RORα Regulates Cholesterol Metabolism of CD8+ T Cells for Anticancer Immunity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
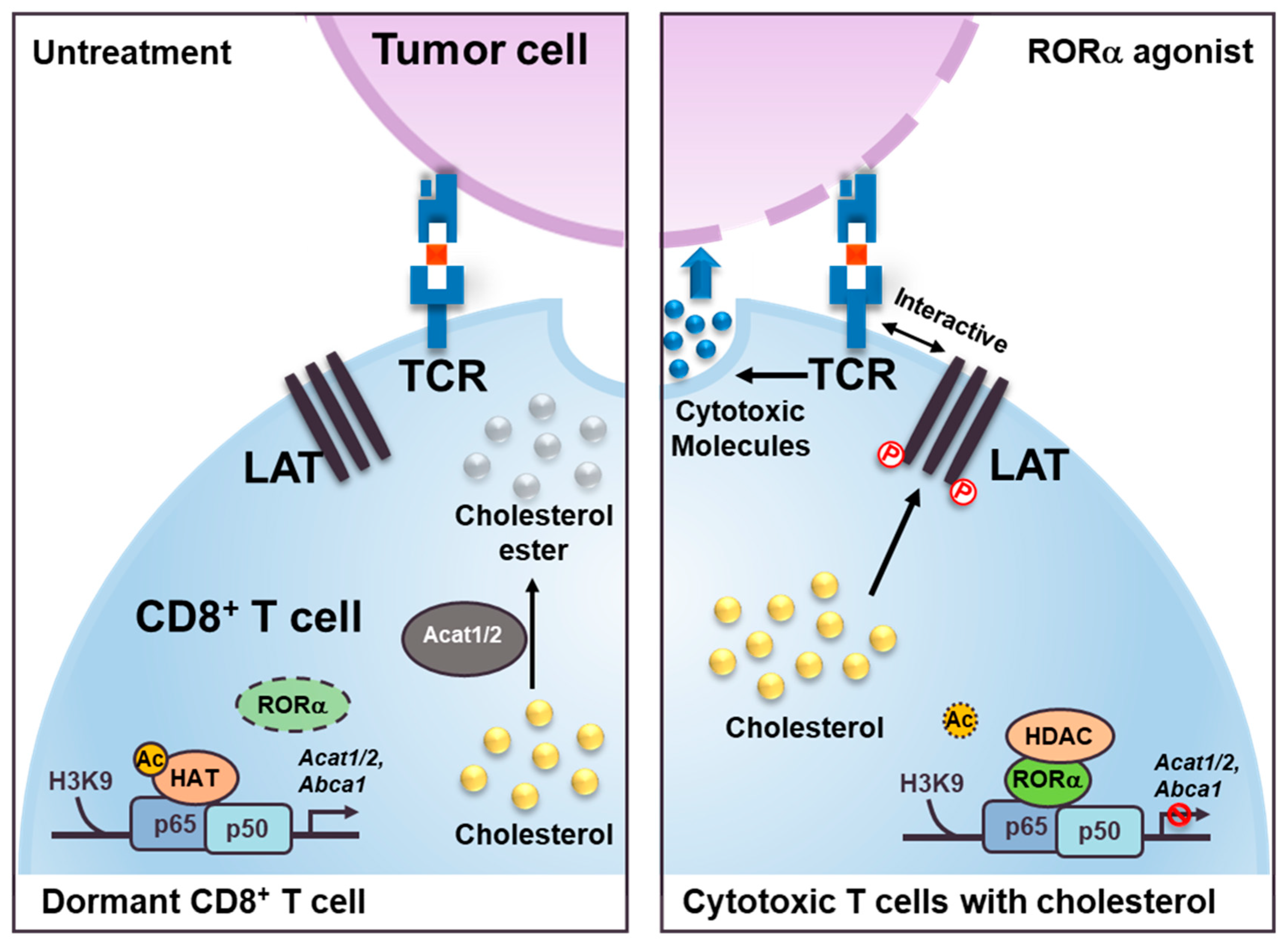
:1. Introduction
2. Results
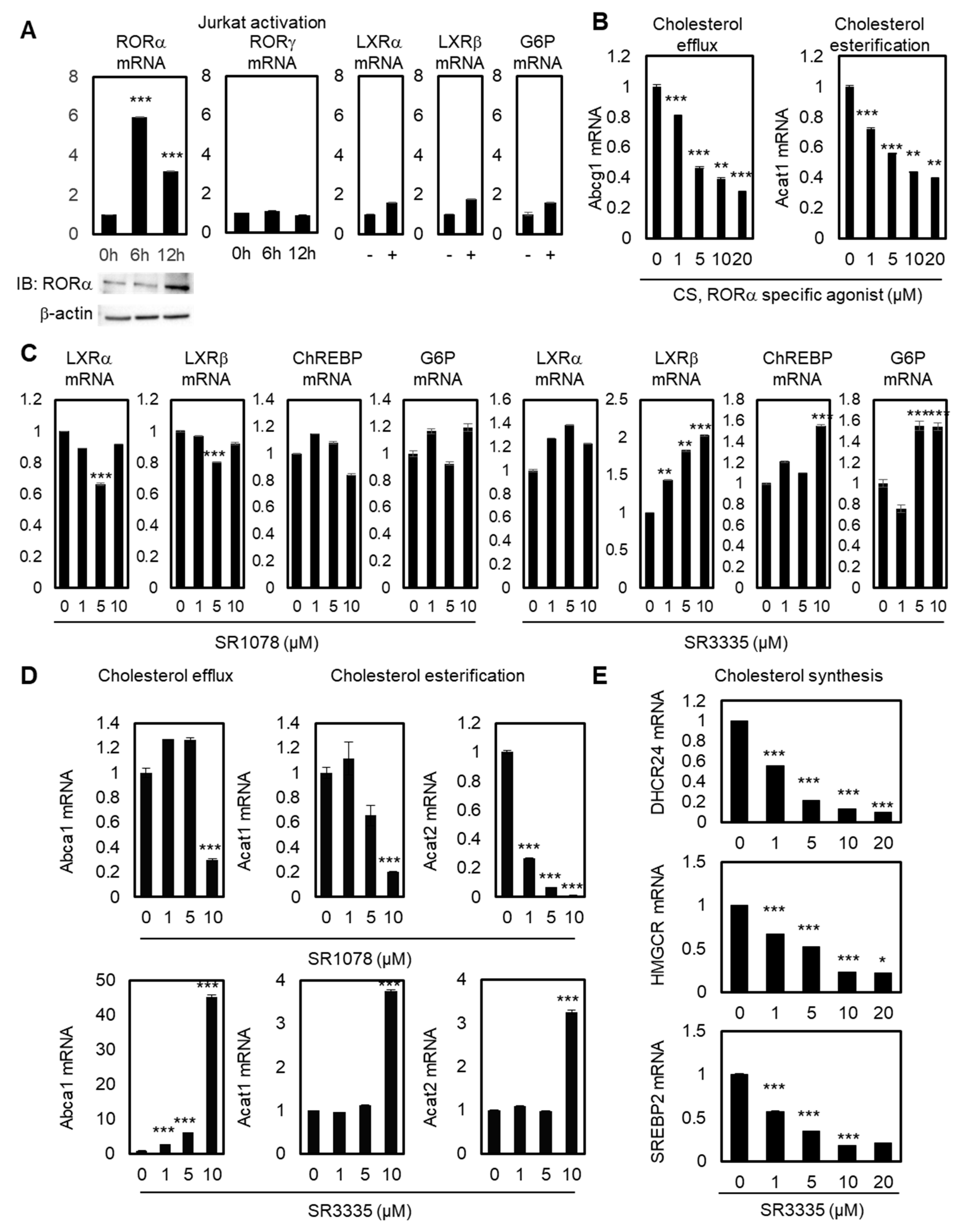
2.1. Enhanced RORα Activities Improves Cholesterol Metabolism in T Cells
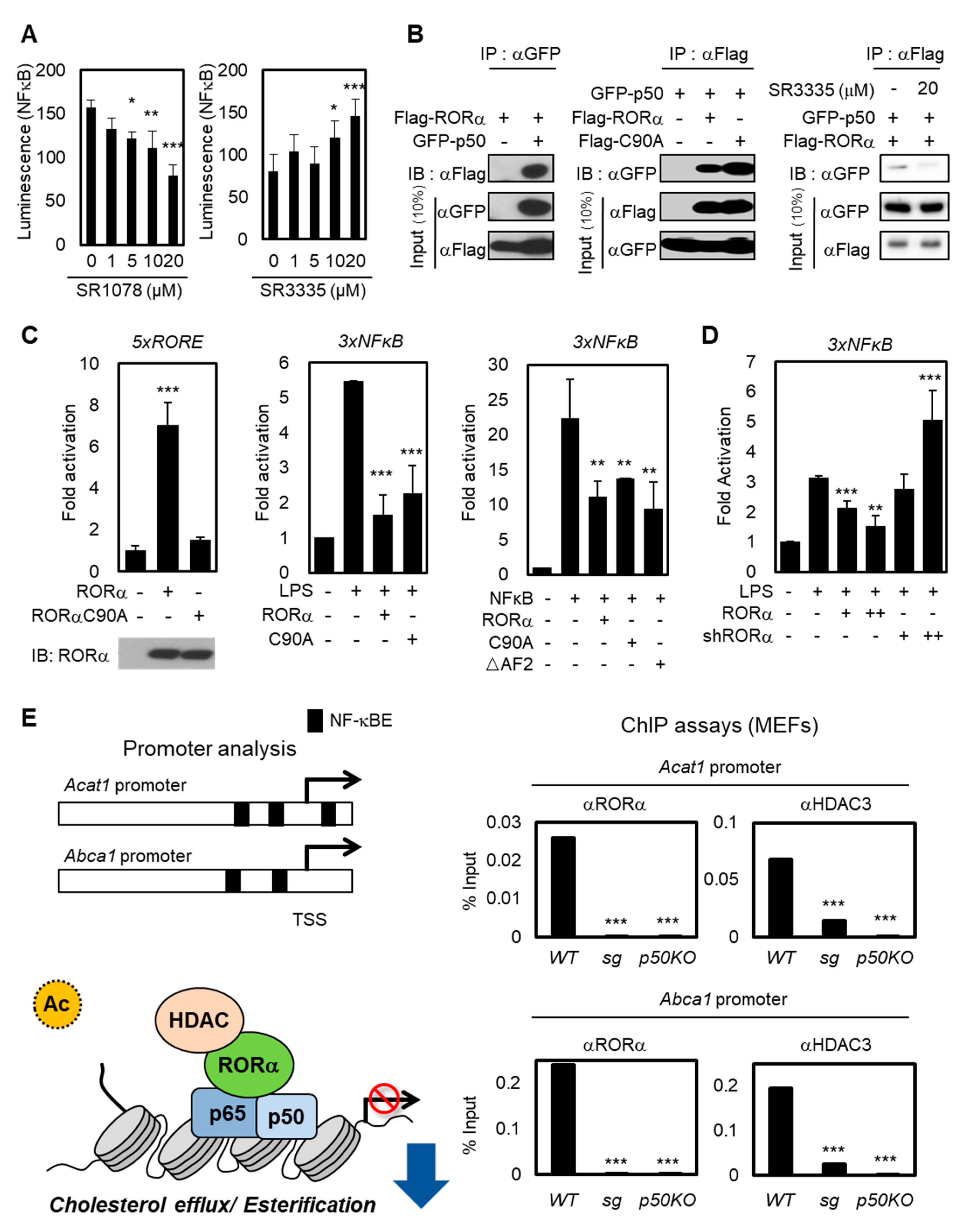
2.2. RORα Acts as a Corepressor for Cholesterol Esterification via NF-κB-Dependent Promoter Regulation
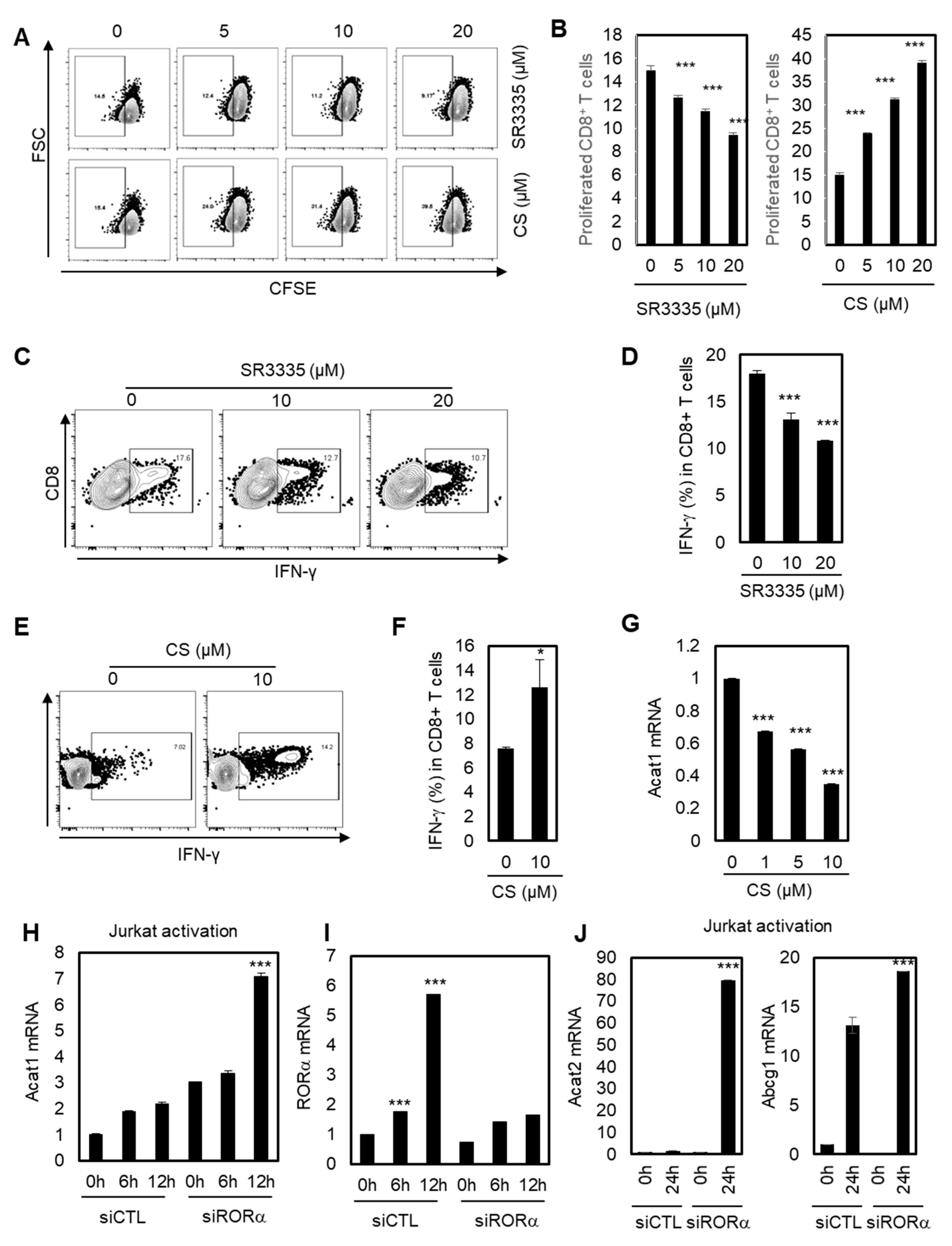
2.3. Stimulating RORα Activities Enhances the Effector Responses of CD8+ T Cells through Cholesterol Esterification
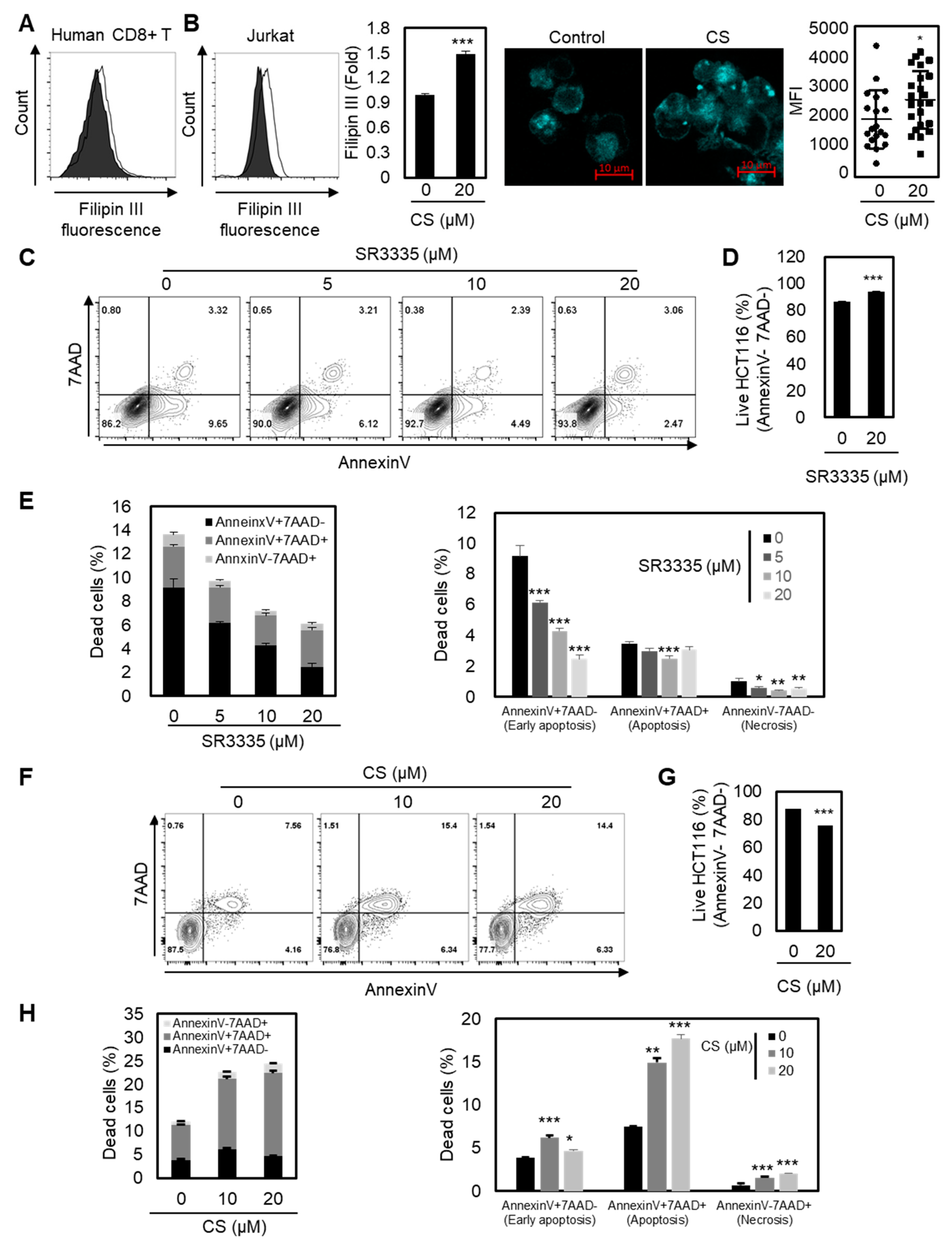
2.4. Elevated Cholesterol Levels in the Plasma Membrane by Increased RORα Activation May Modulate the Function of CD8+ T Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies
4.3. The Measurement of Cholesterol Metabolic Regulation by RORα in T Cells
4.4. The Measurement of NF-κB Regulation by RORα
4.5. Reporter Assays
4.6. RORα Antagonist or Agonist Treatment on CD8+ T Cells
4.7. Cell Proliferation Assay
4.8. Quantitative RT-PCR
4.9. ChIP Assays
4.10. Constructions
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RORα: | Retinoic acid-related orphan receptor α |
| HDAC: | Histone deacetylase |
| PRRs: | Patterns recognition receptors |
| CD8: | Cluster of differentiation 8 |
| TCR: | T cell receptor |
| CS: | Cholesterol sulfate |
References
- Jetten, A.M.; Joo, J.H. Retinoid-Related Orphan Receptors (Rors): Roles in Cellular Differentiation and Development. Adv. Dev. Biol. 2006, 16, 313–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, B.A.; Frankel, W.N.; Kerrebrock, A.W.; Hawkins, T.L.; FitzHugh, W.; Kusumi, K.; Russell, L.B.; Mueller, K.L.; van Berkel, V.; Birren, B.W.; et al. Disruption of the Nuclear Hormone Receptor Roralpha in Staggerer Mice. Nature 1996, 379, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.K.; Kim, D.; Kim, K.; Boo, K.; Yu, Y.S.; Kim, I.S.; Jeon, Y.; Im, S.K.; Lee, S.H.; Lee, J.M.; et al. Roralpha Is Crucial for Attenuated Inflammatory Response to Maintain Intestinal Homeostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 21140–21149. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Lee, J.M.; Yu, Y.S.; Kim, H.; Nam, H.J.; Moon, H.G.; Noh, D.Y.; Kim, K.I.; Fang, S.; Baek, S.H. Roralpha2 Requires Lsd1 to Enhance Tumor Progression in Breast Cancer. Sci. Rep. 2017, 7, 11994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.; Kim, I.S.; Lee, J.M.; Shin, S.Y.; Lee, J.H.; Baek, S.H.; Cho, K.H. The Hidden Switches Underlying Roralpha-Mediated Circuits That Critically Regulate Uncontrolled Cell Proliferation. J. Mol. Cell. Biol. 2014, 6, 338–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Lee, J.S.; Kim, H.; Kim, K.; Park, H.; Kim, J.Y.; Lee, S.H.; Kim, I.S.; Kim, J.; Lee, M.; et al. Ezh2 Generates a Methyl Degron That Is Recognized by the Dcaf1/Ddb1/Cul4 E3 Ubiquitin Ligase Complex. Mol. Cell 2012, 48, 572–586. [Google Scholar] [CrossRef] [Green Version]
- Hwang, E.J.; Lee, J.M.; Jeong, J.; Park, J.H.; Yang, Y.; Lim, J.S.; Kim, J.H.; Baek, S.H.; Kim, K.I. Sumoylation of Roralpha Potentiates Transcriptional Activation Function. Biochem. Biophys. Res. Commun 2009, 378, 513–517. [Google Scholar] [CrossRef]
- Choi, W.S.; Lee, G.; Song, W.H.; Koh, J.T.; Yang, J.; Kwak, J.S.; Kim, H.E.; Kim, S.K.; Son, Y.O.; Nam, H.; et al. The Ch25h-Cyp7b1-Roralpha Axis of Cholesterol Metabolism Regulates Osteoarthritis. Nature 2019, 566, 254–258. [Google Scholar] [CrossRef]
- Kim, H.; Lee, J.M.; Lee, G.; Bhin, J.; Oh, S.K.; Kim, K.; Pyo, K.E.; Lee, J.S.; Yim, H.Y.; Kim, K.I.; et al. DNA Damage-Induced Roralpha Is Crucial for P53 Stabilization and Increased Apoptosis. Mol. Cell 2011, 44, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Kim, I.S.; Kim, H.; Lee, J.S.; Kim, K.; Yim, H.Y.; Jeong, J.; Kim, J.H.; Kim, J.Y.; Lee, H.; et al. Roralpha Attenuates Wnt/Beta-Catenin Signaling by Pkcalpha-Dependent Phosphorylation in Colon Cancer. Mol. Cell 2010, 37, 183–195. [Google Scholar] [CrossRef]
- Wang, Y.; Solt, L.A.; Kojetin, D.J.; Burris, T.P. Regulation of P53 Stability and Apoptosis by a Ror Agonist. PLoS ONE 2012, 7, e34921. [Google Scholar] [CrossRef] [Green Version]
- Kojetin, D.J.; Burris, T.P. Rev-Erb and Ror Nuclear Receptors as Drug Targets. Nat. Rev. Drug Discov. 2014, 13, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plato, A.; Hardison, S.E.; Brown, G.D. Pattern Recognition Receptors in Antifungal Immunity. Semin. Immunopathol. 2015, 37, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Li, Z.; Han, F.; Jia, Y.; Qi, L.; Wu, G.; Cai, W.; Xu, Y.; Li, C.; Zhang, W.; et al. Ror Alpha Protects against Lps-Induced Inflammation by Down-Regulating Sirt1/Nf-Kappa B Pathway. Arch. Biochem. Biophys. 2019, 668, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, L.; Ding, S.; Lin, N.; Ji, Q.; Gao, L.; Su, Y.; He, B.; Pu, J. Novel Protective Role of the Circadian Nuclear Receptor Retinoic Acid-Related Orphan Receptor-Alpha in Diabetic Cardiomyopathy. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef]
- Kumar, N.; Kojetin, D.J.; Solt, L.A.; Kumar, K.G.; Nuhant, P.; Duckett, D.R.; Cameron, M.D.; Butler, A.A.; Roush, W.R.; Griffin, P.R.; et al. Identification of Sr3335 (Ml-176): A Synthetic Roralpha Selective Inverse Agonist. ACS Chem. Biol. 2011, 6, 218–222. [Google Scholar] [CrossRef]
- Kallen, J.; Schlaeppi, J.M.; Bitsch, F.; Delhon, I.; Fournier, B. Crystal Structure of the Human Roralpha Ligand Binding Domain in Complex with Cholesterol Sulfate at 2.2 A. J. Biol. Chem. 2004, 279, 14033–14038. [Google Scholar] [CrossRef] [Green Version]
- Marciano, D.P.; Chang, M.R.; Corzo, C.A.; Goswami, D.; Lam, V.Q.; Pascal, B.D.; Griffin, P.R. The Therapeutic Potential of Nuclear Receptor Modulators for Treatment of Metabolic Disorders: Ppargamma, Rors, and Rev-Erbs. Cell. Metab. 2014, 19, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Kosti, P.; Maher, J.; Arnold, J.N. Perspectives on Chimeric Antigen Receptor T-Cell Immunotherapy for Solid Tumors. Front. Immunol. 2018, 9, 1104. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial Cd8+ Tumor-Infiltrating Lymphocytes and a High Cd8+/Regulatory T Cell Ratio Are Associated with Favorable Prognosis in Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Nakano, O.; Sato, M.; Naito, Y.; Suzuki, K.; Orikasa, S.; Aizawa, M.; Suzuki, Y.; Shintaku, I.; Nagura, H.; Ohtani, H. Proliferative Activity of Intratumoral Cd8(+) T-Lymphocytes as a Prognostic Factor in Human Renal Cell Carcinoma: Clinicopathologic Demonstration of Antitumor Immunity. Cancer Res. 2001, 61, 5132–5136. [Google Scholar] [PubMed]
- Halle, S.; Halle, O.; Forster, R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity in Vivo. Trends Immunol. 2017, 38, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T Cells: The Promise and Challenges of Cancer Immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.M.; Roberti, M.P.; Mordoh, J. Natural Killer Cells in Human Cancer: From Biological Functions to Clinical Applications. J. Biomed. Biotechnol. 2011, 2011, 676198. [Google Scholar] [CrossRef]
- Wu, W.; Shi, X.; Xu, C. Regulation of T Cell Signalling by Membrane Lipids. Nat. Rev. Immunol. 2016, 16, 690–701. [Google Scholar] [CrossRef]
- Tillman, T.S.; Cascio, M. Effects of Membrane Lipids on Ion Channel Structure and Function. Cell. Biochem. Biophys. 2003, 38, 161–190. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the Antitumour Response of Cd8(+) T Cells by Modulating Cholesterol Metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.Y.; Li, B.L.; Chang, C.C.; Urano, Y. Acyl-Coenzyme A:Cholesterol Acyltransferases. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1–9. [Google Scholar] [CrossRef] [Green Version]
- Hanyu, O.; Nakae, H.; Miida, T.; Higashi, Y.; Fuda, H.; Endo, M.; Kohjitani, A.; Sone, H.; Strott, C.A. Cholesterol Sulfate Induces Expression of the Skin Barrier Protein Filaggrin in Normal Human Epidermal Keratinocytes through Induction of Roralpha. Biochem. Biophys. Res. Commun. 2012, 428, 99–104. [Google Scholar] [CrossRef]
- Wang, Y.; Kumar, N.; Nuhant, P.; Cameron, M.D.; Istrate, M.A.; Roush, W.R.; Griffin, P.R.; Burris, T.P. Identification of Sr1078, a Synthetic Agonist for the Orphan Nuclear Receptors Roralpha and Rorgamma. ACS Chem. Biol. 2010, 5, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Becker, C.; Ivanov, I.I.; Hirth, S.; Wirtz, S.; Neufert, C.; Pouly, S.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; et al. Rorgamma-Expressing Th17 Cells Induce Murine Chronic Intestinal Inflammation Via Redundant Effects of Il-17a and Il-17f. Gastroenterology 2009, 136, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Eidenschenk, C.; Kiefer, J.R.; Ouyang, W. Post-Translational Regulation of Rorgammat-a Therapeutic Target for the Modulation of Interleukin-17-Mediated Responses in Autoimmune Diseases. Cytokine Growth Factor Rev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, H.J.; Hoang, M.H.; Yeo, S.K.; Jia, Y.; Lee, S.J. Induction of Abca1 and Abcg1 Expression by the Liver X Receptor Modulator Cineole in Macrophages. Bioorg. Med. Chem Lett. 2013, 23, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Fulford, T.S.; Messina, N.L.; Grumont, R.J. Nf-Kappab Control of T Cell Development. Nat. Immunol. 2014, 15, 15–25. [Google Scholar] [CrossRef]
- Chen, F.E.; Kempiak, S.; Huang, D.B.; Phelps, C.; Ghosh, G. Construction, Expression, Purification and Functional Analysis of Recombinant Nfkappab P50/P65 Heterodimer. Protein. Eng. 1999, 12, 423–428. [Google Scholar] [CrossRef]
- Lei, L.; Xiong, Y.; Chen, J.; Yang, J.B.; Wang, Y.; Yang, X.Y.; Chang, C.C.; Song, B.L.; Chang, T.Y.; Li, B.L. Tnf-Alpha Stimulates the Acat1 Expression in Differentiating Monocytes to Promote the Ce-Laden Cell Formation. J. Lipid. Res. 2009, 50, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wan, Y.; Huang, C. The Biological Functions of Nf-Kappab1 (P50) and Its Potential as an Anti-Cancer Target. Curr. Cancer Drug Targets 2009, 9, 566–571. [Google Scholar] [CrossRef]
- Ge, J.; Zhai, W.; Cheng, B.; He, P.; Qi, B.; Lu, H.; Zeng, Y.; Chen, X. Insulin Induces Human Acyl-Coenzyme A: Cholesterol Acyltransferase1 Gene Expression Via Map Kinases and Ccaat/Enhancer-Binding Protein Alpha. J. Cell Biochem. 2013, 114, 2188–2198. [Google Scholar] [CrossRef]
- Cavelier, L.B.; Qiu, Y.; Bielicki, J.K.; Afzal, V.; Cheng, J.F.; Rubin, E.M. Regulation and Activity of the Human Abca1 Gene in Transgenic Mice. J. Biol. Chem. 2001, 276, 18046–18051. [Google Scholar] [CrossRef] [Green Version]
- Santamarina-Fojo, S.; Peterson, K.; Knapper, C.; Qiu, Y.; Freeman, L.; Cheng, J.F.; Osorio, J.; Remaley, A.; Yang, X.P.; Haudenschild, C.; et al. Complete Genomic Sequence of the Human Abca1 Gene: Analysis of the Human and Mouse Atp-Binding Cassette a Promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 7987–7992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Yang, Y.; Wei, J.; Cun, X.; Lu, Z.; Qiu, Y.; Zhang, Z.; He, Q. Enhanced Chemo-Immunotherapy against Melanoma by Inhibition of Cholesterol Esterification in Cd8(+) T Cells. Nanomedicine 2018, 14, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, J.; Liu, Y.; Kang, L.; Chen, H.; Jin, Y.; Zhao, F.; Feng, J.; Fang, C.; Zhu, B.; et al. Cholesterol Esterification Enzyme Inhibition Enhances Antitumor Effects of Human Chimeric Antigen Receptors Modified T Cells. J. Immunother. 2018, 41, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Guerrero, E.; Sanchez-Abarca, L.I.; Domingo, E.; Ramos, T.L.; Bejarano-Garcia, J.A.; Gonzalez-Campos, J.A.; Caballero-Velazquez, T.; Perez-Simon, J.A. Selection of Tumor-Specific Cytotoxic T Lymphocytes in Acute Myeloid Leukemia Patients through the Identification of T-Cells Capable to Establish Stable Interactions with the Leukemic Cells: "Doublet Technology". Front. Immunol. 2018, 9, 1971. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G. Targeting Cancer Metabolism: A Therapeutic Window Opens. Nat. Rev. Drug. Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. Pd-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. Tigit and Pd-1 Dual Checkpoint Blockade Enhances Antitumor Immunity and Survival in Gbm. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.; Hernandez, J.A.; Patenia, R.; Kim, T.K.; Khalili, J.; Dougherty, M.C.; Hanley, P.J.; Bollard, C.M.; Komanduri, K.V.; et al. Lovastatin Inhibits T-Cell Proliferation While Preserving the Cytolytic Function of Ebv, Cmv, and Mart-1-Specific Ctls. J. Immunother. 2010, 33, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Overton, E.T.; Sterrett, S.; Westfall, A.O.; Kahan, S.M.; Burkholder, G.; Zajac, A.J.; Goepfert, P.A.; Bansal, A. Effects of Atorvastatin and Pravastatin on Immune Activation and T-Cell Function in Antiretroviral Therapy-Suppressed Hiv-1-Infected Patients. AIDS 2014, 28, 2627–2631. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Wang, C.; Evers, B.M.; Zhou, B.P.; Xu, R. Roralpha Suppresses Breast Tumor Invasion by Inducing Sema3f Expression. Cancer Res. 2012, 72, 1728–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, J.K.; Choi, Y.K.; Kang, Y.N.; Jang, B.K.; Kang, K.J.; Jeon, Y.H.; Lee, H.W.; Jeon, J.H.; Koo, S.H.; Jeong, W.I.; et al. Retinoic Acid-Related Orphan Receptor Alpha Reprograms Glucose Metabolism in Glutamine-Deficient Hepatoma Cells. Hepatology 2015, 61, 953–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T Helper 17 Lineage Differentiation Is Programmed by Orphan Nuclear Receptors Ror Alpha and Ror Gamma. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamontova, A.; Seguret-Mace, S.; Esposito, B.; Chaniale, C.; Bouly, M.; Delhaye-Bouchaud, N.; Luc, G.; Staels, B.; Duverger, N.; Mariani, J.; et al. Severe Atherosclerosis and Hypoalphalipoproteinemia in the Staggerer Mouse, a Mutant of the Nuclear Receptor Roralpha. Circulation 1998, 98, 2738–2743. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fan, W.; Xu, J.; Lu, M.; Yamamoto, H.; Auwerx, J.; Sears, D.D.; Talukdar, S.; Oh, D.; Chen, A.; et al. Adipocyte Ncor Knockout Decreases Ppargamma Phosphorylation and Enhances Ppargamma Activity and Insulin Sensitivity. Cell 2011, 147, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Boo, K.; Yu, Y.S.; Oh, S.K.; Kim, H.; Jeon, Y.; Bhin, J.; Hwang, D.; Kim, K.I.; Lee, J.S.; et al. Roralpha Controls Hepatic Lipid Homeostasis Via Negative Regulation of Ppargamma Transcriptional Network. Nat. Commun. 2017, 8, 162. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y. Cancer Immunotherapy: Harnessing the Immune System to Battle Cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, I.K.; Song, H.; Kim, H.; Kim, I.S.; Tran, N.L.; Kim, S.-H.; Oh, S.J.; Lee, J.M. RORα Regulates Cholesterol Metabolism of CD8+ T Cells for Anticancer Immunity. Cancers 2020, 12, 1733. https://doi.org/10.3390/cancers12071733
Lee IK, Song H, Kim H, Kim IS, Tran NL, Kim S-H, Oh SJ, Lee JM. RORα Regulates Cholesterol Metabolism of CD8+ T Cells for Anticancer Immunity. Cancers. 2020; 12(7):1733. https://doi.org/10.3390/cancers12071733
Chicago/Turabian StyleLee, In Kyu, Hyerin Song, Hyerim Kim, Ik Soo Kim, Na Ly Tran, Sang-Heon Kim, Seung Ja Oh, and Ji Min Lee. 2020. "RORα Regulates Cholesterol Metabolism of CD8+ T Cells for Anticancer Immunity" Cancers 12, no. 7: 1733. https://doi.org/10.3390/cancers12071733
APA StyleLee, I. K., Song, H., Kim, H., Kim, I. S., Tran, N. L., Kim, S. -H., Oh, S. J., & Lee, J. M. (2020). RORα Regulates Cholesterol Metabolism of CD8+ T Cells for Anticancer Immunity. Cancers, 12(7), 1733. https://doi.org/10.3390/cancers12071733