibility, C.; Eccles, D.; et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet. 2012, 44, 475–476. [Google Scholar] [CrossRef]
- Arvai, K.J.; Roberts, M.E.; Torene, R.I.; Susswein, L.R.; Marshall, M.L.; Zhang, Z.; Carter, N.J.; Yackowski, L.; Rinella, E.S.; Klein, R.T.; et al. Age-adjusted association of homologous recombination genes with ovarian cancer using clinical exomes as controls. Hered. Cancer Clin. Pract. 2019, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.R.; Rowley, S.M.; Sawyer, S.; Eccles, D.M.; Trainer, A.H.; Mitchell, G.; James, P.A.; Campbell, I.G. Analysis of RAD51D in ovarian cancer patients and families with a history of ovarian or breast cancer. PLoS ONE 2013, 8, e54772. [Google Scholar] [CrossRef]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- AlHilli, M.M.; Becker, M.A.; Weroha, S.J.; Flatten, K.S.; Hurley, R.M.; Harrell, M.I.; Oberg, A.L.; Maurer, M.J.; Hawthorne, K.M.; Hou, X.; et al. In vivo anti-tumor activity of the PARP inhibitor niraparib in homologous recombination deficient and proficient ovarian carcinoma. Gynecol. Oncol. 2016, 143, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis Oncol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Samartzis, E.P.; Zimmermann, A.K.; Fink, D.; Moch, H.; Noske, A.; Dedes, K.J. Lack of MRE11-RAD50-NBS1 (MRN) complex detection occurs frequently in low-grade epithelial ovarian cancer. BMC Cancer 2017, 17, 44. [Google Scholar] [CrossRef] [Green Version]
- Kessous, R.; Octeau, D.; Klein, K.; Tonin, P.N.; Greenwood, C.M.T.; Pelmus, M.; Laskov, I.; Kogan, L.; Salvador, S.; Lau, S.; et al. Distinct homologous recombination gene expression profiles after neoadjuvant chemotherapy associated with clinical outcome in patients with ovarian cancer. Gynecol. Oncol. 2018, 148, 553–558. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, G.; Xue, F.; Edwards, R.; Sood, A.K.; Zhang, W.; Yang, D. Copy number deletion of RAD50 as predictive marker of BRCAness and PARP inhibitor response in BRCA wild type ovarian cancer. Gynecol. Oncol. 2016, 141, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Nepomuceno, T.C.; De Gregoriis, G.; de Oliveira, F.M.B.; Suarez-Kurtz, G.; Monteiro, A.N.; Carvalho, M.A. The Role of PALB2 in the DNA Damage Response and Cancer Predisposition. Int. J. Mol. Sci. 2017, 18, 1886. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkas, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Kluska, A.; Balabas, A.; Piatkowska, M.; Czarny, K.; Paczkowska, K.; Nowakowska, D.; Mikula, M.; Ostrowski, J. PALB2 mutations in BRCA1/2-mutation negative breast and ovarian cancer patients from Poland. BMC Med. Genom. 2017, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef]
- Poti, A.; Gyergyak, H.; Nemeth, E.; Rusz, O.; Toth, S.; Kovacshazi, C.; Chen, D.; Szikriszt, B.; Spisak, S.; Takeda, S.; et al. Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 2019, 20, 240. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Hampton, O.A.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; et al. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr. Blood Cancer 2015, 62, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seal, S.; Thompson, D.; Renwick, A.; Elliott, A.; Kelly, P.; Barfoot, R.; Chagtai, T.; Jayatilake, H.; Ahmed, M.; Spanova, K.; et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 1239–1241. [Google Scholar] [CrossRef]
- Thompson, E.R.; Rowley, S.M.; Li, N.; McInerny, S.; Devereux, L.; Wong-Brown, M.W.; Trainer, A.H.; Mitchell, G.; Scott, R.J.; James, P.A.; et al. Panel Testing for Familial Breast Cancer: Calibrating the Tension Between Research and Clinical Care. J. Clin. Oncol. 2016, 34, 1455–1459. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [Green Version]
- Easton, D.F.; Lesueur, F.; Decker, B.; Michailidou, K.; Li, J.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; et al. No evidence that protein truncating variants in BRIP1 are associated with breast cancer risk: Implications for gene panel testing. J. Med. Genet. 2016, 53, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Slavin, T.P.; Maxwell, K.N.; Lilyquist, J.; Vijai, J.; Neuhausen, S.L.; Hart, S.N.; Ravichandran, V.; Thomas, T.; Maria, A.; Villano, D.; et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer 2017, 3, 22. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Weber-Lassalle, N.; Hauke, J.; Ramser, J.; Richters, L.; Gross, E.; Blumcke, B.; Gehrig, A.; Kahlert, A.K.; Muller, C.R.; Hackmann, K.; et al. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res. 2018, 20, 7. [Google Scholar] [CrossRef]
- Moyer, C.L.; Ivanovich, J.; Gillespie, J.L.; Doberstein, R.; Radke, M.R.; Richardson, M.E.; Kaufmann, S.H.; Swisher, E.M.; Goodfellow, P.J. Rare BRIP1 Missense Alleles Confer Risk for Ovarian and Breast Cancer. Cancer Res. 2020, 80, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Willers, H.; Feng, Z.; Ghosh, J.C.; Kim, S.; Weaver, D.T.; Chung, J.H.; Powell, S.N.; Xia, F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell Biol. 2004, 24, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buisson, R.; Masson, J.Y. PALB2 self-interaction controls homologous recombination. Nucleic Acids Res. 2012, 40, 10312–10323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrenson, K.; Iversen, E.S.; Tyrer, J.; Weber, R.P.; Concannon, P.; Hazelett, D.J.; Li, Q.; Marks, J.R.; Berchuck, A.; Lee, J.M.; et al. Common variants at the CHEK2 gene locus and risk of epithelial ovarian cancer. Carcinogenesis 2015, 36, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Phelan, C.M.; Kuchenbaecker, K.B.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.J.; Dennis, J.; Pirie, A.; Riggan, M.J.; Chornokur, G.; et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017, 49, 680–691. [Google Scholar] [CrossRef] [Green Version]
- Michalska, M.M.; Samulak, D.; Romanowicz, H.; Bienkiewicz, J.; Sobkowski, M.; Ciesielski, K.; Smolarz, B. Single nucleotide polymorphisms (SNPs) of hOGG1 and XRCC1 DNA repair genes and the risk of ovarian cancer in Polish women. Tumour Biol. 2015, 36, 9457–9463. [Google Scholar] [CrossRef]
- Chen, X.; Liu, X.; Wang, J.; Guo, W.; Sun, C.; Cai, Z.; Wu, Q.; Xu, X.; Wang, Y. Functional polymorphisms of the hOGG1 gene confer risk to type 2 epithelial ovarian cancer in Chinese. Int. J. Gynecol. Cancer 2011, 21, 1407–1413. [Google Scholar] [CrossRef]
- Osorio, A.; Milne, R.L.; Kuchenbaecker, K.; Vaclova, T.; Pita, G.; Alonso, R.; Peterlongo, P.; Blanco, I.; de la Hoya, M.; Duran, M.; et al. DNA glycosylases involved in base excision repair may be associated with cancer risk in BRCA1 and BRCA2 mutation carriers. PLoS Genet. 2014, 10, e1004256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xin, X.; Zhang, J.; Li, J.; Chen, B.; Zou, W. Apurinic/apyrimidinic endonuclease 1 polymorphisms are associated with ovarian cancer susceptibility in a Chinese population. Int. J. Gynecol. Cancer 2013, 23, 1393–1399. [Google Scholar] [CrossRef]
- Malisic, E.J.; Krivokuca, A.M.; Boljevic, I.Z.; Jankovic, R.N. Impact of RAD51 G135C and XRCC1 Arg399Gln polymorphisms on ovarian carcinoma risk in Serbian women. Cancer Biomark. 2015, 15, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Zhang, X.; Tang, Q.L.; Wang, X.Y.; Kai, L. Prediction value of XRCC 1 gene polymorphism on the survival of ovarian cancer treated by adjuvant chemotherapy. Asian Pac. J. Cancer Prev. 2012, 13, 5007–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Li, W. Association between polymorphisms of XRCC1 and ADPRT genes and ovarian cancer survival with platinum-based chemotherapy in Chinese population. Mol. Cell Biochem. 2013, 372, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, M.; Gennery, A.R.; Seidel, J.; Concannon, P.; Jeggo, P.A. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair 2004, 3, 1227–1235. [Google Scholar] [CrossRef]
- Sekiguchi, J.M.; Ferguson, D.O. DNA double-strand break repair: A relentless hunt uncovers new prey. Cell 2006, 124, 260–262. [Google Scholar] [CrossRef] [Green Version]
- Bentley, J.; L’Hote, C.; Platt, F.; Hurst, C.D.; Lowery, J.; Taylor, C.; Sak, S.C.; Harnden, P.; Knowles, M.A.; Kiltie, A.E. Papillary and muscle invasive bladder tumors with distinct genomic stability profiles have different DNA repair fidelity and KU DNA-binding activities. Genes Chromosomes Cancer 2009, 48, 310–321. [Google Scholar] [CrossRef]
- Windhofer, F.; Krause, S.; Hader, C.; Schulz, W.A.; Florl, A.R. Distinctive differences in DNA double-strand break repair between normal urothelial and urothelial carcinoma cells. Mutat. Res. 2008, 638, 56–65. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Mufti, G.J.; Rassool, F.V. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002, 62, 2791–2797. [Google Scholar]
- Deriano, L.; Guipaud, O.; Merle-Beral, H.; Binet, J.L.; Ricoul, M.; Potocki-Veronese, G.; Favaudon, V.; Maciorowski, Z.; Muller, C.; Salles, B.; et al. Human chronic lymphocytic leukemia B cells can escape DNA damage-induced apoptosis through the nonhomologous end-joining DNA repair pathway. Blood 2005, 105, 4776–4783. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.; Donoghue, P.; Dixon, M.; O’Sullivan, R.; O’Donnell, R.L.; Murray, J.; Kaufmann, A.; Curtin, N.J.; Edmondson, R.J. Ovarian Cancers Harbor Defects in Nonhomologous End Joining Resulting in Resistance to Rucaparib. Clin. Cancer Res. 2017, 23, 2050–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.E.; van der Burg, M.; IJspeert, H.; Carroll, P.; Wu, Q.; Ochi, T.; Leitch, A.; Miller, E.S.; Kysela, B.; Jawad, A.; et al. Mutations in the NHEJ component XRCC4 cause primordial dwarfism. Am. J. Hum. Genet. 2015, 96, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, S.; Villalobos, V.M.; Gevaert, O.; Abramovitz, M.; Williams, C.; Sikic, B.I.; Leyland-Jones, B. Single Gene Prognostic Biomarkers in Ovarian Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0149183. [Google Scholar] [CrossRef] [PubMed]
- Assis, J.; Pereira, D.; Medeiros, R. Ovarian cancer and DNA repair: DNA ligase IV as a potential key. World J. Clin. Oncol. 2013, 4, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Pearce, C.L.; Near, A.M.; Van Den Berg, D.J.; Ramus, S.J.; Gentry-Maharaj, A.; Menon, U.; Gayther, S.A.; Anderson, A.R.; Edlund, C.K.; Wu, A.H.; et al. Validating genetic risk associations for ovarian cancer through the international Ovarian Cancer Association Consortium. Br. J. Cancer 2009, 100, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary ovarian cancer: Not only BRCA 1 and 2 genes. Biomed. Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Lin, B.; Cowan, A.; Heinen, C.D. ATR-Chk1 activation mitigates replication stress caused by mismatch repair-dependent processing of DNA damage. Proc. Natl. Acad. Sci. USA 2018, 115, 1523–1528. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, E.; Gerrits, B.; Marra, G.; Schlapbach, R.; Jiricny, J. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 2007, 282, 2976–2986. [Google Scholar] [CrossRef] [Green Version]
- Guillotin, D.; Martin, S.A. Exploiting DNA mismatch repair deficiency as a therapeutic strategy. Exp. Cell Res. 2014, 329, 110–115. [Google Scholar] [CrossRef]
- Jonsson, J.M.; Bartuma, K.; Dominguez-Valentin, M.; Harbst, K.; Ketabi, Z.; Malander, S.; Jonsson, M.; Carneiro, A.; Masback, A.; Jonsson, G.; et al. Distinct gene expression profiles in ovarian cancer linked to Lynch syndrome. Fam. Cancer 2014, 13, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Helder-Woolderink, J.M.; Blok, E.A.; Vasen, H.F.; Hollema, H.; Mourits, M.J.; De Bock, G.H. Ovarian cancer in Lynch syndrome; a systematic review. Eur. J. Cancer 2016, 55, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J.P.; Goodheart, M.J.; Sood, A.K.; Holmes, R.J.; Hatterman-Zogg, M.A.; Buller, R.E. Mismatch repair gene expression defects contribute to microsatellite instability in ovarian carcinoma. Cancer 2003, 98, 2199–2206. [Google Scholar] [CrossRef] [PubMed]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Collura, A.; Lefevre, J.H.; Svrcek, M.; Tougeron, D.; Zaanan, A.; Duval, A. Microsatellite instability and cancer: From genomic instability to personalized medicine. Med. Sci. (Paris) 2019, 35, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.A.; Wentzensen, N. Frequency of mismatch repair deficiency in ovarian cancer: A systematic review. This article is a US Government work and, as such, is in the public domain of the United States of America. Int. J. Cancer 2011, 129, 1914–1922. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, H.; Nakayama, K.; Ishikawa, M.; Ishibashi, T.; Nakamura, K.; Sawada, K.; Yoshimura, Y.; Tatsumi, N.; Kurose, S.; Minamoto, T.; et al. Relationship between Microsatellite Instability, Immune Cells Infiltration, and Expression of Immune Checkpoint Molecules in Ovarian Carcinoma: Immunotherapeutic Strategies for the Future. Int. J. Mol. Sci. 2019, 20, 5129. [Google Scholar] [CrossRef] [Green Version]
- Howitt, B.E.; Strickland, K.C.; Sholl, L.M.; Rodig, S.; Ritterhouse, L.L.; Chowdhury, D.; D’Andrea, A.D.; Matulonis, U.A.; Konstantinopoulos, P.A. Clear cell ovarian cancers with microsatellite instability: A unique subset of ovarian cancers with increased tumor-infiltrating lymphocytes and PD-1/PD-L1 expression. Oncoimmunology 2017, 6, e1277308. [Google Scholar] [CrossRef] [Green Version]
- Fraune, C.; Rosebrock, J.; Simon, R.; Hube-Magg, C.; Makrypidi-Fraune, G.; Kluth, M.; Buscheck, F.; Hoflmayer, D.; Schmalfeldt, B.; Muller, V.; et al. High homogeneity of MMR deficiency in ovarian cancer. Gynecol. Oncol. 2020, 156, 669–675. [Google Scholar] [CrossRef]
- Fink, D.; Nebel, S.; Aebi, S.; Zheng, H.; Cenni, B.; Nehme, A.; Christen, R.D.; Howell, S.B. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996, 56, 4881–4886. [Google Scholar]
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum resistance: The role of DNA repair pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Yan, L.; Xiao-Fei, L.; Hai-Yan, S.; Juan, C.; Shan, K. Hypermethylation of mismatch repair gene hMSH2 associates with platinum-resistant disease in epithelial ovarian cancer. Clin. Epigenet. 2019, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Ueda, H.; Etoh, T.; Koike, E.; Fujinami, N.; Mitsuhashi, A.; Hoshiai, H. A change in promoter methylation of hMLH1 is a cause of acquired resistance to platinum-based chemotherapy in epithelial ovarian cancer. Anticancer Res. 2007, 27, 1449–1452. [Google Scholar] [PubMed]
- Zhao, C.; Li, S.; Zhao, M.; Zhu, H.; Zhu, X. Prognostic values of DNA mismatch repair genes in ovarian cancer patients treated with platinum-based chemotherapy. Arch. Gynecol. Obstet. 2018, 297, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.L.; Izumi, T.; Mitra, S. Oxidized base damage and single-strand break repair in mammalian genomes: Role of disordered regions and posttranslational modifications in early enzymes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 123–153. [Google Scholar] [CrossRef] [Green Version]
- D’Errico, M.; Parlanti, E.; Pascucci, B.; Fortini, P.; Baccarini, S.; Simonelli, V.; Dogliotti, E. Single nucleotide polymorphisms in DNA glycosylases: From function to disease. Free Radic. Biol. Med. 2017, 107, 278–291. [Google Scholar] [CrossRef]
- Vodicka, P.; Urbanova, M.; Makovicky, P.; Tomasova, K.; Kroupa, M.; Stetina, R.; Opattova, A.; Kostovcikova, K.; Siskova, A.; Schneiderova, M.; et al. Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients. Int. J. Mol. Sci. 2020, 21, 2473. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Stetina, R.; Polakova, V.; Tulupova, E.; Naccarati, A.; Vodickova, L.; Kumar, R.; Hanova, M.; Pardini, B.; Slyskova, J.; et al. Association of DNA repair polymorphisms with DNA repair functional outcomes in healthy human subjects. Carcinogenesis 2007, 28, 657–664. [Google Scholar] [CrossRef]
- Benitez-Buelga, C.; Vaclova, T.; Ferreira, S.; Urioste, M.; Inglada-Perez, L.; Soberon, N.; Blasco, M.A.; Osorio, A.; Benitez, J. Molecular insights into the OGG1 gene, a cancer risk modifier in BRCA1 and BRCA2 mutations carriers. Oncotarget 2016, 7, 25815–25825. [Google Scholar] [CrossRef] [Green Version]
- Tomasova, K.; Kroupa, M.; Forsti, A.; Vodicka, P.; Vodickova, L. Telomere maintenance in interplay with DNA repair in pathogenesis and treatment of colorectal cancer. Mutagenesis 2020. [Google Scholar] [CrossRef]
- Poulsen, M.L.; Bisgaard, M.L. MUTYH Associated Polyposis (MAP). Curr. Genom. 2008, 9, 420–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, A.K.; Reece, J.C.; Dowty, J.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Southey, M.C.; Young, J.P.; Cleary, S.P.; Kim, H.; et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int. J. Cancer 2016, 139, 1557–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, T.; Ljungquist, S.; Siegert, W.; Nyberg, B.; Sperens, B. DNA N-glycosidases: Properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem. 1977, 252, 3286–3294. [Google Scholar]
- Vodicka, P.; Hemminki, K. Phosphodiester cleavage in apurinic dinucleotides. Chem. Biol. Interact. 1988, 68, 153–164. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Al-Attar, A.; Gossage, L.; Fareed, K.R.; Shehata, M.; Mohammed, M.; Zaitoun, A.M.; Soomro, I.; Lobo, D.N.; Abbotts, R.; Chan, S.; et al. Human apurinic/apyrimidinic endonuclease (APE1) is a prognostic factor in ovarian, gastro-oesophageal and pancreatico-biliary cancers. Br. J. Cancer 2010, 102, 704–709. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Q.; Zhang, Y.; Wang, R.; Zhang, J.; Chen, B.; Wang, J.; Zhang, W.; Xin, X. Prognostic significance of APE1 cytoplasmic localization in human epithelial ovarian cancer. Med. Oncol. 2012, 29, 1265–1271. [Google Scholar] [CrossRef]
- Fan, X.; Wen, L.; Li, Y.; Lou, L.; Liu, W.; Zhang, J. The expression profile and prognostic value of APE/Ref-1 and NPM1 in high-grade serous ovarian adenocarcinoma. APMIS 2017, 125, 857–862. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.; Sultana, R.; Abbotts, R.; Hawkes, C.; Seedhouse, C.; Chan, S.; Madhusudan, S. Clinicopathological and functional significance of XRCC1 expression in ovarian cancer. Int. J. Cancer 2013, 132, 2778–2786. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Lightfoot, M.; Montemorano, L.; Bixel, K. PARP Inhibitors in Gynecologic Cancers: What Is the Next Big Development? Curr. Oncol. Rep. 2020, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Przybycinski, J.; Nalewajska, M.; Marchelek-Mysliwiec, M.; Dziedziejko, V.; Pawlik, A. Poly-ADP-ribose polymerases (PARPs) as a therapeutic target in the treatment of selected cancers. Expert Opin. Ther. Targets 2019, 23, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzese, E.; Centonze, S.; Diana, A.; Carlino, F.; Guerrera, L.P.; Di Napoli, M.; De Vita, F.; Pignata, S.; Ciardiello, F.; Orditura, M. PARP inhibitors in ovarian cancer. Cancer Treat. Rev. 2019, 73, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Tufegdzic Vidakovic, A.; Mitter, R.; Kelly, G.P.; Neumann, M.; Harreman, M.; Rodriguez-Martinez, M.; Herlihy, A.; Weems, J.C.; Boeing, S.; Encheva, V.; et al. Regulation of the RNAPII Pool Is Integral to the DNA Damage Response. Cell 2020, 180, 1245–1261. [Google Scholar] [CrossRef]
- Black, J.O. Xeroderma Pigmentosum. Head Neck Pathol. 2016, 10, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Egly, J.M. Trichothiodystrophy view from the molecular basis of DNA repair/transcription factor TFIIH. Hum. Mol. Genet. 2009, 18, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Zhang, A.; Zhao, Y.; Xiang, J.; Yu, D.; Liang, Z.; Xu, C.; Zhang, Q.; Li, J.; Duan, P. The association of polymorphisms in nucleotide excision repair genes with ovarian cancer susceptibility. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Cao, D.; Ma, X.; Yang, J.; Peng, P.; Yu, M.; Zhou, H.; Zhang, Y.; Li, L.; Huo, X.; et al. Identification of a Prognostic Signature Associated With DNA Repair Genes in Ovarian Cancer. Front. Genet. 2019, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvakumaran, M.; Pisarcik, D.A.; Bao, R.; Yeung, A.T.; Hamilton, T.C. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003, 63, 1311–1316. [Google Scholar] [PubMed]
- Ishibashi, M.; Toyoshima, M.; Zhang, X.; Hasegawa-Minato, J.; Shigeta, S.; Usui, T.; Kemp, C.J.; Grandori, C.; Kitatani, K.; Yaegashi, N. Tyrosine kinase receptor TIE-1 mediates platinum resistance by promoting nucleotide excision repair in ovarian cancer. Sci. Rep. 2018, 8, 13207. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Yang, B.; Zhao, J.; Shen, S.; Gao, J. Role of common ERCC1 polymorphisms in cisplatin-resistant epithelial ovarian cancer patients: A study in Chinese cohort. Int. J. Immunogenet. 2020. [Google Scholar] [CrossRef]
- King, B.S.; Cooper, K.L.; Liu, K.J.; Hudson, L.G. Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J. Biol. Chem. 2012, 287, 39824–39833. [Google Scholar] [CrossRef] [Green Version]
- Ji, P.; Wang, X.; Xie, N.; Li, Y. N6-Methyladenosine in RNA and DNA: An Epitranscriptomic and Epigenetic Player Implicated in Determination of Stem Cell Fate. Stem Cells Int. 2018, 2018, 3256524. [Google Scholar] [CrossRef]
- Soll, J.M.; Sobol, R.W.; Mosammaparast, N. Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends Biochem. Sci. 2017, 42, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, L.; Zhong, D. Photolyase: Dynamics and Mechanisms of Repair of Sun-Induced DNA Damage. Photochem. Photobiol. 2017, 93, 78–92. [Google Scholar] [CrossRef]
- Steurer, B.; Turkyilmaz, Y.; van Toorn, M.; van Leeuwen, W.; Escudero-Ferruz, P.; Marteijn, J.A. Fluorescently-labelled CPD and 6-4PP photolyases: New tools for live-cell DNA damage quantification and laser-assisted repair. Nucleic Acids Res. 2019, 47, 3536–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soll, J.M.; Brickner, J.R.; Mudge, M.C.; Mosammaparast, N. RNA ligase-like domain in activating signal cointegrator 1 complex subunit 1 (ASCC1) regulates ASCC complex function during alkylation damage. J. Biol. Chem. 2018, 293, 13524–13533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, H.J.; Suh, D.S.; Choi, K.U.; Yoo, H.J.; Joo, W.D.; Yoon, M.S. Inactivation of O(6)-methyguanine-DNA methyltransferase by promoter hypermethylation: Association of epithelial ovarian carcinogenesis in specific histological types. J. Obstet. Gynaecol. Res. 2011, 37, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Luo, J.Y.; Tan, H.Z. Associations of MGMT promoter hypermethylation with squamous intraepithelial lesion and cervical carcinoma: A meta-analysis. PLoS ONE 2019, 14, e0222772. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Y.; Liu, F.; Xu, L.; Peng, F.; Zhao, N.; Fu, B.; Zhu, Z.; Shi, Y.; Liu, J.; et al. A systematic review and meta-analysis: Association between MGMT hypermethylation and the clinicopathological characteristics of non-small-cell lung carcinoma. Sci. Rep. 2018, 8, 1439. [Google Scholar] [CrossRef] [Green Version]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 2961–2966. [Google Scholar] [CrossRef] [Green Version]
- Mongan, N.P.; Emes, R.D.; Archer, N. Detection and analysis of RNA methylation. F1000Research 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.H.; Chambers, S.K. Human ALKBH3-induced m (1)A demethylation increases the CSF-1 mRNA stability in breast and ovarian cancer cells. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.H.; Laszlo, C.F.; Greco, S.; Chambers, S.K. Regulation of colony stimulating factor-1 expression and ovarian cancer cell behavior in vitro by miR-128 and miR-152. Mol. Cancer 2012, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Gan, X.; Jiang, X.; Diao, S.; Wu, H.; Hu, J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J. Exp. Clin. Cancer Res. 2019, 38, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Nagel, Z.D.; Chaim, I.A.; Samson, L.D. Inter-individual variation in DNA repair capacity: A need for multi-pathway functional assays to promote translational DNA repair research. DNA Repair 2014, 19, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Melis, J.P.; Luijten, M.; Mullenders, L.H.; van Steeg, H. The role of XPC: Implications in cancer and oxidative DNA damage. Mutat. Res. 2011, 728, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Musak, L.; Frank, C.; Kazimirova, A.; Vymetalkova, V.; Barancokova, M.; Smolkova, B.; Dzupinkova, Z.; Jiraskova, K.; Vodenkova, S.; et al. Interactions of DNA repair gene variants modulate chromosomal aberrations in healthy subjects. Carcinogenesis 2015, 36, 1299–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, J.M.; Popp, O.; Gebhard, D.; Veith, S.; Fischbach, A.; Beneke, S.; Leitenstorfer, A.; Bergemann, J.; Scheffner, M.; Ferrando-May, E.; et al. Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function. FEBS J. 2014, 281, 3625–3641. [Google Scholar] [CrossRef]
- Nagel, Z.D.; Margulies, C.M.; Chaim, I.A.; McRee, S.K.; Mazzucato, P.; Ahmad, A.; Abo, R.P.; Butty, V.L.; Forget, A.L.; Samson, L.D. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 1823–1832. [Google Scholar] [CrossRef] [Green Version]
- Krieger, K.L.; Hu, W.F.; Ripperger, T.; Woods, N.T. Functional Impacts of the BRCA1-mTORC2 Interaction in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 5876. [Google Scholar] [CrossRef] [Green Version]
- Fece de la Cruz, F.; Gapp, B.V.; Nijman, S.M. Synthetic lethal vulnerabilities of cancer. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 513–531. [Google Scholar] [CrossRef]
- Nijman, S.M. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Farolfi, A.; Gurioli, G.; Fugazzola, P.; Burgio, S.L.; Casanova, C.; Ravaglia, G.; Altavilla, A.; Costantini, M.; Amadori, A.; Framarini, M.; et al. Immune System and DNA Repair Defects in Ovarian Cancer: Implications for Locoregional Approaches. Int. J. Mol. Sci. 2019, 20, 2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Minasian, L.; Kohn, E.C. New strategies in ovarian cancer treatment. Cancer 2019, 125, 4623–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, C.D.; VanWye, A.B.; Dowless, M.; Blosser, W.; Falcon, B.L.; Stewart, J.; Stephens, J.; Beckmann, R.P.; Bence Lin, A.; Stancato, L.F. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin. Cancer Res. 2017, 23, 4354–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, R.; Abdel-Fatah, T.; Abbotts, R.; Hawkes, C.; Albarakati, N.; Seedhouse, C.; Ball, G.; Chan, S.; Rakha, E.A.; Ellis, I.O.; et al. Targeting XRCC1 deficiency in breast cancer for personalized therapy. Cancer Res. 2013, 73, 1621–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, R.; McNeill, D.R.; Abbotts, R.; Mohammed, M.Z.; Zdzienicka, M.Z.; Qutob, H.; Seedhouse, C.; Laughton, C.A.; Fischer, P.M.; Patel, P.M.; et al. Synthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitors. Int. J. Cancer 2012, 131, 2433–2444. [Google Scholar] [CrossRef]
- Fang, Y.Y.; Bi, F.F.; Zhou, Y.M.; Sun, W.P.; Li, C.Y.; Liu, Q.; Zhao, Y.; Li, D. Nicotinamide adenine dinucleotide (NAD) may affect DNA methyltransferase 1 through regulation of BRCA1 in ovarian cancer. Am. J. Cancer Res. 2015, 5, 1199–1206. [Google Scholar]
- Jiang, Z.; Lai, Y.; Beaver, J.M.; Tsegay, P.S.; Zhao, M.L.; Horton, J.K.; Zamora, M.; Rein, H.L.; Miralles, F.; Shaver, M.; et al. Oxidative DNA Damage Modulates DNA Methylation Pattern in Human Breast Cancer 1 (BRCA1) Gene via the Crosstalk between DNA Polymerase beta and a de novo DNA Methyltransferase. Cells 2020, 9, 255. [Google Scholar] [CrossRef] [Green Version]
- Chuang, Y.T.; Chang, C.L. Extending platinum-free interval in partially platinum-sensitive recurrent ovarian cancer by a non-platinum regimen: Its possible clinical significance. Taiwan J. Obstet. Gynecol. 2012, 51, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Ghisoni, E.; Giannone, G.; Tuninetti, V.; Genta, S.; Scotto, G.; Aglietta, M.; Sangiolo, D.; Mittica, G.; Valabrega, G. Veliparib: A new therapeutic option in ovarian cancer? Future Oncol. 2019, 15, 1975–1987. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R D 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther 2020, 5, 60. [Google Scholar] [CrossRef]
- Fields, E.C.; McGuire, W.P.; Lin, L.; Temkin, S.M. Radiation Treatment in Women with Ovarian Cancer: Past, Present, and Future. Front. Oncol. 2017, 7, 177. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Bryant, C.; Rawlinson, R.; Massey, A.J. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer 2014, 14, 570. [Google Scholar] [CrossRef] [Green Version]
- Itamochi, H.; Nishimura, M.; Oumi, N.; Kato, M.; Oishi, T.; Shimada, M.; Sato, S.; Naniwa, J.; Sato, S.; Kudoh, A.; et al. Checkpoint kinase inhibitor AZD7762 overcomes cisplatin resistance in clear cell carcinoma of the ovary. Int. J. Gynecol. Cancer 2014, 24, 61–69. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Farkkila, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Zhang, Y.; Chen, S.; Weng, X.; Rao, Y.; Fang, H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed. Pharmacother. 2020, 123, 109661. [Google Scholar] [CrossRef] [PubMed]
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.R.; Lee, J.M. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017, 8, 111026–111040. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. Search NCT IDs: NCT04267939, NCT02627443, NCT04149145, NCT02595892, NCT02487095, NCT04065269, and NCT03462342. Available online: https://clinicaltrials.gov (accessed on 18 March 2020).
- Teng, P.N.; Bateman, N.W.; Darcy, K.M.; Hamilton, C.A.; Maxwell, G.L.; Bakkenist, C.J.; Conrads, T.P. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol. Oncol. 2015, 136, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Dominguez, D.; Chen, S.; Fan, J.; Qin, L.; Long, A.; Li, X.; Zhang, Y.; Shi, H.; Zhang, B. WEE1 inhibition by MK1775 as a single-agent therapy inhibits ovarian cancer viability. Oncol. Lett. 2017, 14, 3580–3586. [Google Scholar] [CrossRef] [Green Version]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients with Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Brandsma, I.; Fleuren, E.D.G.; Williamson, C.T.; Lord, C.J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2017, 26, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant BRCA1 Mutant Ovarian Cells. Diagnostics 2020, 10, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlin, J.; Allen, J.; Ahmad, S.F.; Hughes, G.; Sheridan, V.; Odedra, R.; Farrington, P.; Cadogan, E.B.; Riches, L.C.; Garcia-Trinidad, A.; et al. Orally Bioavailable and Blood-Brain Barrier-Penetrating ATM Inhibitor (AZ32) Radiosensitizes Intracranial Gliomas in Mice. Mol. Cancer Ther. 2018, 17, 1637–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [Green Version]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Slipicevic, A.; Holth, A.; Hellesylt, E.; Trope, C.G.; Davidson, B.; Florenes, V.A. Wee1 is a novel independent prognostic marker of poor survival in post-chemotherapy ovarian carcinoma effusions. Gynecol. Oncol. 2014, 135, 118–124. [Google Scholar] [CrossRef]
- Saini, P.; Li, Y.; Dobbelstein, M. Wee1 is required to sustain ATR/Chk1 signaling upon replicative stress. Oncotarget 2015, 6, 13072–13087. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Shao, F.; Martin, S.; Xu, X.; Deng, C.X. WEE1 inhibition targets cell cycle checkpoints for triple negative breast cancers to overcome cisplatin resistance. Sci. Rep. 2017, 7, 43517. [Google Scholar] [CrossRef] [Green Version]
- Parsels, L.A.; Karnak, D.; Parsels, J.D.; Zhang, Q.; Velez-Padilla, J.; Reichert, Z.R.; Wahl, D.R.; Maybaum, J.; O’Connor, M.J.; Lawrence, T.S.; et al. PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization with Combined WEE1 and PARP Inhibitors. Mol. Cancer Res. 2018, 16, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slyskova, J.; Cordero, F.; Pardini, B.; Korenkova, V.; Vymetalkova, V.; Bielik, L.; Vodickova, L.; Pitule, P.; Liska, V.; Matejka, V.M.; et al. Post-treatment recovery of suboptimal DNA repair capacity and gene expression levels in colorectal cancer patients. Mol. Carcinog. 2015, 54, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Slyskova, J.; Korenkova, V.; Collins, A.R.; Prochazka, P.; Vodickova, L.; Svec, J.; Lipska, L.; Levy, M.; Schneiderova, M.; Liska, V.; et al. Functional, genetic, and epigenetic aspects of base and nucleotide excision repair in colorectal carcinomas. Clin. Cancer Res. 2012, 18, 5878–5887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodenkova, S.; Jiraskova, K.; Urbanova, M.; Kroupa, M.; Slyskova, J.; Schneiderova, M.; Levy, M.; Buchler, T.; Liska, V.; Vodickova, L.; et al. Base excision repair capacity as a determinant of prognosis and therapy response in colon cancer patients. DNA Repair 2018, 72, 77–85. [Google Scholar] [CrossRef]
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly (ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Ke, G.; Bi, R.; Wu, X. Clinicopathological and survival characteristic of mismatch repair status in ovarian clear cell carcinoma. J. Surg. Oncol. 2020. [Google Scholar] [CrossRef]
- Bonadona, V.; Bonaiti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Kawashima, N.; Yoshida, H.; Miwa, M.; Fujiwara, K. MLH1 Is a Prognostic Biomarker for Serous Ovarian Cancer Treated With Platinum- and Taxane-based Chemotherapy. Anticancer Res. 2019, 39, 5505–5513. [Google Scholar] [CrossRef]
- Guo, X.; Wu, W.; Gao, H.; Li, X.; He, Q.; Zhu, Y.; Liu, N. PMS2 germline mutation c.943C>T (p.Arg315*)-induced Lynch syndrome-associated ovarian cancer. Mol. Genet. Genom. Med. 2019, 7, e721. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xiang, Q.; Mu, G.; Xie, Q.; Chen, S.; Zhou, S.; Hu, K.; Cui, Y.M. XRCC1 polymorphism and overall survival in ovarian cancer patients treated with platinum-based chemotherapy: A systematic review and MOOSE-compliant meta-analysis. Medicine (Baltimore) 2018, 97, e12996. [Google Scholar] [CrossRef]
- Cheng, C.X.; Xue, M.; Li, K.; Li, W.S. Predictive value of XRCC1 and XRCC3 gene polymorphisms for risk of ovarian cancer death after chemotherapy. Asian Pac. J. Cancer Prev. 2012, 13, 2541–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, N.D.; Agadjanian, H.; Nassanian, H.; Miller, C.W.; Orsulic, S.; Karlan, B.Y.; Walsh, C.S. Xeroderma pigmentosum complementation group C single-nucleotide polymorphisms in the nucleotide excision repair pathway correlate with prolonged progression-free survival in advanced ovarian cancer. Cancer 2012, 118, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.M.; Samulak, D.; Romanowicz, H.; Sobkowski, M.; Smolarz, B. An Association between Single Nucleotide Polymorphisms of Lys751Gln ERCC2 Gene and Ovarian Cancer in Polish Women. Adv. Med. 2015, 2015, 109593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peethambaram, P.; Fridley, B.L.; Vierkant, R.A.; Larson, M.C.; Kalli, K.R.; Elliott, E.A.; Oberg, A.L.; White, K.L.; Rider, D.N.; Keeney, G.L.; et al. Polymorphisms in ABCB1 and ERCC2 associated with ovarian cancer outcome. Int. J. Mol. Epidemiol. Genet. 2011, 2, 185–195. [Google Scholar]
- Khrunin, A.V.; Moisseev, A.; Gorbunova, V.; Limborska, S. Genetic polymorphisms and the efficacy and toxicity of cisplatin-based chemotherapy in ovarian cancer patients. Pharmacogenom. J. 2010, 10, 54–61. [Google Scholar] [CrossRef]
- Krivak, T.C.; Darcy, K.M.; Tian, C.; Bookman, M.; Gallion, H.; Ambrosone, C.B.; Deloia, J.A. Single nucleotide polypmorphisms in ERCC1 are associated with disease progression, and survival in patients with advanced stage ovarian and primary peritoneal carcinoma; a Gynecologic Oncology Group study. Gynecol. Oncol. 2011, 122, 121–126. [Google Scholar] [CrossRef]
- Fukumoto, T.; Zhu, H.; Nacarelli, T.; Karakashev, S.; Fatkhutdinov, N.; Wu, S.; Liu, P.; Kossenkov, A.V.; Showe, L.C.; Jean, S.; et al. N(6)-Methylation of Adenosine of FZD10 mRNA Contributes to PARP Inhibitor Resistance. Cancer Res. 2019, 79, 2812–2820. [Google Scholar] [CrossRef] [Green Version]









| Gene | SNP | Functionality | Effect | Odds Ratio (OR), Hazard Ratio (HR), Confidence Interval (CI) | Population | Reference |
|---|---|---|---|---|---|---|
| CHEK2 | rs17507066 | Intron variant | ↑ risk of serous EOC | OR: 0.86; 95% CI: 0.81–0.91 | 15,397 patients, 30,816 controls | [88] |
| rs6005807 | Intron variant | ↑ risk of EOC | OR: 1.12, 95% CI: 1.07–1.18 | 15,397 patients, 30,816 controls | [88] | |
| ↑ risk of serous EOC | OR: 1.17, 95% CI: 1.11–1.23 | 25,509 patients, 40,941 controls | [89] | |||
| OGG1 | rs1052133 | Missense variant, Ser326Cys | ↑ risk | OR: 2.89; 95% CI: 2.47–3.38 | 720 patients, 720 controls | [90] |
| ↑ risk type II EOC | OR: 1.66; 95% CI: 1.26–2.17 | 420 patients, 840 controls | [91] | |||
| rs2304277 | Intron variant | ↑ risk for BRCA1/2 carriers | HR: 1.12, 95% CI: 1.03–1.21 | Stage I 1782 mutations carriers Stage II 23,463 mutations carriers | [92] | |
| APE1 | rs1130409 | Missense variant, Asp148Glu | ↓ risk | OR: 0.486; 95% CI: 0.344–0.688 | 124 patients, 141 controls | [93] |
| XRCC1 | rs25487 | Missense variant, Arg399Gln | ↑ risk | OR: 2.54; 95% CI: 1.22–5.29 | 50 patients, 78 controls | [94] |
| ↑ risk of death | HR: 1.98; 95% CI: 1.09–3.93 | 195 patients | [95] | |||
| rs1799782 | Missense variant, Arg194Trp | ↑ OS | HR: 0.61, 95% CI: 0.34–0.96 | 229 patients | [96] |
| DNA Repair Pathway | Gene | Predisposition Impact | Prognostic Impact | Therapeutic Potential (or Use) |
|---|---|---|---|---|
| Homologous recombination repair | BRCA1 | Mutations associated with ↑ risk [45] and earlier onset [46] | ↑ OS vs. non-carriers [50] | Better response to platinum-based chemotherapeutics [50,53], response to PARPi [55,229] |
| BRCA2 | Mutations associated with ↑ risk [45] and earlier onset [46] | ↑ OS vs. non-carriers [50] | Better response to platinum-based chemotherapy [50,53], response to PARPi [55,229] | |
| RAD51C | Mutations associated with ↑ risk [59,60] and earlier onset [60] | N/A | Response to PARPi (in vivo and in vitro evidence) [64,65] | |
| RAD51D | Mutations associated with ↑ risk [9,61,62,63] and earlier onset [60] | N/A | Response to PARPi (in vivo and in vitro evidence) [65] | |
| RAD50 | Mutated in about 0.12% of tumors [66] | Copy number deletion associated with ↑ OS and PFS [69] | In vitro knock-down associated with better response to PARPi [69] | |
| PALB2 | Mutations associated with ↑ risk [73] | N/A | Response to PARPi (in vivo and in vitro evidence) [74,75] | |
| BRIP1 | Mutations associated with ↑ risk [62,82,83,84] | N/A | Likely to predispose the response to PARPi and platinum [55]–needs further evaluation | |
| Non-homologous end joining | XRCC4 | N/A | ↑ expression associated with ↓ OS [106] | N/A |
| LIG4 | Possible involvement of SNPs needs further evaluation | N/A | N/A | |
| Mismatch repair | MSH6 | N/A | N/A | Deficiency predisposes to platinum sensitivity in clear cell carcinoma [230] |
| MLH1 | Mutations associated with ↑ risk of Lynch syndrome-associated OvC [231] | ↓ expression associated with ↑ OS and PFS [232] | N/A | |
| PMS2 | Germline mutation associated with ↑ risk of Lynch syndrome-associated OvC [233] | N/A | N/A | |
| Base excision repair | OGG1 | SNPs associated with ↑ risk [90,91,132] | N/A | N/A |
| MUTYH | Biallelic mutation associated with ↑ risk [135] | N/A | N/A | |
| APE1 | SNP associated with ↑ risk [93] | ↑ expression [139] and cytoplasmatic localization [140,141] have ↓ prognosis and OS | N/A | |
| XRCC1 | SNP associated with ↑ risk [94] | SNPs [95,96,234,235] and ↑ expression [142] associated with ↓ prognosis | N/A | |
| PARP1 | N/A | N/A | PARPi approved application for patients with germline BRCA1/2 mutations, with germline or somatic mutation BRCA1/2 with relapsed illness or with relapsed illness sensitive to platin-derivate chemotherapy regardless to BRCA status (FDA and EMA guidlines) | |
| Nucleotide excision repair | XPC | N/A | SNPs associated with ↑ PFS [236] | N/A |
| XPD/ERCC2 | SNP associated with ↑ risk [237] | SNPs associated with prognosis [238] | SNP associated with severe neutropenia in patients treated by cisplatin-based chemotherapy [239] | |
| ERCC1 | N/A | SNPs associated with ↑ OS [240] | SNP associated with ↑ risk of nephrotoxicity in patients treated by cisplatin-based chemotherapy [239] | |
| Direct repair | MGMT | N/A | N/A | Likely to drive chemoresistance [170] |
| ALKB | N/A | N/A | ALKBH5 downregulation contributes to PARPi resistance in BRCA-deficient EOC [241] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomasova, K.; Cumova, A.; Seborova, K.; Horak, J.; Koucka, K.; Vodickova, L.; Vaclavikova, R.; Vodicka, P. DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome. Cancers 2020, 12, 1713. https://doi.org/10.3390/cancers12071713
Tomasova K, Cumova A, Seborova K, Horak J, Koucka K, Vodickova L, Vaclavikova R, Vodicka P. DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome. Cancers. 2020; 12(7):1713. https://doi.org/10.3390/cancers12071713
Chicago/Turabian StyleTomasova, Kristyna, Andrea Cumova, Karolina Seborova, Josef Horak, Kamila Koucka, Ludmila Vodickova, Radka Vaclavikova, and Pavel Vodicka. 2020. "DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome" Cancers 12, no. 7: 1713. https://doi.org/10.3390/cancers12071713
APA StyleTomasova, K., Cumova, A., Seborova, K., Horak, J., Koucka, K., Vodickova, L., Vaclavikova, R., & Vodicka, P. (2020). DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome. Cancers, 12(7), 1713. https://doi.org/10.3390/cancers12071713