A Multiplex Assay for the Stratification of Patients with Primary Central Nervous System Lymphoma Using Targeted Mass Spectrometry

Abstract
:1. Introduction
2. Results
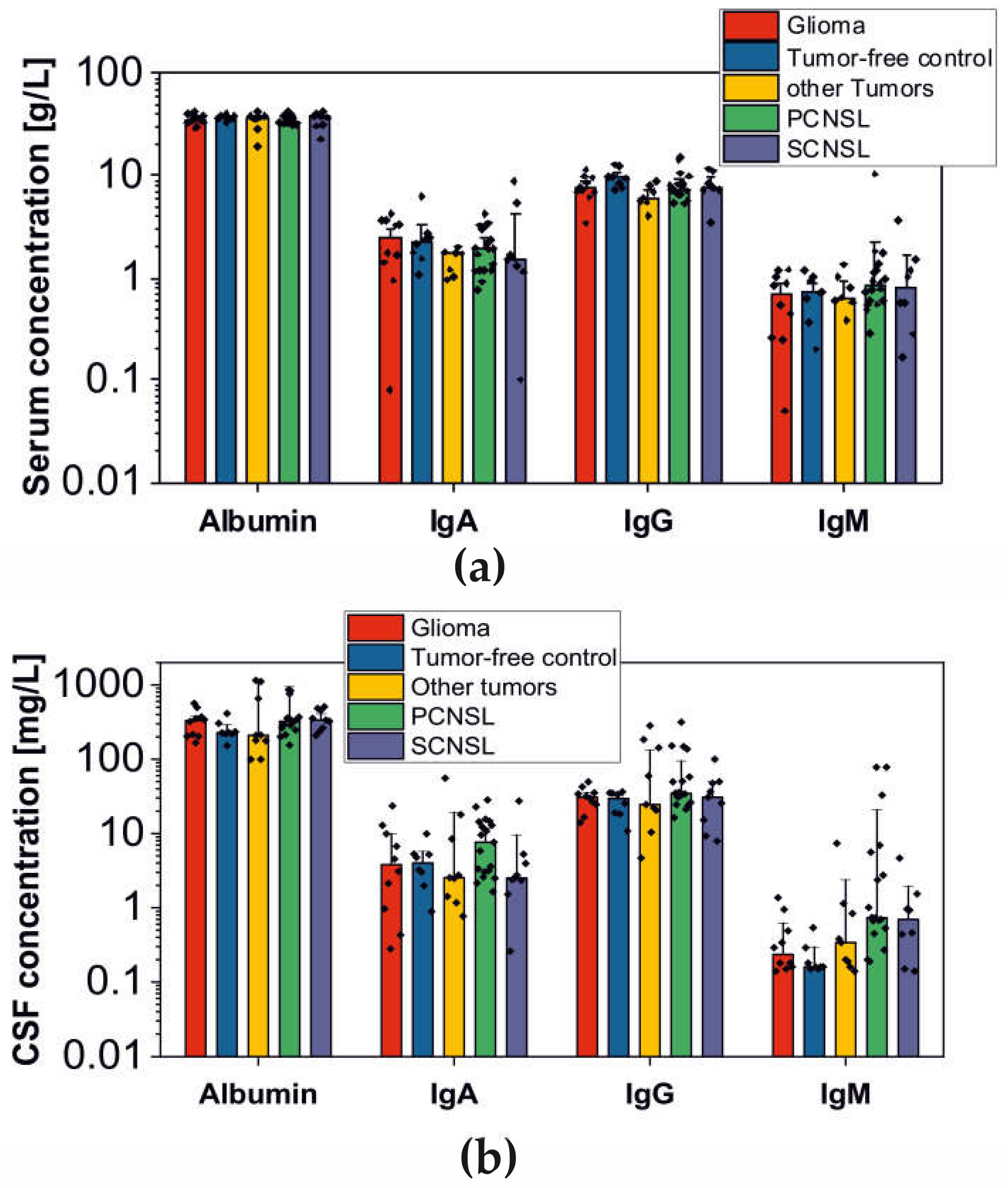
2.1. Analysis of BBB Dysfunction in the Discovery Cohort
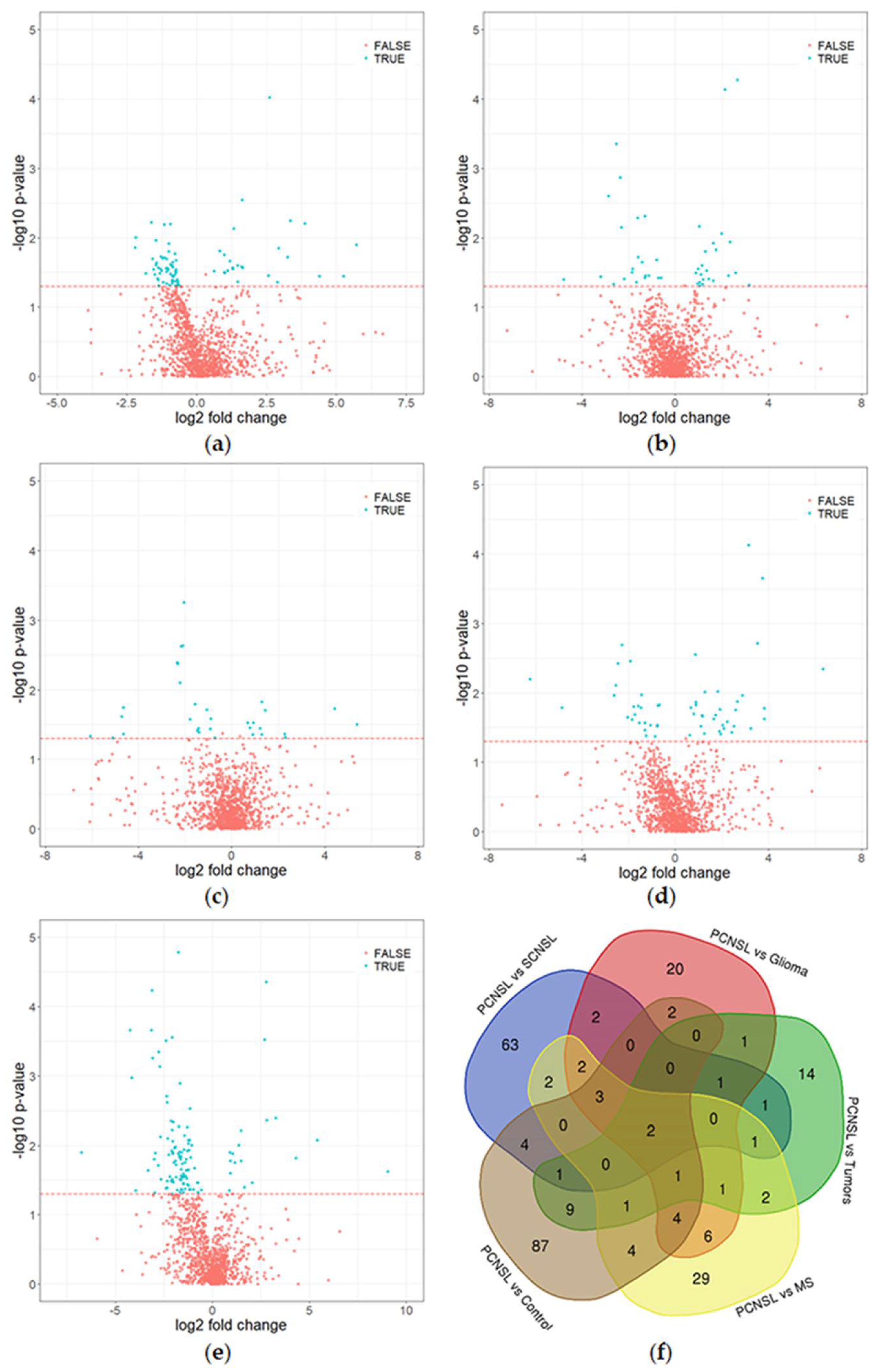
2.2. Discovery Study: Identification of Candidate Biomarkers by Quantitative Mass Spectrometry
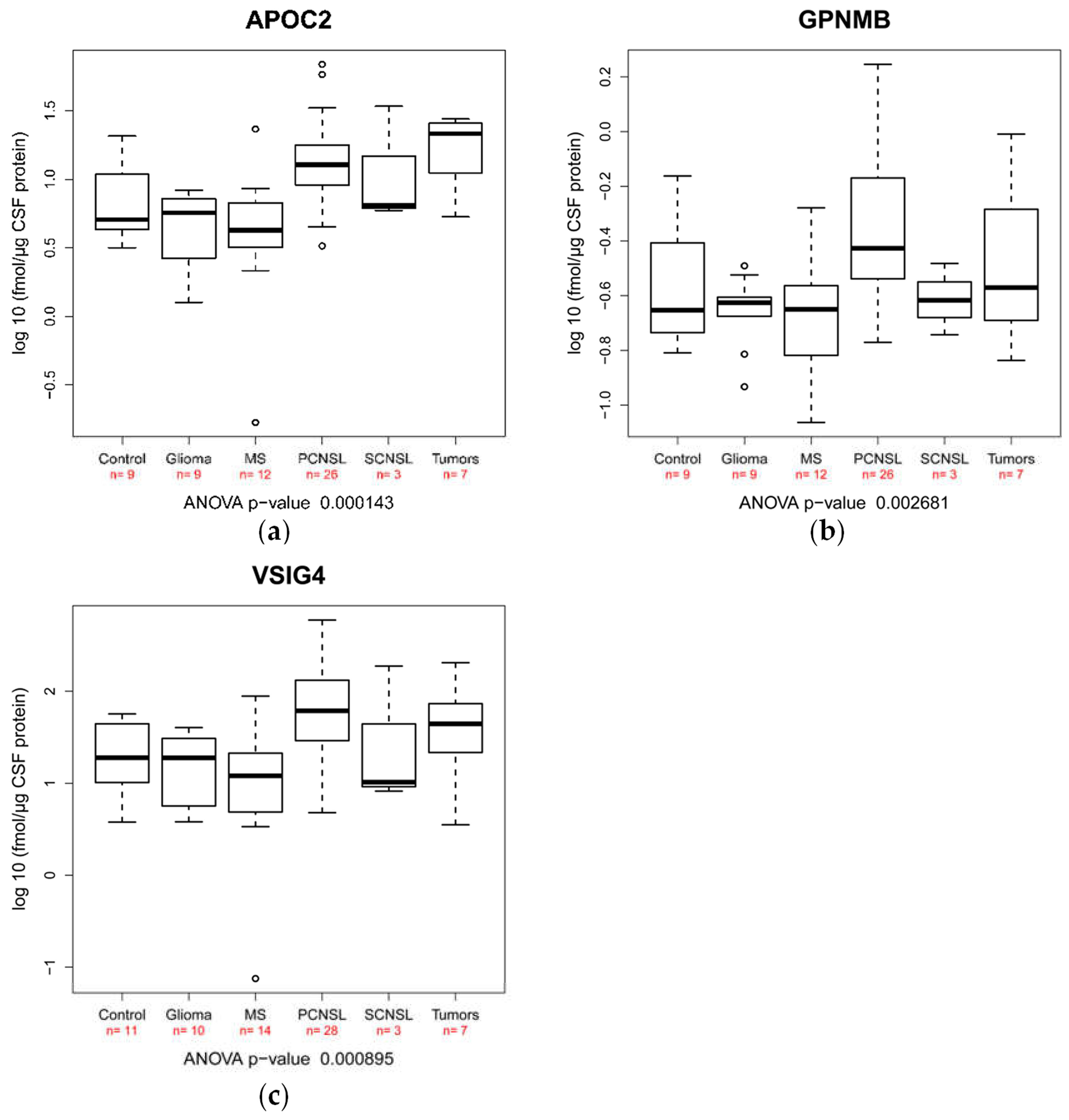
2.3. Validation of Candidate Peptide Biomarker in an Independent Patient Cohort Using SRM
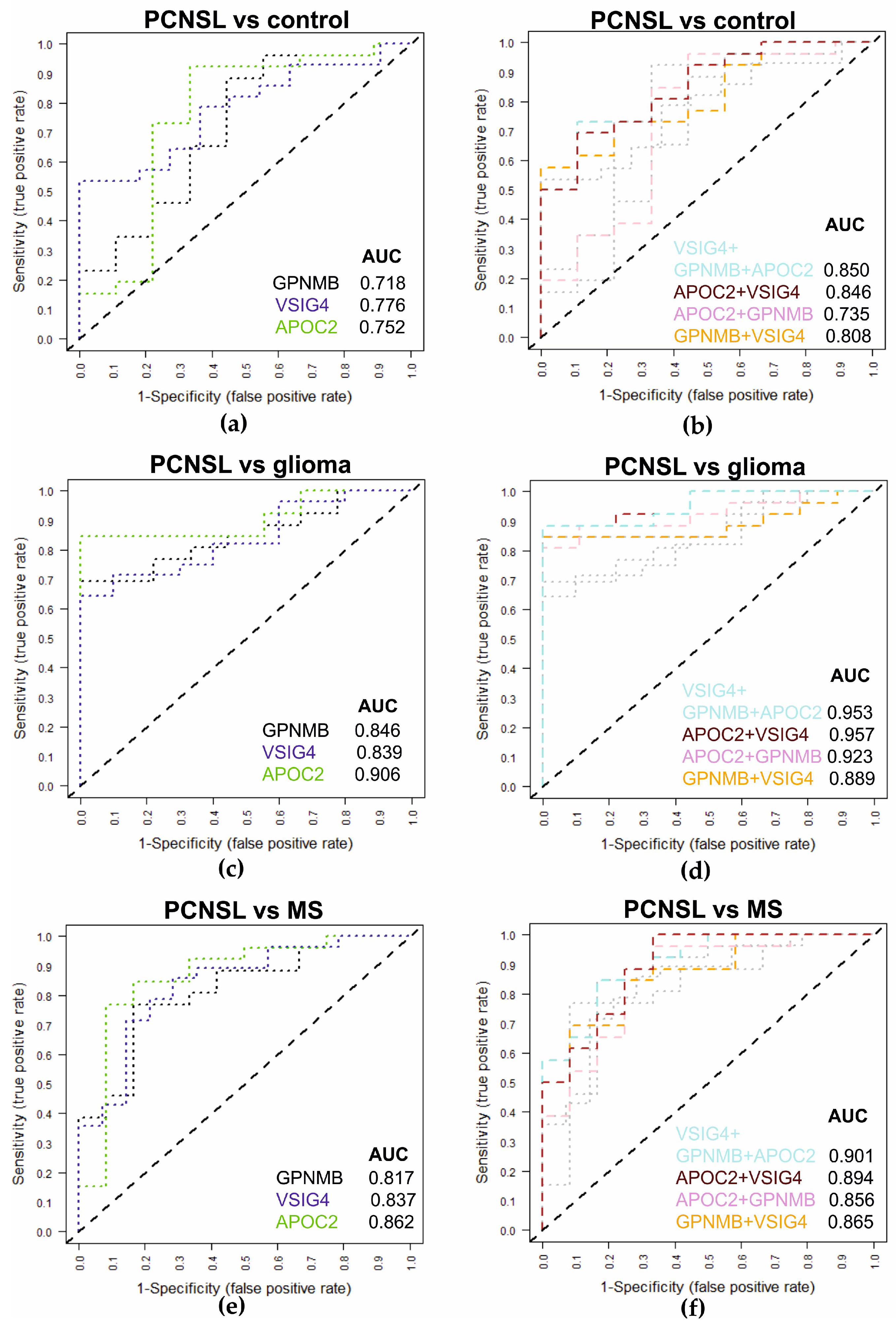
2.4. Multiplexed SRM Assay for the Diagnosis of PCNSL Using CSF Samples
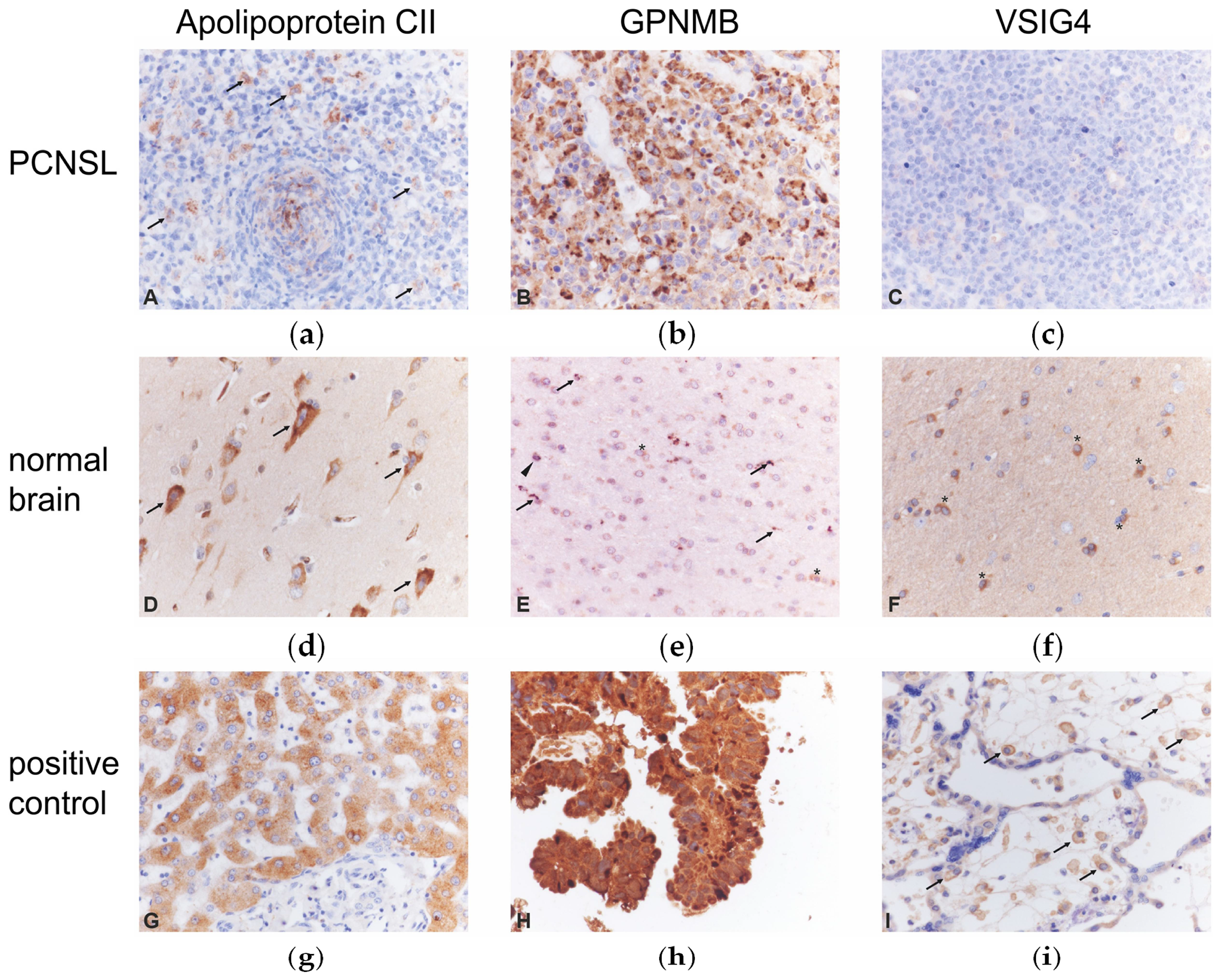
2.5. Immunohistochemistry of Candidate Biomarkers
3. Discussion
4. Materials and Methods
4.1. Patients, Clinical Data and CSF Collection
4.2. Sample Preparation
4.3. Liquid Chromatography Coupled Mass Spectrometric Analysis - Discovery
4.4. Identification and Quantification
4.5. Statistical Analysis
4.6. SRM Analysis—Validation
4.7. Immunohistochemistry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Korfel, A.; Schlegel, U. Diagnosis and treatment of primary CNS lymphoma. Nat. Rev. Neurol. 2013, 9, 317–327. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.P.; Decker, P.A.; Tieu, C.; Cerhan, J.R. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkin’s lymphoma. Am. J. Hematol. 2013, 88, 997–1000. [Google Scholar]
- Camilleri-Broet, S.; Martin, A.; Moreau, A.; Angonin, R.; Henin, D.; Gontier, M.F.; Rousselet, M.C.; Caulet-Maugendre, S.; Cuilliere, P.; Lefrancq, T.; et al. Primary central nervous system lymphomas in 72 immunocompetent patients: Pathologic findings and clinical correlations. Groupe Ouest Est d’etude des Leucenies et Autres Maladies du Sang (GOELAMS). Am. J. Clin. Pathol. 1998, 110, 607–612. [Google Scholar] [CrossRef]
- Batchelor, T.; Loeffler, J.S. Primary CNS lymphoma. J. Clin. Oncol. 2006, 24, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.; Schlegel, U. Have treatment protocols for primary CNS lymphoma advanced in the past 10 years. Expert Rev. Anticancer Ther. 2019, 19, 909–915. [Google Scholar] [CrossRef]
- Jimenez de la Pena, M.D.; Vicente, L.G.; Alonso, R.C.; Cabero, S.F.; Suarez, A.M.; de Vega, V.M. The Multiple Faces of Nervous System Lymphoma. Atypical Magnetic Resonance Imaging Features and Contribution of the Advanced Imaging. Curr. Probl. Diagn. Radiol. 2017, 46, 136–145. [Google Scholar] [CrossRef]
- Deckert, M.; Engert, A.; Bruck, W.; Ferreri, A.J.; Finke, J.; Illerhaus, G.; Klapper, W.; Korfel, A.; Kuppers, R.; Maarouf, M.; et al. Modern concepts in the biology, diagnosis, differential diagnosis and treatment of primary central nervous system lymphoma. Leukemia 2011, 25, 1797–1807. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Wong, V.S.; Kadoch, C.; Gao, H.X.; Barajas, R.; Chen, L.; Josephson, S.A.; Scott, B.; Douglas, V.; Maiti, M.; et al. CXCL13 plus interleukin 10 is highly specific for the diagnosis of CNS lymphoma. Blood 2013, 121, 4740–4748. [Google Scholar] [CrossRef] [Green Version]
- Sasayama, T.; Nakamizo, S.; Nishihara, M.; Kawamura, A.; Tanaka, H.; Mizukawa, K.; Miyake, S.; Taniguchi, M.; Hosoda, K.; Kohmura, E. Cerebrospinal fluid interleukin-10 is a potentially useful biomarker in immunocompetent primary central nervous system lymphoma (PCNSL). Neuro Oncol. 2012, 14, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Nacinovic-Duletic, A.; Stifter, S.; Dvornik, S.; Skunca, Z.; Jonjic, N. Correlation of serum IL-6, IL-8 and IL-10 levels with clinicopathological features and prognosis in patients with diffuse large B-cell lymphoma. Int. J. Lab. Hematol. 2008, 30, 230–239. [Google Scholar] [CrossRef]
- Kersten, M.J.; Evers, L.M.; Dellemijn, P.L.; van den Berg, H.; Portegies, P.; Hintzen, R.Q.; van Lier, R.A.; von dem Borne, A.E.; van Oers, R.H. Elevation of cerebrospinal fluid soluble CD27 levels in patients with meningeal localization of lymphoid malignancies. Blood 1996, 87, 1985–1989. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.; Korfel, A.; Pfeiffer, S.; Kiewe, P.; Volk, H.D.; Cakiroglu, H.; Widmann, T.; Thiel, E. CXCL13 and CXCL12 in central nervous system lymphoma patients. Clin. Cancer Res. 2009, 15, 5968–5973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldera-Lupa, D.M.; Etemad-Parishanzadeh, O.; Brocksieper, M.; Kirchgaessler, N.; Seidel, S.; Kowalski, T.; Montesinos-Rongen, M.; Deckert, M.; Schlegel, U.; Stuhler, K. Proteomic changes in cerebrospinal fluid from primary central nervous system lymphoma patients are associated with protein ectodomain shedding. Oncotarget 2017, 8, 110118–110132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murase, S.; Saio, M.; Andoh, H.; Takenaka, K.; Shinoda, J.; Nishimura, Y.; Sakai, N.; Takami, T. Diagnostic utility of CSF soluble CD27 for primary central nervous system lymphoma in immunocompetent patients. Neurol. Res. 2000, 22, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Murase, S.; Saio, M.; Takenaka, K.; Shinoda, J.; Nishimura, Y.; Sakai, N.; Takami, T. Increased levels of CSF soluble CD27 in patients with primary central nervous system lymphoma. Cancer Lett. 1998, 132, 181–186. [Google Scholar] [CrossRef]
- Plotkin, S.R.; Batchelor, T.T. Primary nervous-system lymphoma. Lancet Oncol. 2001, 2, 354–365. [Google Scholar] [CrossRef]
- Zetterberg, H.; Andreasson, U.; Blennow, K. CSF antithrombin III and disruption of the blood-brain barrier. J. Clin. Oncol. 2009, 27, 2302–2303. [Google Scholar] [CrossRef]
- Magliozzi, R.; Cross, A.H. Can CSF biomarkers predict future MS disease activity and severity? Mult. Scler. J. 2020, 26, 582–590. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Tomita, T.; Vogelstein, J.T.; Zhou, S.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Selected reaction monitoring approach for validating peptide biomarkers. Proc. Natl. Acad. Sci. USA 2017, 114, 13519–13524. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C. Maximizing an ROC-type measure via linear combination of markers when the gold reference is continuous. Stat. Med. 2013, 32, 1893–1903. [Google Scholar] [CrossRef]
- Roy, S.; Josephson, S.A.; Fridlyand, J.; Karch, J.; Kadoch, C.; Karrim, J.; Damon, L.; Treseler, P.; Kunwar, S.; Shuman, M.A.; et al. Protein biomarker identification in the CSF of patients with CNS lymphoma. J. Clin. Oncol. 2008, 26, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandari, P.S.; Qian, J.; Yehia, G.; Joshi, D.D.; Maloof, P.B.; Potian, J.; Oh, H.S.; Gascon, P.; Harrison, J.S.; Rameshwar, P. Hematopoietic growth factor inducible neurokinin-1 type: A transmembrane protein that is similar to neurokinin 1 interacts with substance P. Regul. Pept. 2003, 111, 169–178. [Google Scholar] [CrossRef]
- Kuan, C.T.; Wakiya, K.; Dowell, J.M.; Herndon, J.E., 2nd; Reardon, D.A.; Graner, M.W.; Riggins, G.J.; Wikstrand, C.J.; Bigner, D.D. Glycoprotein nonmetastatic melanoma protein B, a potential molecular therapeutic target in patients with glioblastoma multiforme. Clin. Cancer Res. 2006, 12, 1970–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, L.; Schmitz, N.; Kurrer, M.O.; Bauer, M.; Hinton, H.I.; Behnke, S.; Gatto, D.; Sebbel, P.; Beerli, R.R.; Sonderegger, I.; et al. VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J. Clin. Investig. 2006, 116, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Kei, A.A.; Filippatos, T.D.; Tsimihodimos, V.; Elisaf, M.S. A review of the role of apolipoprotein C-II in lipoprotein metabolism and cardiovascular disease. Metabolism 2012, 61, 906–921. [Google Scholar] [CrossRef]
- Martin, N.A.; Nawrocki, A.; Molnar, V.; Elkjaer, M.L.; Thygesen, E.K.; Palkovits, M.; Acs, P.; Sejbaek, T.; Nielsen, H.H.; Hegedus, Z.; et al. Orthologous proteins of experimental de-and remyelination are differentially regulated in the CSF proteome of multiple sclerosis subtypes. PLoS ONE 2018, 13, e0202530. [Google Scholar] [CrossRef] [Green Version]
- Huttenhain, R.; Choi, M.; Martin de la Fuente, L.; Oehl, K.; Chang, C.V.; Zimmermann, A.K.; Malander, S.; Olsson, H.; Surinova, S.; Clough, T.; et al. A Targeted Mass Spectrometry Strategy for Developing Proteomic Biomarkers: A Case Study of Epithelial Ovarian Cancer. Mol. Cell Proteom. 2019, 18, 1836–1850. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | |||||
|---|---|---|---|---|---|
| Discovery Cohort | |||||
| Group | Total | Male | Female | Age (Range) | Age (Median) |
| PCNSL | 19 | 9 | 10 | 49–74 | 60 |
| SCNSL | 9 | 4 | 5 | 51–63 | 60 |
| Multiple sclerosis | 9 | 4 | 5 | 36–70 | 51 |
| Gliomas | 10 | 6 | 4 | 38–76 | 56 |
| Other tumors | 10 | 5 | 5 | 41–76 | 62 |
| Tumor-free control | 8 | 4 | 4 | 52–69 | 60 |
| b | |||||
| Validation Cohort | |||||
| Group | Total | Male | Female | Age (Range) | Age (Median) |
| PCNSL | 28 | 10 | 18 | 38–83 | 65 |
| SCNSL | 3 | 2 | 1 | 69–79 | 73 |
| Multiple sclerosis | 14 | 3 | 11 | 18–43 | 27 |
| Gliomas * | 10 | 6 | 4 | 38–76 | 56 |
| Other tumors | 7 | 2 | 5 | 42–79 | 48 |
| Tumor-free control | 11 | 5 | 6 | 55–66 | 60 |
| c | |||||
| IHC Cohort | |||||
| Group | Total | Male | Female | Age (Range) | Age (Median) |
| PCNSL | 20 | 10 | 10 | 47–86 | 71 |
| Tumor-free control | 5 | 2 | 3 | 54–60 | 55 |
| Group | Total Number of Patients | Patients with BBB-Disruption | Proportion | Range of CSF/Serum Ratio (mean) |
|---|---|---|---|---|
| PCNSL | 19 | 11 | 58% | Alb.: 4.7–26.3 (10.9) IgG: 2.9–17.6 (6.4) IgA: 1.6–13.2 (4.3) IgM: 0.2–68.6 (6.5) |
| SCNSL | 9 | 5 | 56% | Alb.: 5.6–15.2 (10.6) IgG: 0.7–9.0 (4.3) IgA: 0.3–5.1 (2.3) IgM: 0.4–1.3 (0.8) |
| Gliomas | 10 | 6 | 60% | Alb.: 3.9–17.0 (9.3) IgG: 1.9–8.7 (4.2) IgA: 0.1–5.6 (2.6) IgM: 0.1–3.8 (1.1) |
| Other tumors | 10 | 4 | 40% | Alb.: 3.5–85.5 (20.6) IgG: 1.2–49.4 (15.3) IgA: 0.4–31.1 (6.7) IgM: 0.1–12.3 (2.1) |
| Tumor-free control | 8 | 1 | 13% | Alb.: 4.1–10.7 (6.7) IgG: 1.1–4.6 (2.8) IgA: 0.5–2.8 (1.7) IgM: 0.2–0.8 (0.4) |
| Patient Group | Proteins (Total) | CNS Proteins | Pro-Portion of CNS Proteins | Membrane Proteins | Proportion of Membrane Proteins | Secreted Proteins | Proportion of Secreted Proteins |
|---|---|---|---|---|---|---|---|
| PCNSL vs. SCNSL | 71 | 36 | 51% | 46 | 65% | 31 | 44% |
| PCNSL vs. Glioma | 41 | 23 | 56% | 23 | 56% | 20 | 49% |
| PCNSL vs. Tumors | 31 | 17 | 55% | 15 | 48% | 17 | 55% |
| PCNSL vs. multiple sclerosis | 50 | 26 | 52% | 30 | 60% | 33 | 66% |
| PCNSL vs. Control | 88 | 57 | 65% | 57 | 65% | 62 | 70% |
| Feature | Sequence | Accession | Gene | Description | p-Value | AUC a (Discovery Cohort) | Diff. Analysis | Tissue | Abundance Range |
|---|---|---|---|---|---|---|---|---|---|
| 909 | DYESGQLYGLEK b | Q659C4 | LARP1B | La-related protein 1B | 0.03610 | 0.81 | PS | High | |
| 1347 | TALALEVGELVK b | P46108 | CRK | Adapter molecule crk | 0.00631 | 0.83 | PS | Medium | |
| 2253 | LEDKVK a | P17275 | JUNB | Transcription factor jun-B | 0.02260 | 0.82 | PS | Medium | |
| 7149 | VFINLLDSYSSGNIGK b | Q96M32 | AK7 | Adenylate kinase 7 | 0.01150 | 0.78 | PG | Medium | |
| 8945 | TAAQNLYEK | P02655 | APOC2 | Apolipoprotein C-II | 0.02180 | 0.83 | PM | Medium | |
| 10516 | AGADLSLLDR b | P19838 | NFKB1 | Nuclear factor NF-kappa-B p105 subunit | 0.00279 | 0.86 | PM | Medium | |
| 12738 | WVQWFGDGK c | Q9UBC3 | DNMT3B | DNA (cytosine-5)-methyltransferase 3B | 0.02850 | 0.81 | PS | Brain | Medium |
| 19446 | GSDPVTIFLR | Q9Y279 | VSIG4 | V-set and immunoglobulin domain-containing protein 4 | 0.01580 | 0.74 | PG | Brain | Medium |
| 23266 | DSFPYLEPLGAIPDVQK | Q15111 | PLCL1 | Inactive phospholipase C-like protein 1 | 0.02440 | 0.83 | PS | Brain | Medium |
| 23650 | ADLIAYLK | P99999 | CYCS | Cytochrome c | 0.01900 | 0.85 | PG | Brain | Medium |
| 29307 | DGLILTSR | Q99497 | PARK7 | Protein deglycase DJ-1 | 0.00492 | 0.82 | PG | Brain | Medium |
| 30944 | VEFLRPSFTDGTIR c | Q15223 | NECTIN1 | Nectin-1 | 0.02080 | 0.72 | PG | Brain/Plasma | Medium |
| 36323 | SSGLVSNAPGVQIR | P04180 | LCAT | Phosphatidylcholine-sterol acyltransferase | 0.00055 | 0.91 | PC | Brain/Plasma | Medium |
| 36464 | NGEEFSFLK | Q99574 | SERPINI1 | Neuroserpin | 0.04050 | 0.75 | PT/PC | Brain | Medium |
| 37006 | AYVPIAQVK | Q14956 | GPNMB | Transmembrane glycoprotein NMB | 0.00862 | 0.77 | PG/PM | Brain/Liver | Medium |
| 37088 | EVLPAIR b | O00391 | QSOX1 | Sulfhydryl oxidase 1 | 0.02580 | 0.73 | PT | Brain/Plasma | Medium |
| 38690 | ALGFEAAESSLTK b | Q8TDL5 | BPIFB1 | BPI fold-containing family B member 1 | 0.00045 | 0.93 | PC | Medium | |
| 38843 | VNHAVLAVGYGEK b | P09668 | CTSH | Pro-cathepsin H | 0.04180 | 0.75 | PG | Liver | Medium |
| 39626 | DQVANSAFVER b | P07900 | HSP90AA1 | Heat shock protein HSP 90-alpha | 0.03180 | 0.73 | PG | Brain/Liver | Medium |
| 40050 | DGLIPLEIR c | Q13228 | SELENBP1 | Selenium-binding protein 1 | 0.00031 | 0.94 | PC | Brain | Medium |
| 45513 | EMDPVTQLYTMTSTLEYK a | Q13740 | ALCAM | CD166 antigen | 0.00516 | 0.84 | PG/PC | Brain/Plasma/Liver | Low |
| 50614 | WVGDLPNGR b | Q96DR8 | MUCL1 | Mucin-like protein 1 | 0.01760 | 0.82 | PM | Low | |
| 57938 | ATYIQNYR c | Q01459 | CTBS | Di-N-acetylchitobiase | 0.02710 | 0.81 | PM | Brain/Liver | Medium |
| 61240 | FDAPPEAVAAK b | O95969 | SCGB1D2 | Secretoglobin family 1D member 2 | 0.00203 | 0.83 | PM | Brain | Low |
| 62517 | FRDLEEDPYLPGNPR b | P22304 | IDS | Iduronate 2-sulfatase | 0.01920 | 0.78 | PT | Liver | Medium |
| 68357 | AYLEVTDVIADRPPPVIR b | Q9Y6N7 | ROBO1 | Roundabout homolog 1 | 0.01630 | 0.81 | PM | Low | |
| 73987 | DLAEVPASIPVNTR | Q9NT99 | LRRC4B | Leucine-rich repeat-containing protein 4B | 0.03940 | 0.71 | PG | Brain | Low |
| 89726 | ISGLIYEETR b | P62805 | HIST1H4A | Histone H4 | 0.01910 | 0.82 | PS | Brain | Medium |
| 137254 | VTDANYGELQEHKAQAYLK a | Q8TAG5 | VSTM2A | V-set and transmembrane domain-containing protein 2A | 0.04850 | 0.74 | PT | Brain | Low |
| 142941 | ILSGRPPLGFLNPR b | O14773 | TPP1 | Tripeptidyl-peptidase 1 | 0.03580 | 0.82 | PS | Brain/Liver | Low |
| 177384 | ESYNVQLQLPAR b | Q14112 | NID2 | Nidogen-2 | 0.04400 | 0.75 | PT | Liver | Medium |
| 199236 | SQLEAIFLR b | Q8IV08 | PLD3 | Phospholipase D3 | 0.00249 | 0.85 | PG | Brain/Liver | Low |
| Protein | Accession | Peptide | Control [fmol/µg] | Glioma [fmol/µg] | Multiple Sclerosis [fmol/µg] | PCNSL [fmol/µg] | SCNSL [fmol/µg] | Tumors [fmol/µg] |
|---|---|---|---|---|---|---|---|---|
| LRRC4B | Q9NT99 | DLAEVPASIPVNTR | 106.75 (92.08) | 47.70 (40.22) | 41.41 (40.33) | 71.08 (60.99) | 64.46 (98.25) | 46.54 (50.42) |
| PARK7 | Q99497 | DGLILTSR | 6.66 (2.76) | 6.15 (2.69) | 6.66 (5.49) | 7.46 (3.45) | 5.30 (2.24) | 8.22 (8.94) |
| SERPINI1 | Q99574 | NGEEFSFLK | 1.70 (0.84) | 1.00 (0.56) | 1.04 (0.58) | 1.00 (0.70) | 0.69 (0.61) | 0.53 (0.36) |
| VSIG4 | Q9Y279 | GSDPVTIFLR | 26.12 (19.35) | 19.18 (13.56) | 21.77 (27.56) | 104.10 (132.02) | 68.75 (103.03) | 63.12 (67.76) |
| APOC2 | P02655 | TAAQNLYEK | 8.44 (6.77) | 5.19 (2.58) | 5.96 (5.92) | 16.84 (15.17) | 15.48 (16.12) | 18.72 (9.25) |
| CYCS | P99999 | ADLIAYLK | 5.10 (3.19) | 3.12 (1.60) | 3.62 (2.92) | 3.46 (2.22) | 2.04 (0.97) | 7.29 (12.64) |
| GPNMB | Q14956 | AYVPIAQVK | 0.33 (0.20) | 0.23 (0.06) | 0.24 (0.12) | 0.55 (0.41) | 0.25 (0.07) | 0.41 (0.30) |
| LCAT | P04180 | SSGLVSNAPGVQIR | 3.55 (2.15) | 2.29 (1.13) | 2.87 (1.94) | 2.95 (1.58) | 1.82 (0.25) | 2.17 (1.12) |
| Protein | Accession | Peptide | ANOVA p-Value | PCNSL vs. Control | PCNSL vs. Glioma | PCNSL vs. Multiple Sclerosis | PCNSL vs. SCNSL | PCNSL vs. Tumors |
|---|---|---|---|---|---|---|---|---|
| LRRC4B | Q9NT99 | DLAEVPASIPVNTR | 0.1603 | 0.8443 | 0.9988 | 0.5417 | 0.8747 | 0.8312 |
| PARK7 | Q99497 | DGLILTSR | 0.6558 | 0.9997 | 0.9866 | 0.5067 | 0.9845 | 0.9983 |
| SERPINI1 | Q99574 | NGEEFSFLK | 0.0298 | 0.1350 | 0.9982 | 0.9976 | 0.9386 | 0.6346 |
| VSIG4 | Q9Y279 | GSDPVTIFLR | 0.0009 | 0.1540 | 0.0339 | 0.0006 | 0.8844 | 0.9495 |
| APOC2 | P02655 | TAAQNLYEK | 0.0001 | 0.2592 | 0.0124 | 0.0008 | 0.9991 | 0.9924 |
| CYCS | P99999 | ADLIAYLK | 0.8286 | 0.9319 | 1.0000 | 0.9995 | 0.9667 | 1.0000 |
| GPNMB | Q14956 | AYVPIAQVK | 0.0027 | 0.2994 | 0.0191 | 0.0049 | 0.4793 | 0.7925 |
| LCAT | P04180 | SSGLVSNAPGVQIR | 0.7455 | 0.9989 | 0.9902 | 0.8298 | 0.9889 | 0.9743 |
| Case | Sex | Age | Apolipoprotein CII | GPNMB | VSIG4 |
|---|---|---|---|---|---|
| 1 | f | 77 | − | − | − |
| 2 | m | 79 | − | − | − |
| 3 | f | 71 | − | ++ | − |
| 4 | m | 66 | − | − | − |
| 5 | m | 86 | − | ++ | − |
| 6 | f | 66 | ++ | ++ | − |
| 7 | f | 70 | − | ++ | − |
| 8 | f | 80 | − | − | − |
| 9 | f | 76 | − | − | − |
| 10 | f | 69 | − | + | − |
| 11 | m | 60 | − | − | − |
| 12 | m | 47 | − | − | − |
| 13 | m | 79 | − | − | − |
| 14 | m | 69 | − | − | − |
| 15 | f | 76 | − | − | − |
| 16 | m | 78 | − | − | − |
| 17 | f | 75 | − | +++ | − |
| 18 | f | 60 | − | + | − |
| 19 | m | 70 | + | − | − |
| 20 | m | 70 | − | − | − |
| 21 | f | 54 | neurons + | neurons +, astrocytes +, oligodendrocytes weakly + | astrocytes + |
| 22 | f | 55 | neurons + | astrocytes + microglial cells + | astrocytes + |
| 23 | f | 60 | neurons + | astrocytes + microglial cells + | astrocytes + |
| 24 | m | 54 | neurons + | microglial cells + | astrocytes + |
| 25 | m | 58 | neurons + | microglial cells + | astrocytes + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waldera-Lupa, D.M.; Poschmann, G.; Kirchgaessler, N.; Etemad-Parishanzadeh, O.; Baberg, F.; Brocksieper, M.; Seidel, S.; Kowalski, T.; Brunn, A.; Haghikia, A.; et al. A Multiplex Assay for the Stratification of Patients with Primary Central Nervous System Lymphoma Using Targeted Mass Spectrometry. Cancers 2020, 12, 1732. https://doi.org/10.3390/cancers12071732
Waldera-Lupa DM, Poschmann G, Kirchgaessler N, Etemad-Parishanzadeh O, Baberg F, Brocksieper M, Seidel S, Kowalski T, Brunn A, Haghikia A, et al. A Multiplex Assay for the Stratification of Patients with Primary Central Nervous System Lymphoma Using Targeted Mass Spectrometry. Cancers. 2020; 12(7):1732. https://doi.org/10.3390/cancers12071732
Chicago/Turabian StyleWaldera-Lupa, Daniel M., Gereon Poschmann, Nina Kirchgaessler, Omid Etemad-Parishanzadeh, Falk Baberg, Mareike Brocksieper, Sabine Seidel, Thomas Kowalski, Anna Brunn, Aiden Haghikia, and et al. 2020. "A Multiplex Assay for the Stratification of Patients with Primary Central Nervous System Lymphoma Using Targeted Mass Spectrometry" Cancers 12, no. 7: 1732. https://doi.org/10.3390/cancers12071732
APA StyleWaldera-Lupa, D. M., Poschmann, G., Kirchgaessler, N., Etemad-Parishanzadeh, O., Baberg, F., Brocksieper, M., Seidel, S., Kowalski, T., Brunn, A., Haghikia, A., Gold, R., Stefanski, A., Deckert, M., Schlegel, U., & Stühler, K. (2020). A Multiplex Assay for the Stratification of Patients with Primary Central Nervous System Lymphoma Using Targeted Mass Spectrometry. Cancers, 12(7), 1732. https://doi.org/10.3390/cancers12071732