Cancer Nanomedicine Special Issue Review Anticancer Drug Delivery with Nanoparticles: Extracellular Vesicles or Synthetic Nanobeads as Therapeutic Tools for Conventional Treatment or Immunotherapy




Abstract
:1. Introduction
2. EV as Drug Delivery Vehicles in Cancer
2.1. Extracellular Vesicles in the Physiopathology of Intercellular Communication
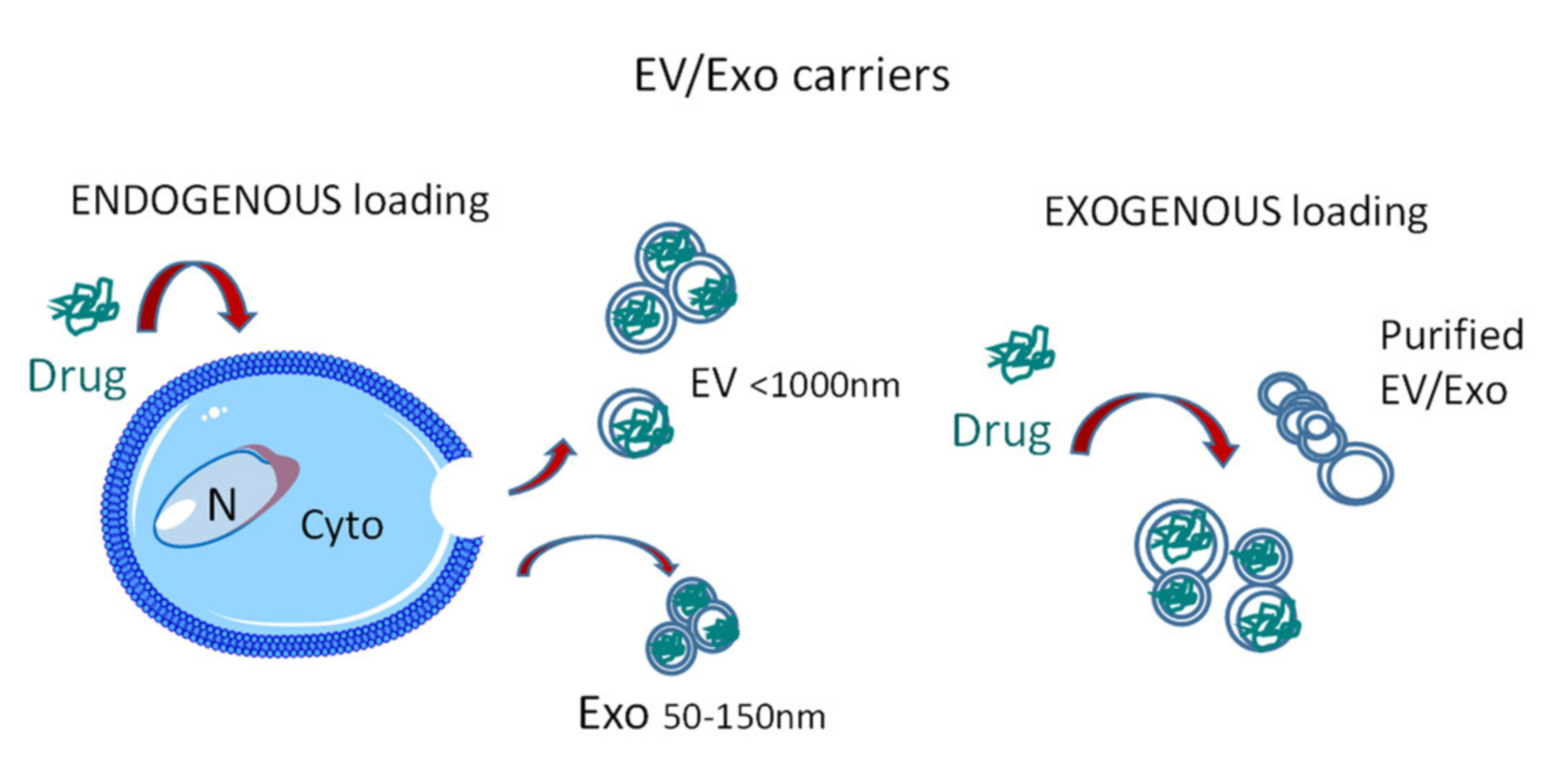
2.2. EV Sources and Loading
2.3. Further EV Modifications for Therapeutic Purposes
3. Exo as Drug Carriers to Enhance Anti-Tumor Immune Response
3.1. EV and Exo in Tumor Immunity
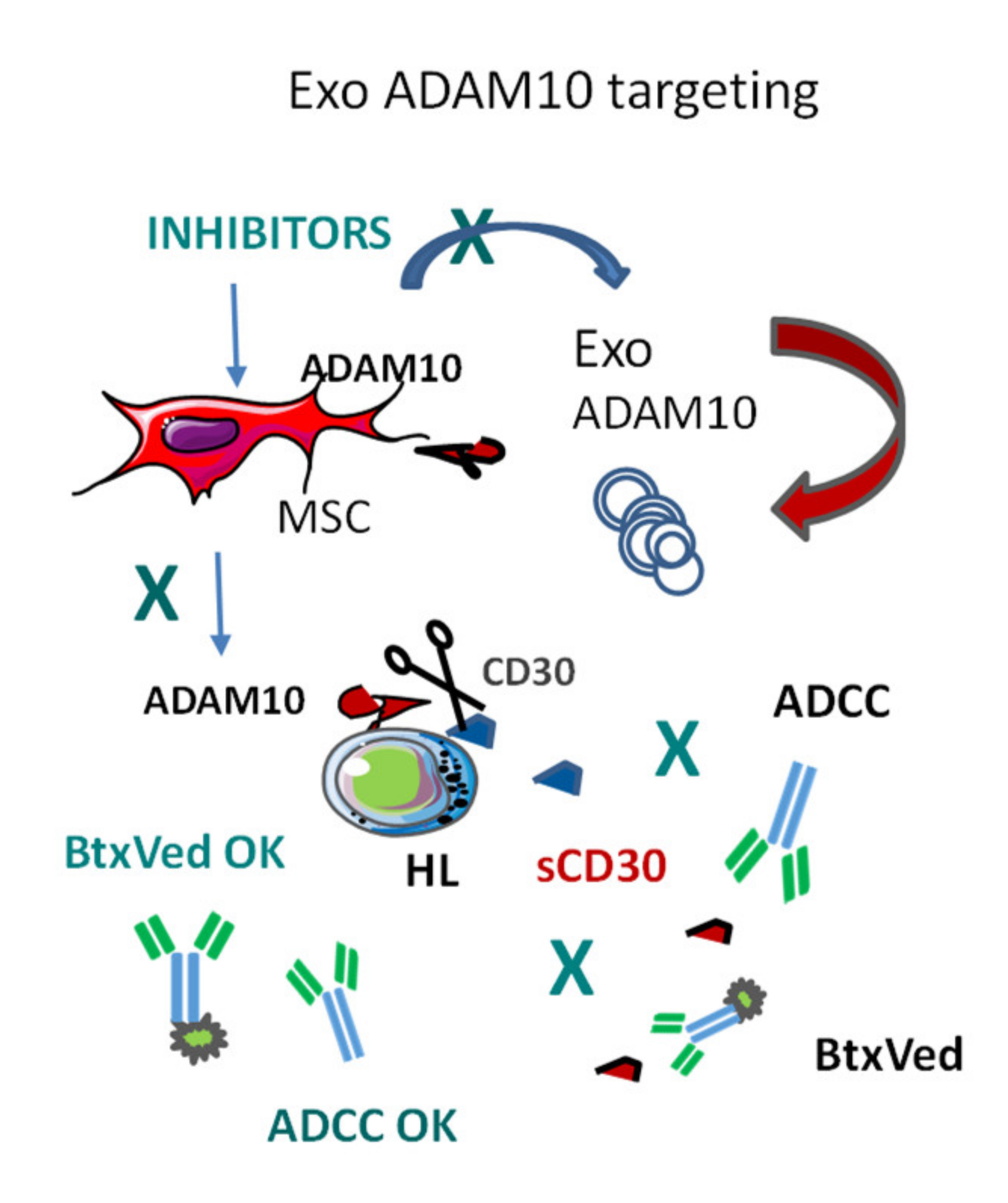
3.2. Exosomal ADAM10 and ADAM10 Inhibitors in Cancer
4. NP in Anti-Cancer Theranostic Strategies
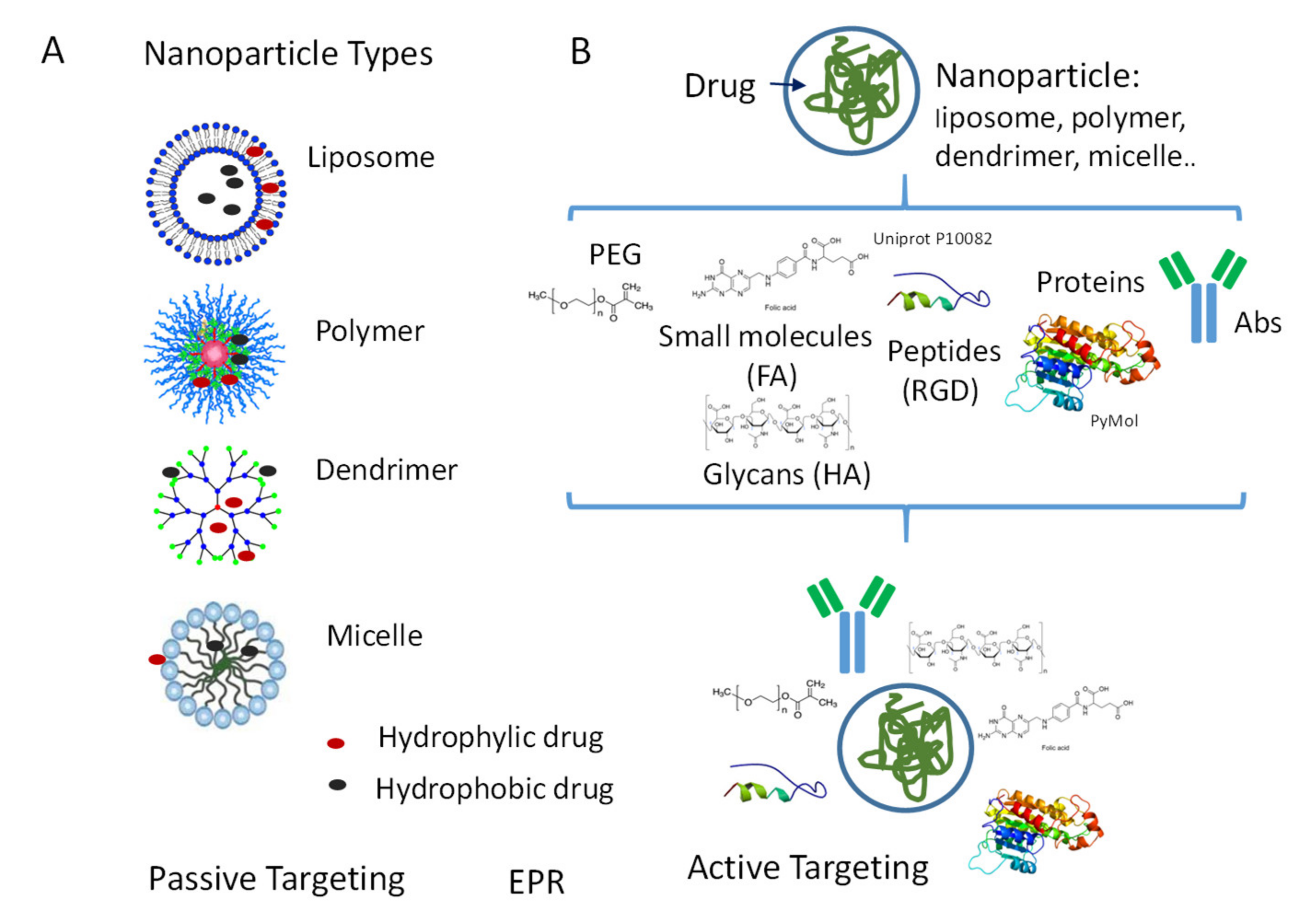
4.1. NP Types
4.2. NP Surface Modifications for Passive or Active Targeting
4.3. Nanoformulations of Drugs to Increase Anti-Tumor Immune Response
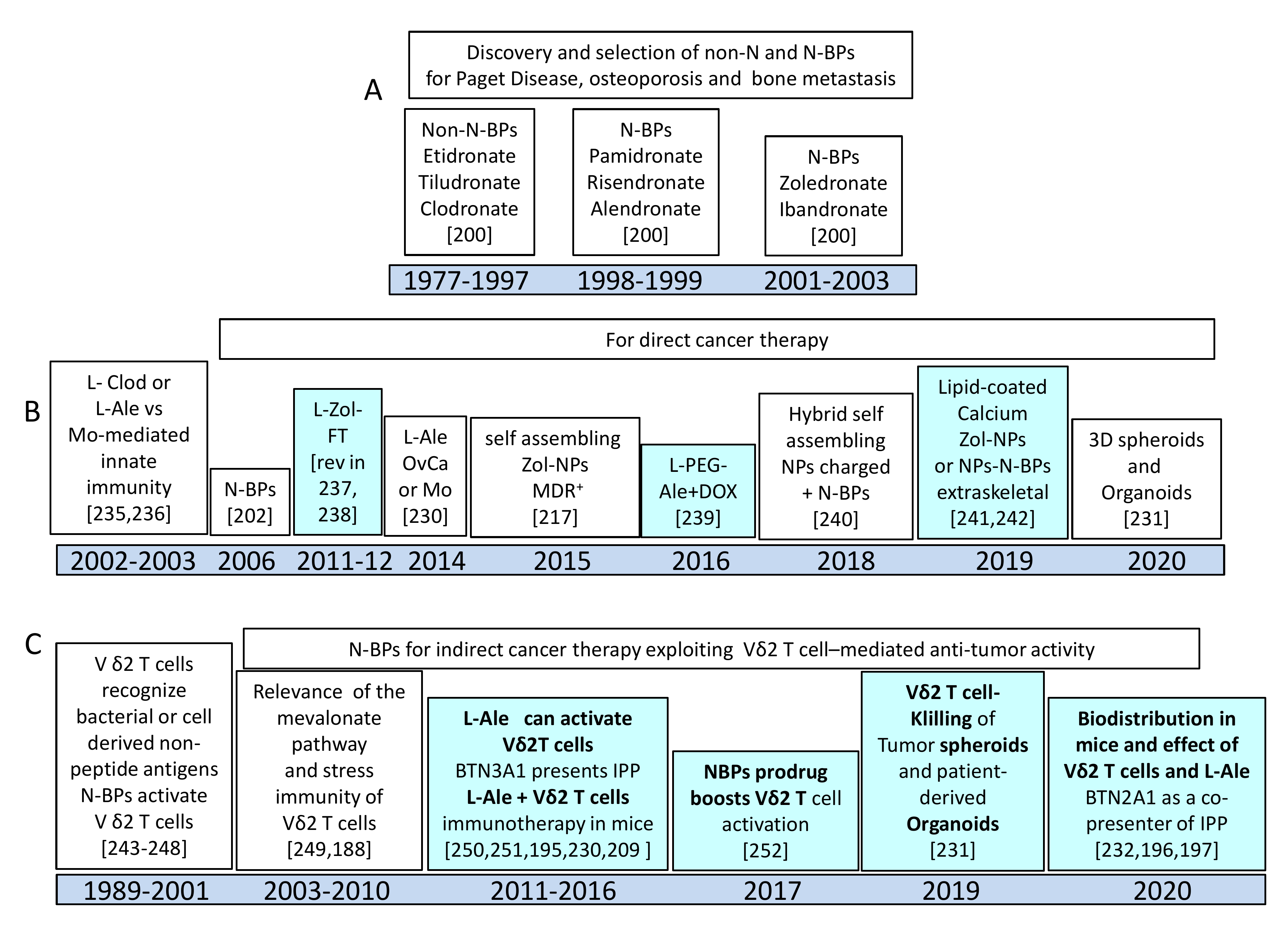
5. Nanoformulated Aminobisphosphonates (N-BPs) as Anti-Cancer Immunostimulators
5.1. N-BPs as Stimulators of Anti-Tumour γδT Cell Response
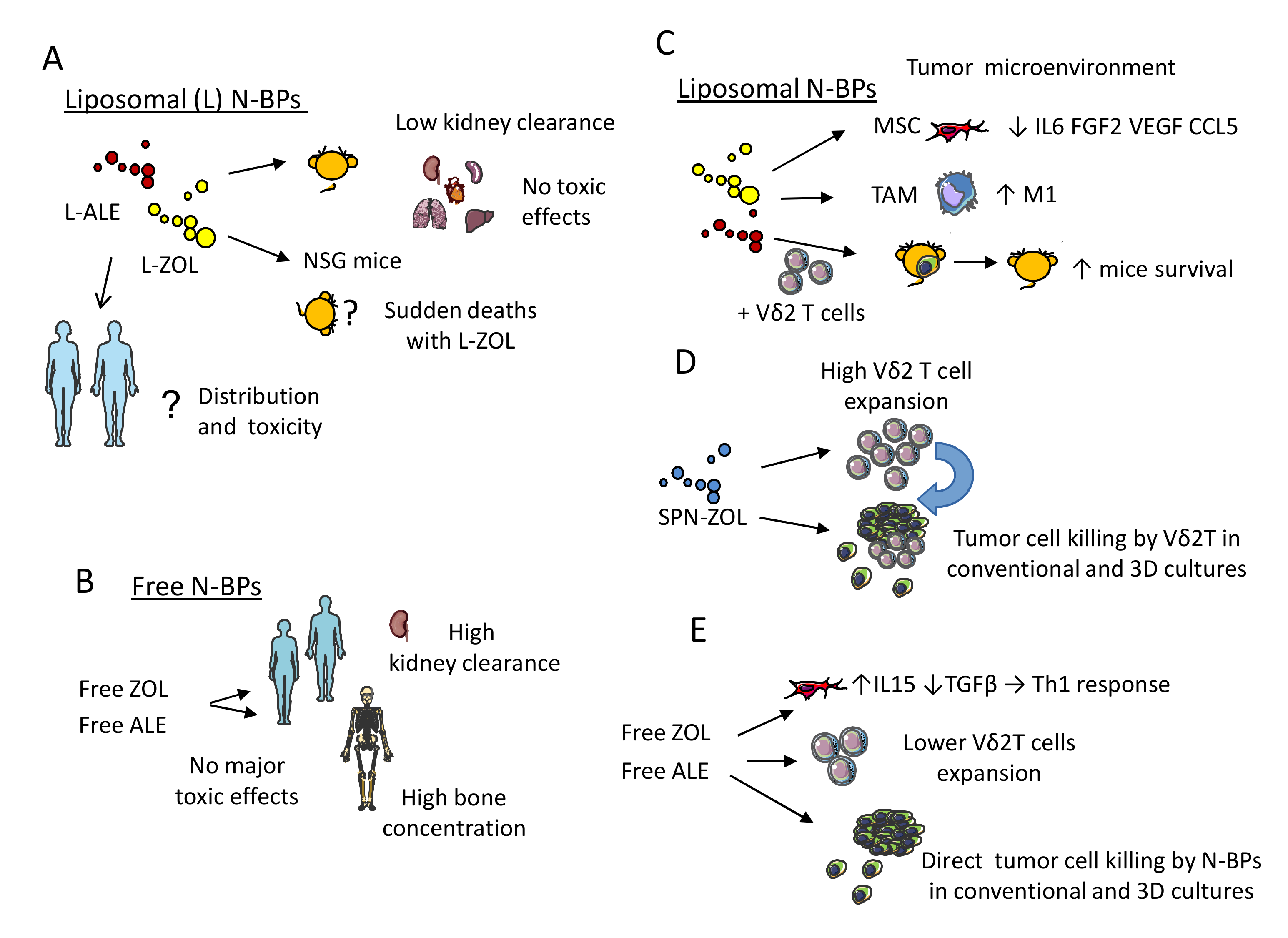
5.2. Nanoformulations of N-BPs: Pharmacokinetics and Toxic Effects in Murine Models
5.3. Functional Behavior of N-BPs Nanoformulations: Direct and Indirect Anti-Tumor Effect
6. Challenges for Clinical Implementation
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mukerjee, A.; Ranjan, A.P.; Vishwanatha, J.K. Combinatorial nanoparticles for cancer diagnosis and therapy. Curr. Med. Chem. 2012, 19, 3714–3721. [Google Scholar] [CrossRef]
- De Jong, W.H.; Borm, P.J.A. Drug delivery and nanoparticles: Applications and hazards. Int. J. Nanomedicine 2008, 3, 133–149. [Google Scholar] [CrossRef] [Green Version]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug. Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Zaimy, M.A.; Saffarzadeh, N.; Mohammadi, A.; Pourghadamyari, H.; Izadi, P.; Sarli, A.; Moghaddam, L.K.; Paschepari, S.R.; Azizi, H.; Torkamandi, S.; et al. New methods in the diagnosis of cancer and gene therapy of cancer based on nanoparticles. Cancer Gene Ther. 2017, 24, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Baetke, S.C.; Lammers, T.; Kiessling, F. Applications of nanoparticles for diagnosis and therapy of cancer. Br. J. Radiol. 2015, 88, 20150207. [Google Scholar] [CrossRef] [PubMed]
- Phillips, E.; Penate-Medina, O.; Zanzonico, P.B.; Carvajal, R.D.; Mohan, P.; Ye, Y.; Humm, J.; Gönen, M.; Kalaigian, H.; Schöder, H.; et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Sci. Transl. Med. 2014, 6, 260ra149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashir, M.R.; Bhatti, L.; Marin, D.; Nelson, R.C. Emerging applications for ferumoxytol as a contrast agent in MRI. J. Magn. Reson. Imaging 2015, 41, 884–898. [Google Scholar] [CrossRef] [PubMed]
- Kipp, J.E. The role of solid nanoparticle technology in the parenteral delivery of poorly water-soluble drugs. Int. J. Pharm. 2004, 284, 109–122. [Google Scholar] [CrossRef]
- LaVan, D.A.; McGuire, T.; Langer, R. Small-scale systems for in vivo drug delivery. Nat. Biotechnol. 2003, 21, 1184–1191. [Google Scholar] [CrossRef]
- Gibaud, S.; Demoy, M.; Andreux, J.P.; Weingarten, C.; Gouritin, B.; Couvreur, P. Cells involved in the capture of nanoparticles in hematopoietic organs. J. Pharm. Sci. 1996, 85, 944–950. [Google Scholar] [CrossRef]
- Demoy, M.; Gibaud, S.; Andreux, J.P.; Weingarten, C.; Gouritin, B.; Couvreur, P. Splenic trapping of nanoparticles: Complementary approaches for in situ studies. Pharm. Res. 1997, 14, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Simberg, D.; Skotland, T.; Yaghmur, A.; Hunter, A.C. The Interplay Between Blood Proteins, Complement, and Macrophages on Nanomedicine Performance and Responses. J. Pharmacol. Exp. Ther. 2019, 370, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Niidome, T.; Yamagata, M.; Okamoto, Y.; Akiyama, Y.; Takahashi, H.; Kawano, T.; Katayama, Y.; Niidome, Y. PEG-modified gold nanorods with a stealth character for in vivo applications. J. Control. Release 2006, 114, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Roy, A.; Dwarakanath, B.S. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front. Oncol. 2017, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.W.; Ferry, D.R.; Kerr, D.J.; Rea, D.; Whitlock, M.; Poyner, R.; Boivin, C.; Hesslewood, S.; Twelves, C.; Blackie, R.; et al. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int. J. Oncol. 2009, 34, 1629–1636. [Google Scholar] [CrossRef] [Green Version]
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [Green Version]
- De Wever, O.; Hendrix, A. A supporting ecosystem to mature extracellular vesicles into clinical application. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Sancho-Albero, M.; Navascués, N.; Mendoza, G.; Sebastián, V.; Arruebo, M.; Martín-Duque, P.; Santamaría, J. Exosome origin determines cell targeting and the transfer of therapeutic nanoparticles towards target cells. J. Nanobiotechnology 2019, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Quarto, R.; Tasso, R. Extracellular Vesicles as Natural, Safe and Efficient Drug Delivery Systems. Pharmaceutics 2019, 11, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M.; Qiao, Y.; Concepcion, W.; Thakor, A.S. Stem cell-derived extracellular vesicles: Role in oncogenic processes, bioengineering potential, and technical challenges. Stem Cell Res. Ther. 2019, 10, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Vakhshiteh, F.; Atyabi, F.; Ostad, S.N. Mesenchymal stem cell exosomes: A two-edged sword in cancer therapy. Int. J. Nanomedicine 2019, 14, 2847–2859. [Google Scholar] [CrossRef] [Green Version]
- Dinh, P.-U.C.; Paudel, D.; Brochu, H.; Popowski, K.D.; Gracieux, M.C.; Cores, J.; Huang, K.; Hensley, M.T.; Harrell, E.; Vandergriff, A.C.; et al. Inhalation of lung spheroid cell secretome and exosomes promotes lung repair in pulmonary fibrosis. Nat. Commun. 2020, 11, 1064. [Google Scholar] [CrossRef]
- Vulpis, E.; Soriani, A.; Cerboni, C.; Santoni, A.; Zingoni, A. Cancer Exosomes as Conveyors of Stress-Induced Molecules: New Players in the Modulation of NK Cell Response. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Federici, C.; Shahaj, E.; Cecchetti, S.; Camerini, S.; Casella, M.; Iessi, E.; Camisaschi, C.; Paolino, G.; Calvieri, S.; Ferro, S.; et al. Natural-Killer-Derived Extracellular Vesicles: Immune Sensors and Interactors. Front. Immunol. 2020, 11, 262. [Google Scholar] [CrossRef] [Green Version]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Park, G.B.; Kim, D. Insulin-like growth factor-1 activates different catalytic subunits p110 of PI3K in a cell-type-dependent manner to induce lipogenesis-dependent epithelial-mesenchymal transition through the regulation of ADAM10 and ADAM17. Mol. Cell. Biochem. 2018, 439, 199–211. [Google Scholar] [CrossRef]
- Alabi, R.O.; Farber, G.; Blobel, C.P. Intriguing Roles for Endothelial ADAM10/Notch Signaling in the Development of Organ-Specific Vascular Beds. Physiol. Rev. 2018, 98, 2025–2061. [Google Scholar] [CrossRef] [PubMed]
- Sépult, C.; Bellefroid, M.; Rocks, N.; Donati, K.; Gérard, C.; Gilles, C.; Ludwig, A.; Duysinx, B.; Noël, A.; Cataldo, D. ADAM10 mediates malignant pleural mesothelioma invasiveness. Oncogene 2019, 38, 3521–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal Stem Cells for Regenerative Medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-Y.; Tao, Y.-W.; Gao, S.; Li, P.; Zheng, J.-M.; Zhang, S.-E.; Liang, J.; Zhang, Y. Cancer-associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome-mediated paracrine miR-34a-5p. EbioMedicine 2018, 36, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- Nanou, A.; Miller, M.C.; Zeune, L.L.; de Wit, S.; Punt, C.J.A.; Groen, H.J.M.; Hayes, D.F.; de Bono, J.S.; Terstappen, L.W.M.M. Tumour-derived extracellular vesicles in blood of metastatic cancer patients associate with overall survival. Br. J. Cancer 2020, 122, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17. [Google Scholar] [CrossRef]
- Pascucci, L.; Coccè, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control Release 2014, 192, 262–270. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, Y.; Zhang, H.; Xu, P.; Liu, J.; Ma, J.; Lv, M.; Li, D.; Katirai, F.; Shen, G.-X.; et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat. Commun. 2012, 3, 1282. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Du, Y.; Jing, L.; Liang, X.; Li, Y.; Li, X.; Dai, Z.; Tian, J. Infra Red Dye and Endostar Loaded Poly Lactic Acid Nano Particles as a Novel Theranostic Nanomedicine for Breast Cancer. J. Biomed. Nanotechnol. 2016, 12, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Collino, F.; Deregibus, M.C.; Grange, C.; Tetta, C.; Camussi, G. Microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev. 2013, 22, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.-T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Invest. 2013, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.-C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Invest. 2016, 126, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Eitan, E.; Green, J.; Bodogai, M.; Mode, N.A.; Bæk, R.; Jørgensen, M.M.; Freeman, D.W.; Witwer, K.W.; Zonderman, A.B.; Biragyn, A.; et al. Age-Related Changes in Plasma Extracellular Vesicle Characteristics and Internalization by Leukocytes. Sci. Rep. 2017, 7, 1342. [Google Scholar] [CrossRef] [Green Version]
- Akbar, N.; Azzimato, V.; Choudhury, R.P.; Aouadi, M. Extracellular vesicles in metabolic disease. Diabetologia 2019, 62, 2179–2187. [Google Scholar] [CrossRef] [Green Version]
- Shaimardanova, A.A.; Solovyeva, V.V.; Chulpanova, D.S.; James, V.; Kitaeva, K.V.; Rizvanov, A.A. Extracellular vesicles in the diagnosis and treatment of central nervous system diseases. Neural Regen. Res. 2020, 15, 586–596. [Google Scholar] [CrossRef]
- Eichelser, C.; Stückrath, I.; Müller, V.; Milde-Langosch, K.; Wikman, H.; Pantel, K.; Schwarzenbach, H. Increased serum levels of circulating exosomal microRNA-373 in receptor-negative breast cancer patients. Oncotarget 2014, 5, 9650–9663. [Google Scholar] [CrossRef] [Green Version]
- Saari, H.; Lázaro-Ibáñez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control Release 2015, 220, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.-G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, A.J.; Mäger, I.; de Jong, O.G.; Varela, M.A.; Schiffelers, R.M.; El Andaloussi, S.; Wood, M.J.A.; Vader, P. Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol. Ther. 2017, 25, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomari, H.; Forouzandeh Moghadam, M.; Soleimani, M. Targeted cancer therapy using engineered exosome as a natural drug delivery vehicle. Onco. Targets Ther. 2018, 11, 5753–5762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalimuthu, S.; Gangadaran, P.; Rajendran, R.L.; Zhu, L.; Oh, J.M.; Lee, H.W.; Gopal, A.; Baek, S.H.; Jeong, S.Y.; Lee, S.-W.; et al. A New Approach for Loading Anticancer Drugs Into Mesenchymal Stem Cell-Derived Exosome Mimetics for Cancer Therapy. Front. Pharmacol. 2018, 9, 1116. [Google Scholar] [CrossRef] [PubMed]
- Kanchanapally, R.; Deshmukh, S.K.; Chavva, S.R.; Tyagi, N.; Srivastava, S.K.; Patel, G.K.; Singh, A.P.; Singh, S. Drug-loaded exosomal preparations from different cell types exhibit distinctive loading capability, yield, and antitumor efficacies: A comparative analysis. Int. J. Nanomedicine 2019, 14, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Kolluri, K.K.; Gowers, K.H.C.; Janes, S.M. TRAIL delivery by MSC-derived extracellular vesicles is an effective anticancer therapy. J. Extracell. Vesicles 2017, 6, 1265291. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Shimbo, K.; Miyaki, S.; Ishitobi, H.; Kato, Y.; Kubo, T.; Shimose, S.; Ochi, M. Exosome-formed synthetic microRNA-143 is transferred to osteosarcoma cells and inhibits their migration. Biochem. Biophys. Res. Commun. 2014, 445, 381–387. [Google Scholar] [CrossRef]
- Garofalo, M.; Saari, H.; Somersalo, P.; Crescenti, D.; Kuryk, L.; Aksela, L.; Capasso, C.; Madetoja, M.; Koskinen, K.; Oksanen, T.; et al. Antitumor effect of oncolytic virus and paclitaxel encapsulated in extracellular vesicles for lung cancer treatment. J. Control Release 2018, 283, 223–234. [Google Scholar] [CrossRef]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.P.; Kim, E.Y.; Badr, C.E.; Weissleder, R.; Mempel, T.R.; Tannous, B.A.; Breakefield, X.O. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat. Commun. 2015, 6, 7029. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Rocco, G.; Baldari, S.; Toietta, G. Towards Therapeutic Delivery of Extracellular Vesicles: Strategies for In Vivo Tracking and Biodistribution Analysis. Stem Cells Int. 2016, 2016, 5029619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida-Aoki, N.; Tominaga, N.; Kosaka, N.; Ochiya, T. Altered biodistribution of deglycosylated extracellular vesicles through enhanced cellular uptake. J. Extracell. Vesicles 2020, 9, 1713527. [Google Scholar] [CrossRef] [PubMed]
- Charoenviriyakul, C.; Takahashi, Y.; Morishita, M.; Nishikawa, M.; Takakura, Y. Role of Extracellular Vesicle Surface Proteins in the Pharmacokinetics of Extracellular Vesicles. Mol. Pharm. 2018, 15, 1073–1080. [Google Scholar] [CrossRef]
- Kooijmans, S.a.A.; Fliervoet, L.a.L.; van der Meel, R.; Fens, M.H.a.M.; Heijnen, H.F.G.; van Bergen En Henegouwen, P.M.P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomedicine 2018, 14, 195–204. [Google Scholar] [CrossRef]
- Liu, J.; Ye, Z.; Xiang, M.; Chang, B.; Cui, J.; Ji, T.; Zhao, L.; Li, Q.; Deng, Y.; Xu, L.; et al. Functional extracellular vesicles engineered with lipid-grafted hyaluronic acid effectively reverse cancer drug resistance. Biomaterials 2019, 223, 119475. [Google Scholar] [CrossRef]
- Garinchesa, P.; Sakamoto, J.; Welt, S.; Real, F.; Rettig, W.; Old, L. Organ-specific expression of the colon cancer antigen A33, a cell surface target for antibody-based therapy. Int. J. Oncol. 1996, 9, 465–471. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Gong, C.; Wang, Z.; Xia, Q.; Gu, F.; Hu, C.; Zhang, L.; Guo, H.; Gao, S. A33 antibody-functionalized exosomes for targeted delivery of doxorubicin against colorectal cancer. Nanomedicine 2018, 14, 1973–1985. [Google Scholar] [CrossRef]
- Shi, J.; Heegaard, C.W.; Rasmussen, J.T.; Gilbert, G.E. Lactadherin binds selectively to membranes containing phosphatidyl-L-serine and increased curvature. Biochim. Biophys. Acta 2004, 1667, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooijmans, S.A.A.; Gitz-Francois, J.J.J.M.; Schiffelers, R.M.; Vader, P. Recombinant phosphatidylserine-binding nanobodies for targeting of extracellular vesicles to tumor cells: A plug-and-play approach. Nanoscale 2018, 10, 2413–2426. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-H.; Forterre, A.V.; Zhao, J.; Frimannsson, D.O.; Delcayre, A.; Antes, T.J.; Efron, B.; Jeffrey, S.S.; Pegram, M.D.; Matin, A.C. Anti-HER2 scFv-Directed Extracellular Vesicle-Mediated mRNA-Based Gene Delivery Inhibits Growth of HER2-Positive Human Breast Tumor Xenografts by Prodrug Activation. Mol. Cancer Ther. 2018, 17, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, D.; Raimondo, S.; Calabrese, G.; Forte, S.; Cristaldi, M.; Patinella, A.; Memeo, L.; Manno, M.; Raccosta, S.; Diana, P.; et al. Interleukin 3- receptor targeted exosomes inhibit in vitro and in vivo Chronic Myelogenous Leukemia cell growth. Theranostics 2017, 7, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Pi, F.; Binzel, D.W.; Lee, T.J.; Li, Z.; Sun, M.; Rychahou, P.; Li, H.; Haque, F.; Wang, S.; Croce, C.M.; et al. Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat. Nanotechnol. 2018, 13, 82–89. [Google Scholar] [CrossRef]
- Février, B.; Raposo, G. Exosomes: Endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 2004, 16, 415–421. [Google Scholar] [CrossRef]
- Robbins, P.D.; Dorronsoro, A.; Booker, C.N. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J. Clin. Invest. 2016, 126, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Hansen, H.P.; Paes Leme, A.F.; Hallek, M. Role of ADAM10 as a CD30 Sheddase in Classical Hodgkin Lymphoma. Front. Immunol. 2020, 11, 398. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Borghese, C.; Casagrande, N. Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Tableros, V.; Gomez, Y.; Camussi, G.; Brizzi, M.F. Extracellular Vesicles: New Players in Lymphomas. Int. J. Mol. Sci. 2018, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Xie, F.; Wang, L.; Zhang, L.; Zhang, S.; Fang, M.; Zhou, F. The function and clinical application of extracellular vesicles in innate immune regulation. Cell. Mol. Immunol. 2020, 17, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Daassi, D.; Mahoney, K.M.; Freeman, G.J. The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 2020, 20, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, A.; Singh, S.; Ahmad, M.; Khanna, K.; Ahmad, T.; Agrawal, A.; Ghosh, B. Simvastatin mediates inhibition of exosome synthesis, localization and secretion via multicomponent interventions. Sci. Rep. 2019, 9, 16373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, W.; Hao, Y.; He, C.; Li, L.; Zhu, G. Exosome-orchestrated hypoxic tumor microenvironment. Mol. Cancer 2019, 18, 57. [Google Scholar] [CrossRef] [Green Version]
- Logozzi, M.; Spugnini, E.; Mizzoni, D.; Di Raimo, R.; Fais, S. Extracellular acidity and increased exosome release as key phenotypes of malignant tumors. Cancer Metastasis Rev. 2019, 38, 93–101. [Google Scholar] [CrossRef]
- Aung, T.; Chapuy, B.; Vogel, D.; Wenzel, D.; Oppermann, M.; Lahmann, M.; Weinhage, T.; Menck, K.; Hupfeld, T.; Koch, R.; et al. Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proc. Natl. Acad. Sci. USA 2011, 108, 15336–15341. [Google Scholar] [CrossRef] [Green Version]
- Tosetti, F.; Vene, R.; Camodeca, C.; Nuti, E.; Rossello, A.; D’Arrigo, C.; Galante, D.; Ferrari, N.; Poggi, A.; Zocchi, M.R. Specific ADAM10 inhibitors localize in exosome-like vesicles released by Hodgkin lymphoma and stromal cells and prevent sheddase activity carried to bystander cells. Oncoimmunology 2018, 7, e1421889. [Google Scholar] [CrossRef] [Green Version]
- Woan, K.V.; Miller, J.S. Harnessing Natural Killer Cell Antitumor Immunity: From the Bench to Bedside. Cancer Immunol. Res. 2019, 7, 1742–1747. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Petroni, G.; Kroemer, G. Immunogenicity of cell death driven by immune effectors. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingoni, A.; Vulpis, E.; Loconte, L.; Santoni, A. NKG2D Ligand Shedding in Response to Stress: Role of ADAM10. Front. Immunol. 2020, 11, 447. [Google Scholar] [CrossRef]
- Zocchi, M.R.; Camodeca, C.; Nuti, E.; Rossello, A.; Venè, R.; Tosetti, F.; Dapino, I.; Costa, D.; Musso, A.; Poggi, A. ADAM10 new selective inhibitors reduce NKG2D ligand release sensitizing Hodgkin lymphoma cells to NKG2D-mediated killing. OncoImmunology 2016, 5, e1123367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrecht, B.N.; Vanderkerken, M.; Hammad, H. The emerging role of ADAM metalloproteinases in immunity. Nat. Rev. Immunol. 2018, 18, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Noy, P.J.; Yang, J.; Reyat, J.S.; Matthews, A.L.; Charlton, A.E.; Furmston, J.; Rogers, D.A.; Rainger, G.E.; Tomlinson, M.G. TspanC8 Tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) Interact via Their Extracellular Regions: EVIDENCE FOR DISTINCT BINDING MECHANISMS FOR DIFFERENT TspanC8 PROTEINS. J. Biol. Chem. 2016, 291, 3145–3157. [Google Scholar] [CrossRef] [Green Version]
- Harada, Y.; Suzuki, T.; Fukushige, T.; Kizuka, Y.; Yagi, H.; Yamamoto, M.; Kondo, K.; Inoue, H.; Kato, K.; Taniguchi, N.; et al. Generation of the heterogeneity of extracellular vesicles by membrane organization and sorting machineries. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 681–691. [Google Scholar] [CrossRef]
- Ludwig, A.; Hundhausen, C.; Lambert, M.H.; Broadway, N.; Andrews, R.C.; Bickett, D.M.; Leesnitzer, M.A.; Becherer, J.D. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef]
- Witters, L.; Scherle, P.; Friedman, S.; Fridman, J.; Caulder, E.; Newton, R.; Lipton, A. Synergistic inhibition with a dual epidermal growth factor receptor/HER-2/neu tyrosine kinase inhibitor and a disintegrin and metalloprotease inhibitor. Cancer Res. 2008, 68, 7083–7089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camodeca, C.; Nuti, E.; Tosetti, F.; Poggi, A.; D’Arrigo, C.; Zocchi, M.R.; Rossello, A. Synthesis and in vitro Evaluation of ADAM10 and ADAM17 Highly Selective Bioimaging Probes. ChemMedChem 2018. [Google Scholar] [CrossRef]
- Ebsen, H.; Lettau, M.; Kabelitz, D.; Janssen, O. Subcellular localization and activation of ADAM proteases in the context of FasL shedding in T lymphocytes. Molecular Immunology 2015, 65, 416–428. [Google Scholar] [CrossRef]
- Mathews, J.A.; Gibb, D.R.; Chen, B.H.; Scherle, P.; Conrad, D.H. CD23 Sheddase A disintegrin and metalloproteinase 10 (ADAM10) is also required for CD23 sorting into B cell-derived exosomes. J. Biol. Chem. 2010, 285, 37531–37541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Tan, S.; Li, S.; Shen, Q.; Wang, K. Cancer drug delivery in the nano era: An overview and perspectives (Review). Oncol. Rep. 2017, 38, 611–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Cruz, M.; Delgado, Y.; Castillo, B.; Figueroa, C.M.; Molina, A.M.; Torres, A.; Milián, M.; Griebenow, K. Smart Targeting To Improve Cancer Therapeutics. Drug Des. Devel. Ther. 2019, 13, 3753–3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Xiong, M.; Gong, J.; Zhang, Y.; Bai, N.; Luo, Y.; Li, L.; Wei, Y.; Liu, Y.; Tan, X.; et al. Legumain protease-activated TAT-liposome cargo for targeting tumours and their microenvironment. Nat. Commun 2014, 5, 4280. [Google Scholar] [CrossRef] [PubMed]
- Janib, S.M.; Moses, A.S.; MacKay, J.A. Imaging and drug delivery using theranostic nanoparticles. Adv. Drug Deliv. Rev. 2010, 62, 1052–1063. [Google Scholar] [CrossRef] [Green Version]
- Grossman, J.H.; McNeil, S.E. Nanotechnology in Cancer Medicine. Physics Today 2012, 65, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; De, A.; Mozumdar, S. Synthesis of thermoresponsive polymers for drug delivery. Methods Mol. Biol. 2014, 1141, 77–101. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, N.; Cetin, M. Chitosan-poly (lactide-co-glycolide) (CS-PLGA) nanoparticles containing metformin HCl: Preparation and in vitro evaluation. Pak. J. Pharm. Sci 2014, 27, 1923–1929. [Google Scholar] [PubMed]
- Tajmir-Riahi, H.A.; Nafisi, S.; Sanyakamdhorn, S.; Agudelo, D.; Chanphai, P. Applications of chitosan nanoparticles in drug delivery. Methods Mol. Biol. 2014, 1141, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tang, H.; Wu, P. Aqueous Solutions of Poly(ethylene oxide)-Poly(N-isopropylacrylamide): Thermosensitive Behavior and Distinct Multiple Assembly Processes. Langmuir 2015, 31, 6497–6506. [Google Scholar] [CrossRef] [PubMed]
- Buhleier, E.; Wehner, W.; Voegtle, F. ′Cascade′- and ′non-skid-chain-like′ syntheses of molecular cavity topologies. Synthesis 1978, 2, 155–158. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. A New Class of Polymers: Starburst-Dendritic Macromolecules. Polym. J. 1985, 17, 117–132. [Google Scholar] [CrossRef] [Green Version]
- Gillies, E.R.; Fréchet, J.M.J. Dendrimers and dendritic polymers in drug delivery. Drug Discov. Today 2005, 10, 35–43. [Google Scholar] [CrossRef]
- Kesharwani, P.; Jain, K.; Jain, N.K. Dendrimer as nanocarrier for drug delivery. Prog. Polym. Sci. 2014, 39, 268–307. [Google Scholar] [CrossRef]
- Khopade, A.J.; Caruso, F.; Tripathi, P.; Nagaich, S.; Jain, N.K. Effect of dendrimer on entrapment and release of bioactive from liposomes. Int. J. Pharm. 2002, 232, 157–162. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Y.; Wang, M.; Xiao, J.; Cheng, Y. Fluorinated poly(propylenimine) dendrimers as gene vectors. Biomaterials 2014, 35, 5407–5413. [Google Scholar] [CrossRef]
- Rabanel, J.-M.; Faivre, J.; Tehrani, S.F.; Lalloz, A.; Hildgen, P.; Banquy, X. Effect of the Polymer Architecture on the Structural and Biophysical Properties of PEG-PLA Nanoparticles. ACS Appl. Mater Interfaces 2015, 7, 10374–10385. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.K.; Jung, M.; Sym, S.J.; Shin, D.B.; Kang, S.M.; Kyung, S.Y.; Park, J.-W.; Jeong, S.H.; Cho, E.K. A phase II trial of Cremorphor EL-free paclitaxel (Genexol-PM) and gemcitabine in patients with advanced non-small cell lung cancer. Cancer Chemother. Pharmacol. 2014, 74, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.C.; Mwakwari, S.C.; Oyelere, A.K. Gold nanoparticles: From nanomedicine to nanosensing. Nanotechnol. Sci. Appl. 2008, 1, 45–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namiki, Y.; Fuchigami, T.; Tada, N.; Kawamura, R.; Matsunuma, S.; Kitamoto, Y.; Nakagawa, M. Nanomedicine for cancer: Lipid-based nanostructures for drug delivery and monitoring. Acc. Chem. Res. 2011, 44, 1080–1093. [Google Scholar] [CrossRef]
- Libutti, S.K.; Paciotti, G.F.; Byrnes, A.A.; Alexander, H.R.; Gannon, W.E.; Walker, M.; Seidel, G.D.; Yuldasheva, N.; Tamarkin, L. Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin. Cancer Res. 2010, 16, 6139–6149. [Google Scholar] [CrossRef] [Green Version]
- Benezra, M.; Penate-Medina, O.; Zanzonico, P.B.; Schaer, D.; Ow, H.; Burns, A.; DeStanchina, E.; Longo, V.; Herz, E.; Iyer, S.; et al. Multimodal silica nanoparticles are effective cancer-targeted probes in a model of human melanoma. J. Clin. Invest. 2011, 121, 2768–2780. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.; Belleville, P.; Popall, M.; Nicole, L. Applications of advanced hybrid organic-inorganic nanomaterials: From laboratory to market. Chem. Soc. Rev. 2011, 40, 696–753. [Google Scholar] [CrossRef]
- Kim, K.Y. Nanotechnology platforms and physiological challenges for cancer therapeutics. Nanomedicine 2007, 3, 103–110. [Google Scholar] [CrossRef]
- Chan, J.M.; Valencia, P.M.; Zhang, L.; Langer, R.; Farokhzad, O.C. Polymeric nanoparticles for drug delivery. Methods Mol. Biol. 2010, 624, 163–175. [Google Scholar] [CrossRef]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Ipe, B.I.; Bawendi, M.G.; Frangioni, J.V. Renal Clearance of Nanoparticles. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Ilinskaya, A.N.; Dobrovolskaia, M.A. Nanoparticles and the blood coagulation system. Part II: Safety concerns. Nanomedicine (Lond.) 2013, 8, 969–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.G. Small-molecule delivery by nanoparticles for anticancer therapy. Trends Mol. Med. 2010, 16, 594–602. [Google Scholar] [CrossRef] [Green Version]
- Morachis, J.M.; Mahmoud, E.A.; Almutairi, A. Physical and Chemical Strategies for Therapeutic Delivery by Using Polymeric Nanoparticles. Pharmacol. Rev. 2012, 64, 505–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Cho, K.; Wang, X.; Nie, S.; Chen, Z.G.; Shin, D.M. Therapeutic nanoparticles for drug delivery in cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, R.; Madanmohan, S.; Kesavan, A.; Baskar, G.; Krishnamoorthy, Y.R.; Santosham, R.; Ponraju, D.; Rayala, S.K.; Venkatraman, G. Nanomedicine: Towards development of patient-friendly drug-delivery systems for oncological applications. Int. J. Nanomedicine 2012, 7, 1043–1060. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Tsukigawa, K.; Fang, J. A Retrospective 30 Years After Discovery of the Enhanced Permeability and Retention Effect of Solid Tumors: Next-Generation Chemotherapeutics and Photodynamic Therapy--Problems, Solutions, and Prospects. Microcirculation 2016, 23, 173–182. [Google Scholar] [CrossRef]
- Islam, W.; Fang, J.; Imamura, T.; Etrych, T.; Subr, V.; Ulbrich, K.; Maeda, H. Augmentation of the Enhanced Permeability and Retention Effect with Nitric Oxide-Generating Agents Improves the Therapeutic Effects of Nanomedicines. Mol. Cancer Ther. 2018, 17, 2643–2653. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.B.; Vercellotti, G.M.; White, J.G.; Fegan, A.; Wagner, C.R.; Bischof, J.C. Blood-nanoparticle interactions and in vivo biodistribution: Impact of surface PEG and ligand properties. Mol. Pharm. 2012, 9, 2146–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majtan, T.; Bublil, E.M.; Park, I.; Arning, E.; Bottiglieri, T.; Glavin, F.; Kraus, J.P. Pharmacokinetics and pharmacodynamics of PEGylated truncated human cystathionine beta-synthase for treatment of homocystinuria. Life Sci. 2018, 200, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Miele, E.; Spinelli, G.P.; Miele, E.; Tomao, F.; Tomao, S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int. J. Nanomedicine 2009, 4, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Mita, M.M.; Ramanathan, R.K.; Weiss, G.J.; Mita, A.C.; LoRusso, P.M.; Burris, H.A.; Hart, L.L.; Low, S.C.; Parsons, D.M.; et al. Phase I Study of PSMA-Targeted Docetaxel-Containing Nanoparticle BIND-014 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 3157–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar] [PubMed]
- Poiroux, G.; Barre, A.; van Damme, E.J.M.; Benoist, H.; Rougé, P. Plant Lectins Targeting O-Glycans at the Cell Surface as Tools for Cancer Diagnosis, Prognosis and Therapy. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Nobili, S.; Landini, I.; Mazzei, T.; Mini, E. Overcoming tumor multidrug resistance using drugs able to evade P-glycoprotein or to exploit its expression. Med. Res. Rev. 2012, 32, 1220–1262. [Google Scholar] [CrossRef]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef]
- Shimomura, O.; Oda, T.; Tateno, H.; Ozawa, Y.; Kimura, S.; Sakashita, S.; Noguchi, M.; Hirabayashi, J.; Asashima, M.; Ohkohchi, N. A Novel Therapeutic Strategy for Pancreatic Cancer: Targeting Cell Surface Glycan Using rBC2LC-N Lectin-Drug Conjugate (LDC). Mol. Cancer Ther. 2018, 17, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Fernández, M.; Javaid, F.; Chudasama, V. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem. Sci. 2018, 9, 790–810. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Hu, Y.; Putt, K.S.; Singhal, S.; Han, H.; Visscher, D.W.; Murphy, L.M.; Low, P.S. Assessment of folate receptor alpha and beta expression in selection of lung and pancreatic cancer patients for receptor targeted therapies. Oncotarget 2018, 9, 4485–4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Cruz, M.; Cruz-Montañez, A.; Figueroa, C.M.; González-Robles, T.; Davila, J.; Inyushin, M.; Loza-Rosas, S.A.; Molina, A.M.; Muñoz-Perez, L.; Kucheryavykh, L.Y.; et al. Combining Stimulus-Triggered Release and Active Targeting Strategies Improves Cytotoxicity of Cytochrome c Nanoparticles in Tumor Cells. Mol. Pharm. 2016, 13, 2844–2854. [Google Scholar] [CrossRef] [PubMed]
- Lokeshwar, V.B.; Mirza, S.; Jordan, A. Targeting Hyaluronic Acid Family for Cancer Chemoprevention and Therapy. Adv. Cancer Res. 2014, 123, 35–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karousou, E.; Misra, S.; Ghatak, S.; Dobra, K.; Götte, M.; Vigetti, D.; Passi, A.; Karamanos, N.K.; Skandalis, S.S. Roles and targeting of the HAS/hyaluronan/CD44 molecular system in cancer. Matrix Biol. 2017, 59, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Choi, H.; Choi, E.S.; Park, M.-H.; Ryu, J.-H. Hyaluronic Acid-Coated Nanomedicine for Targeted Cancer Therapy. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafino, A.; Zonfrillo, M.; Andreola, F.; Psaila, R.; Mercuri, L.; Moroni, N.; Renier, D.; Campisi, M.; Secchieri, C.; Pierimarchi, P. CD44-targeting for antitumor drug delivery: A new SN-38-hyaluronan bioconjugate for locoregional treatment of peritoneal carcinomatosis. Curr. Cancer Drug Targets 2011, 11, 572–585. [Google Scholar] [CrossRef]
- Zhang, D.; Jia, H.; Wang, Y.; Li, W.-M.; Hou, Y.-C.; Yin, S.-W.; Wang, T.D.; He, S.-X.; Lu, S.-Y. A CD44 specific peptide developed by phage display for targeting gastric cancer. Biotechnol. Lett. 2015, 37, 2311–2320. [Google Scholar] [CrossRef]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.J.; Schwaiger, M.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9. [Google Scholar] [CrossRef]
- Sadatmousavi, P.; Soltani, M.; Nazarian, R.; Jafari, M.; Chen, P. Self-assembling Peptides: Potential Role in Tumor Targeting. Curr.Pharm.Biotechnol. 2011, 12. [Google Scholar] [CrossRef]
- Wynne, J.; Wright, D.; Stock, W. Inotuzumab: From Preclinical Development to Success in B-cell Acute Lymphoblastic Leukemia. Blood advances 2019, 3. [Google Scholar] [CrossRef]
- Connors, J.; Jurczak, W.; Straus, D.; Ansell, S.; Kim, W.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin With Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl.J. Med. 2018, 378. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.; Scott, C. Antibody Conjugated Nanoparticles as a Novel Form of Antibody Drug Conjugate Chemotherapy. Drug Discover. Today. Technol. 2018, 30. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Tagliamonte, M.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Nanoparticles to Improve the Efficacy of Peptide-Based Cancer Vaccines. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodman, C.; Vundu, G.; George, A.; Wilson, C.M. Applications and strategies in nanodiagnosis and nanotherapy in lung cancer. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving cancer immunotherapy using nanomedicines: Progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef]
- Lee, K.; Jeong, D.; Na, K. Doxorubicin Loading Fucoidan Acetate Nanoparticles for Immune and Chemotherapy in Cancer Treatment. Carbohydr.Polym. 2013, 94. [Google Scholar] [CrossRef]
- Vanitha, S.; Chaubey, N.; Ghosh, S.; Sanpui, P. Recombinant Human Granulocyte Macrophage Colony Stimulating Factor (hGM-CSF): Possibility of Nanoparticle-Mediated Delivery in Cancer Immunotherapy. Bioengineered 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Qiao, S.; Dai, Y.; Xu, G.; Dai, B.; Lu, L.; Yu, X.; Luo, Q.; Zhang, Z. Molecular-Targeted Immunotherapeutic Strategy for Melanoma via Dual-Targeting Nanoparticles Delivering Small Interfering RNA to Tumor-Associated Macrophages. ACS Nano 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.; Levy, J.; Reed, R.; Petchpud, W.; Liu, L.; Wendler, C.; Hall, F. Targeting Metastatic Cancer from the Inside: A New Generation of Targeted Gene Delivery Vectors Enables Personalized Cancer Vaccination in Situ. Int.J. Oncol. 2008, 33. [Google Scholar] [CrossRef] [Green Version]
- Young, S.W.S.; Stenzel, M.; Yang, J.-L. Nanoparticle-siRNA: A potential cancer therapy? Crit. Rev. Oncol. Hematol. 2016, 98, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Hu, C.-M.J.; Luk, B.T.; Gao, W.; Copp, J.A.; Tai, Y.; O’Connor, D.E.; Zhang, L. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef]
- Neek, M.; Kim, T.I.; Wang, S.-W. Protein-based nanoparticles in cancer vaccine development. Nanomedicine 2019, 15, 164–174. [Google Scholar] [CrossRef]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, T.; Xu, J. Therapeutic Development of Immune Checkpoint Inhibitors. In Regulation of Cancer Immune Checkpoints; Xu, J., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2020; pp. 619–649. ISBN 9789811532665. [Google Scholar]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Boland, J.L.; Zhou, Q.; Martin, M.; Callahan, M.K.; Konner, J.; O’Cearbhaill, R.E.; Friedman, C.F.; Tew, W.; Makker, V.; Grisham, R.N.; et al. Early disease progression and treatment discontinuation in patients with advanced ovarian cancer receiving immune checkpoint blockade. Gynecol. Oncol. 2019, 152, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Gurung, S.; Khan, F.; Gunassekaran, G.R.; Yoo, J.D.; Poongkavithai Vadevoo, S.M.; Permpoon, U.; Kim, S.-H.; Kim, H.-J.; Kim, I.-S.; Han, H.; et al. Phage display-identified PD-L1-binding peptides reinvigorate T-cell activity and inhibit tumor progression. Biomaterials 2020, 247, 119984. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Church, S.E.; Galon, J. Tumor Microenvironment and Immunotherapy: The Whole Picture Is Better Than a Glimpse. Immunity 2015, 43, 631–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef]
- Oh, S.; Lee, J.-H.; Kwack, K.; Choi, S.-W. Natural Killer Cell Therapy: A New Treatment Paradigm for Solid Tumors. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. Gammadelta T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Musso, A.; Dapino, I.; Zocchi, M. Mechanisms of Tumor Escape from Immune System: Role of Mesenchymal Stromal Cells. Immunol. Lett. 2014, 159. [Google Scholar] [CrossRef]
- Hayday, A.C. γδ T Cell Update: Adaptate Orchestrators of Immune Surveillance. J. Immunol. 2019, 203, 311–320. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kobayashi, H.; Terasaki, T.; Toma, H.; Aruga, A.; Uchiyama, T.; Mizutani, K.; Mikami, B.; Morita, C.T.; Minato, N. Synthesis of pyrophosphate-containing compounds that stimulate Vgamma2Vdelta2 T cells: Application to cancer immunotherapy. Med. Chem. 2007, 3, 85–99. [Google Scholar] [CrossRef]
- Corvaisier, M.; Moreau-Aubry, A.; Diez, E.; Bennouna, J.; Mosnier, J.-F.; Scotet, E.; Bonneville, M.; Jotereau, F. V gamma 9V delta 2 T cell response to colon carcinoma cells. J. Immunol. 2005, 175, 5481–5488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, J.; Kabelitz, D. γδ T cells and epigenetic drugs: A useful merger in cancer immunotherapy? Oncoimmunology 2015, 4, e1006088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabelitz, D.; Wesch, D.; He, W. Perspectives of γδ T Cells in Tumor Immunology. Cancer Res. 2007, 67, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavassori, S.; Kumar, A.; Wan, G.S.; Ramanjaneyulu, G.S.; Cavallari, M.; El Daker, S.; Beddoe, T.; Theodossis, A.; Williams, N.K.; Gostick, E.; et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human γδ T cells. Nat. Immunol. 2013, 14, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, A.K.; et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nöhren, A.; Begley, C.R.; Berwick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vγ9Vδ2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 2020, 52, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Cole, D.K.; Sewell, A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015, 12, 656–668. [Google Scholar] [CrossRef]
- Santini, D.; Vespasiani Gentilucci, U.; Vincenzi, B.; Picardi, A.; Vasaturo, F.; La Cesa, A.; Onori, N.; Scarpa, S.; Tonini, G. The antineoplastic role of bisphosphonates: From basic research to clinical evidence. Ann. Oncol. 2003, 14, 1468–1476. [Google Scholar] [CrossRef]
- La-Beck, N.M.; Liu, X.; Shmeeda, H.; Shudde, C.; Gabizon, A.A. Repurposing amino-bisphosphonates by liposome formulation for a new role in cancer treatment. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Barrett, J.; Worth, E.; Bauss, F.; Epstein, S. Ibandronate: A clinical pharmacological and pharmacokinetic update. J. Clin. Pharmacol. 2004, 44, 951–965. [Google Scholar] [CrossRef]
- Caraglia, M.; Santini, D.; Marra, M.; Vincenzi, B.; Tonini, G.; Budillon, A. Emerging anti-cancer molecular mechanisms of aminobisphosphonates. Endocr. Relat. Cancer 2006, 13, 7–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, P.D. Management of severe osteoporosis. Expert. Opin. Pharmacother. 2016, 17, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Reid, I.R.; Green, J.R.; Lyles, K.W.; Reid, D.M.; Trechsel, U.; Hosking, D.J.; Black, D.M.; Cummings, S.R.; Russell, R.G.G.; Eriksen, E.F. Zoledronate. Bone 2020, 137, 115390. [Google Scholar] [CrossRef] [PubMed]
- von Moos, R.; Costa, L.; Gonzalez-Suarez, E.; Terpos, E.; Niepel, D.; Body, J.-J. Management of bone health in solid tumours: From bisphosphonates to a monoclonal antibody. Cancer Treat. Rev. 2019, 76, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzano, G.; Zappavigna, S.; Luce, A.; D’Onofrio, N.; Balestrieri, M.L.; Grimaldi, A.; Lusa, S.; Ingrosso, D.; Artuso, S.; Porru, M.; et al. Transferrin-Targeted Nanoparticles Containing Zoledronic Acid as a Potential Tool to Inhibit Glioblastoma Growth. J. Biomed. Nanotechnol. 2016, 12, 811–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porru, M.; Zappavigna, S.; Salzano, G.; Luce, A.; Stoppacciaro, A.; Balestrieri, M.L.; Artuso, S.; Lusa, S.; De Rosa, G.; Leonetti, C.; et al. Medical treatment of orthotopic glioblastoma with transferrin-conjugated nanoparticles encapsulating zoledronic acid. Oncotarget 2014, 5, 10446–10459. [Google Scholar] [CrossRef] [Green Version]
- Kolmas, J.; Pajor, K.; Pajchel, L.; Przekora, A.; Ginalska, G.; Oledzka, E.; Sobczak, M. Fabrication and physicochemical characterization of porous composite microgranules with selenium oxyanions and risedronate sodium for potential applications in bone tumors. Int. J. Nanomedicine. 2017, 12, 5633–5642. [Google Scholar] [CrossRef] [Green Version]
- Hodgins, N.O.; Al-Jamal, W.T.; Wang, J.T.-W.; Parente-Pereira, A.C.; Liu, M.; Maher, J.; Al-Jamal, K.T. In vitro potency, in vitro and in vivo efficacy of liposomal alendronate in combination with γδ T cell immunotherapy in mice. J. Control Release 2016, 241, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Hodgins, N.O.; Wang, J.T.-W.; Al-Jamal, K.T. Nano-technology based carriers for nitrogen-containing bisphosphonates delivery as sensitisers of γδ T cells for anticancer immunotherapy. Adv. Drug Deliv. Rev. 2017, 114, 143–160. [Google Scholar] [CrossRef] [Green Version]
- Shmeeda, H.; Amitay, Y.; Tzemach, D.; Gorin, J.; Gabizon, A. Liposome encapsulation of zoledronic acid results in major changes in tissue distribution and increase in toxicity. J. Control Release 2013, 167, 265–275. [Google Scholar] [CrossRef]
- Marra, M.; Salzano, G.; Leonetti, C.; Porru, M.; Franco, R.; Zappavigna, S.; Liguori, G.; Botti, G.; Chieffi, P.; Lamberti, M.; et al. New self-assembly nanoparticles and stealth liposomes for the delivery of zoledronic acid: A comparative study. Biotechnol. Adv. 2012, 30, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Borghese, C.; Casagrande, N.; Pivetta, E.; Colombatti, A.; Boccellino, M.; Amler, E.; Normanno, N.; Caraglia, M.; De Rosa, G.; Aldinucci, D. Self-assembling nanoparticles encapsulating zoledronic acid inhibit mesenchymal stromal cells differentiation, migration and secretion of proangiogenic factors and their interactions with prostate cancer cells. Oncotarget 2017, 8, 42926–42938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mascolo, D.; Varesano, S.; Benelli, R.; Mollica, H.; Salis, A.; Zocchi, M.R.; Decuzzi, P.; Poggi, A. Nanoformulated Zoledronic Acid Boosts the Vδ2 T Cell Immunotherapeutic Potential in Colorectal Cancer. Cancers (Basel) 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.; Di Mascolo, D.; Francardi, M.; Piccardi, F.; Bandiera, T.; Decuzzi, P. Spherical polymeric nanoconstructs for combined chemotherapeutic and anti-inflammatory therapies. Nanomedicine 2016, 12, 2139–2147. [Google Scholar] [CrossRef]
- Salzano, G.; Marra, M.; Porru, M.; Zappavigna, S.; Abbruzzese, A.; La Rotonda, M.I.; Leonetti, C.; Caraglia, M.; De Rosa, G. Self-assembly nanoparticles for the delivery of bisphosphonates into tumors. Int. J. Pharm. 2011, 403, 292–297. [Google Scholar] [CrossRef]
- Schiraldi, C.; Zappavigna, S.; D’ Agostino, A.; Porto, S.; Gaito, O.; Lusa, S.; Lamberti, M.; De Rosa, M.; De Rosa, G.; Caraglia, M. Nanoparticles for the delivery of zoledronic acid to prostate cancer cells: A comparative analysis through time lapse video-microscopy technique. Cancer Biol. Ther. 2014, 15, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P. Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab. Pharmacokinet. 2011, 26, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Benyettou, F.; Alhashimi, M.; O’Connor, M.; Pasricha, R.; Brandel, J.; Traboulsi, H.; Mazher, J.; Olsen, J.-C.; Trabolsi, A. Sequential Delivery of Doxorubicin and Zoledronic Acid to Breast Cancer Cells by CB[7]-Modified Iron Oxide Nanoparticles. ACS Appl. Mater Interfaces 2017, 9, 40006–40016. [Google Scholar] [CrossRef]
- Kopecka, J.; Porto, S.; Lusa, S.; Gazzano, E.; Salzano, G.; Pinzòn-Daza, M.L.; Giordano, A.; Desiderio, V.; Ghigo, D.; De Rosa, G.; et al. Zoledronic acid-encapsulating self-assembling nanoparticles and doxorubicin: A combinatorial approach to overcome simultaneously chemoresistance and immunoresistance in breast tumors. Oncotarget 2016, 7, 20753–20772. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, C.; Schyschka, L.; Kronbach, Z.; Wottge, A.; van Griensven, M.; Wildemann, B.; Vester, H. Signaling pathway STAT1 is strongly activated by IFN-β in the pathogenesis of osteoporosis. Eur. J. Med. Res. 2015, 20. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Xu, Z.; Ding, S.; Yi, G.; Wang, Q. Alendronate promotes osteoblast differentiation and bone formation in ovariectomy-induced osteoporosis through interferon-β/signal transducer and activator of transcription 1 pathway. Exp. Ther. Med. 2018, 15, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Cameron, D.; Dodwell, D.; Bell, R.; Wilson, C.; Rathbone, E.; Keane, M.; Gil, M.; Burkinshaw, R.; Grieve, R.; et al. Adjuvant zoledronic acid in patients with early breast cancer: Final efficacy analysis of the AZURE (BIG 01/04) randomised open-label phase 3 trial. Lancet Oncol. 2014, 15, 997–1006. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Woodward, J.K.; Lefley, D.V.; Evans, C.A.; Coleman, R.E.; Holen, I. Anticancer mechanisms of doxorubicin and zoledronic acid in breast cancer tumor growth in bone. Mol. Cancer Ther. 2009, 8, 2821–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabizon, A.; Horowitz, A.T.; Goren, D.; Tzemach, D.; Mandelbaum-Shavit, F.; Qazen, M.M.; Zalipsky, S. Targeting folate receptor with folate linked to extremities of poly(ethylene glycol)-grafted liposomes: In vitro studies. Bioconjug. Chem. 1999, 10, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Shmeeda, H.; Horowitz, A.T.; Zalipsky, S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv. Drug Deliv. Rev. 2004, 56, 1177–1192. [Google Scholar] [CrossRef]
- Parker, N.; Turk, M.J.; Westrick, E.; Lewis, J.D.; Low, P.S.; Leamon, C.P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 2005, 338, 284–293. [Google Scholar] [CrossRef]
- Shmeeda, H.; Amitay, Y.; Gorin, J.; Tzemach, D.; Mak, L.; Ogorka, J.; Kumar, S.; Zhang, J.A.; Gabizon, A. Delivery of zoledronic acid encapsulated in folate-targeted liposome results in potent in vitro cytotoxic activity on tumor cells. J. Control Release 2010, 146, 76–83. [Google Scholar] [CrossRef]
- Pang, Y.; Fu, Y.; Li, C.; Wu, Z.; Cao, W.; Hu, X.; Sun, X.; He, W.; Cao, X.; Ling, D.; et al. Metal-Organic Framework Nanoparticles for Ameliorating Breast Cancer-Associated Osteolysis. Nano Lett. 2020, 20, 829–840. [Google Scholar] [CrossRef]
- Musso, A.; Catellani, S.; Canevali, P.; Tavella, S.; Venè, R.; Boero, S.; Pierri, I.; Gobbi, M.; Kunkl, A.; Ravetti, J.-L.; et al. Aminobisphosphonates prevent the inhibitory effects exerted by lymph node stromal cells on anti-tumor Vδ 2 T lymphocytes in non-Hodgkin lymphomas. Haematologica 2014, 99, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Guo, X.; Wang, M.; Liu, K. Tumor Microenvironment-Induced Structure Changing Drug/Gene Delivery System for Overcoming Delivery-Associated Challenges. J. Control. Release Off. J. Control. Release Soc. 2020, 323. [Google Scholar] [CrossRef]
- Parente-Pereira, A.; Shmeeda, H.; Whilding, L.; Zambirinis, C.; Foster, J.; van der Stegen, S.; Beatson, R.; Zabinski, T.; Brewig, N.; Sosabowski, J.; et al. Adoptive Immunotherapy of Epithelial Ovarian Cancer With Vγ9Vδ2 T Cells, Potentiated by Liposomal Alendronic Acid. J. Immunol. 2014, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hodgins, N.; Wt, A.-J.; Maher, J.; Sosabowski, J.; Al-Jamal, K. Organ Biodistribution of Radiolabelled γδ T Cells Following Liposomal Alendronate Administration in Different Mouse Tumour Models. Nanotheranostics 2020, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Shi, J.-B.; Li, C. Small extracellular vesicle loading systems in cancer therapy: Current status and the way forward. Cytotherapy 2019, 21, 1122–1136. [Google Scholar] [CrossRef] [PubMed]
- van Rooijen, N.; van Kesteren-Hendrikx, E. Clodronate liposomes: Perspectives in research and therapeutics. J. Liposome Res. 2002, 12, 81–94. [Google Scholar] [CrossRef]
- Danenberg, H.D.; Golomb, G.; Groothuis, A.; Gao, J.; Epstein, H.; Swaminathan, R.V.; Seifert, P.; Edelman, E.R. Liposomal alendronate inhibits systemic innate immunity and reduces in-stent neointimal hyperplasia in rabbits. Circulation 2003, 108, 2798–2804. [Google Scholar] [CrossRef] [Green Version]
- Marra, M.; Salzano, G.; Leonetti, C.; Tassone, P.; Scarsella, M.; Zappavigna, S.; Calimeri, T.; Franco, R.; Liguori, G.; Cigliana, G.; et al. Nanotechnologies to use bisphosphonates as potent anticancer agents: The effects of zoledronic acid encapsulated into liposomes. Nanomedicine 2011, 7, 955–964. [Google Scholar] [CrossRef]
- Gou, J.; Zhang, K.; Tang, X. Self-assembling nanoparticles for the release of bisphosphonates in the treatment of human cancers [WO2012042024]. Expert Opin. Ther. Pat. 2012, 22, 1367–1375. [Google Scholar] [CrossRef]
- Shmeeda, H.; Amitay, Y.; Gorin, J.; Tzemach, D.; Mak, L.; Stern, S.T.; Barenholz, Y.; Gabizon, A. Coencapsulation of alendronate and doxorubicin in pegylated liposomes: A novel formulation for chemoimmunotherapy of cancer. J. Drug Target 2016, 24, 878–889. [Google Scholar] [CrossRef]
- Ristori, S.; Grillo, I.; Lusa, S.; Thamm, J.; Valentino, G.; Campani, V.; Caraglia, M.; Steiniger, F.; Luciani, P.; De Rosa, G. Structural Characterization of Self-Assembling Hybrid Nanoparticles for Bisphosphonate Delivery in Tumors. Mol. Pharm. 2018, 15, 1258–1265. [Google Scholar] [CrossRef]
- Li, X.; Valdes, S.A.; Alzhrani, R.F.; Hufnagel, S.; Hursting, S.D.; Cui, Z. Zoledronic Acid-containing Nanoparticles with Minimum Premature Release Show Enhanced Activity Against Extraskeletal Tumor. ACS Appl. Mater. Interfaces 2019, 11, 7311–7319. [Google Scholar] [CrossRef]
- Zang, X.; Zhang, X.; Hu, H.; Qiao, M.; Zhao, X.; Deng, Y.; Chen, D. Targeted Delivery of Zoledronate to Tumor-Associated Macrophages for Cancer Immunotherapy. Mol. Pharm. 2019, 16, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Porcelli, S.; Fabbi, M.; Lanier, L.L.; Picker, L.J.; Anderson, T.; Warnke, R.A.; Bhan, A.K.; Strominger, J.L.; Brenner, M.B. Human lymphocytes bearing T cell receptor gamma/delta are phenotypically diverse and evenly distributed throughout the lymphoid system. J. Exp. Med. 1989, 169, 1277–1294. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Morita, C.T.; Tanaka, Y.; Nieves, E.; Brenner, M.B.; Bloom, B.R. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature 1995, 375, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Poquet, Y.; Kroca, M.; Halary, F.; Stenmark, S.; Peyrat, M.A.; Bonneville, M.; Fournié, J.J.; Sjöstedt, A. Expansion of Vgamma9 Vdelta2 T cells is triggered by Francisella tularensis-derived phosphoantigens in tularemia but not after tularemia vaccination. Infect. Immun. 1998, 66, 2107–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzmann, V.; Bauer, E.; Wilhelm, M. Gamma/delta T-cell stimulation by pamidronate. N. Engl. J. Med. 1999, 340, 737–738. [Google Scholar] [CrossRef]
- Das, H.; Wang, L.; Kamath, A.; Bukowski, J.F. Vgamma2Vdelta2 T-cell receptor-mediated recognition of aminobisphosphonates. Blood 2001, 98, 1616–1618. [Google Scholar] [CrossRef]
- Espinosa, E.; Belmant, C.; Pont, F.; Luciani, B.; Poupot, R.; Romagné, F.; Brailly, H.; Bonneville, M.; Fournié, J.J. Chemical synthesis and biological activity of bromohydrin pyrophosphate, a potent stimulator of human gamma delta T cells. J. Biol. Chem. 2001, 276, 18337–18344. [Google Scholar] [CrossRef] [Green Version]
- Gober, H.-J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef]
- Agrati, C.; Marianecci, C.; Sennato, S.; Carafa, M.; Bordoni, V.; Cimini, E.; Tempestilli, M.; Pucillo, L.P.; Turchi, F.; Martini, F.; et al. Multicompartment vectors as novel drug delivery systems: Selective activation of Tγδ lymphocytes after zoledronic acid delivery. Nanomedicine 2011, 7, 153–161. [Google Scholar] [CrossRef]
- Gutman, D.; Epstein-Barash, H.; Tsuriel, M.; Golomb, G. Alendronate liposomes for antitumor therapy: Activation of γδ T cells and inhibition of tumor growth. Adv. Exp. Med. Biol. 2012, 733, 165–179. [Google Scholar] [CrossRef]
- Tanaka, Y.; Iwasaki, M.; Murata-Hirai, K.; Matsumoto, K.; Hayashi, K.; Okamura, H.; Sugie, T.; Minato, N.; Morita, C.T.; Toi, M. Anti-Tumor Activity and Immunotherapeutic Potential of a Bisphosphonate Prodrug. Sci. Rep. 2017, 7, 5987. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vesicle Type ± Cargo | Source | Size | Experimental Model/ Condition/Disease | Molecular/Cellular/ Tissue Target | Therapeutic Potential | Ref. |
|---|---|---|---|---|---|---|
| EV/Exo native/ autologous/ allogenic | Tissue/stem cells/MSC | 30 nm–1 µm 8–12 nm | Tissue injury/tissue repair cardioprotection/ neurodegeneration/ immune response | Injured tissues/stem cells/inflammatory cells/immune cells | Cardiac-lung-liver repair/regeneration/ immune regulation | [17,18,19,20,21,25,26,27,28,35] |
| EV native | Adipose tissue | 50 nm–1 µm | Type 2 diabetes | Vasculature/ immune cells | Metabolic/ immune regulation | [47] |
| Exo+curcumin | EL4 mouse lymphoma/RAW 264.7 murine macrophage cell line and others | 30–100 nm | In vivo LPS-induced septic shock mouse model | Activated myeloid cells/inflammatory cells | Improve anti-inflammatory activity of curcumin | [51] |
| Exo+paclitaxel | PC3/LNCaP prostate tumor cell lines | 50 nm–1µm | In vitro model of EV uptake in prostate tumors | PC3/LNCaP prostate tumor cell lines death | Improve paclitaxel cytotoxicity | [50] |
| AA-PEG-exo PTX | Macrophages | 30–100 nm | In vivo mouse model pulmonary metastases | Metastatic cell death | Improve therapeutic outcome | [67] |
| EV+cc-siRNAs | Neuro 2A cell line/mouse DCs | 50 nm–1 µm | Transfection of cc-siRNA loaded-EV in HEK293/ Neuro2A murine neuroblastoma/SH-SY5Y human neuroblastoma/ GM04281 Huntington’s disease fibroblasts | Human antigen R Tumor cell death | Specific delivery of anticancer drugs to neuroblastoma and glioma | [52] |
| Exo-Dox | MSC transduced with HER ligands | 30–100 nm | In vitro uptake by HER+ BT474/SKBR3 breast tumor cell line | Specific targeting of doxorubicin to HER+ breast tumor cells | Improve doxorubicin efficacy | [53] |
| A33Ab-US/Exo/Dox | LIM1215 cells | 30–100 nm | CRC cell lines/in vivo CRC mouse model | Tumor growth inhibition | Improve doxorubicin efficacy/better outcome/survival | [70] |
| NP Type | Size | Biodistribution | Benefits | Drawbacks | Ref. |
|---|---|---|---|---|---|
| Liposomes | 200 nm–1 µm | Variable, poorly predictable | Good biocompatibility and stability, reduced drug toxicity, reduced drug degradation | Variable and slow drug delivery, liposome degradation | [105,107] |
| Natural polymers (peptides, glycans) | 20–300 nm | Good distribution and organ targeting | Good biocompatibility, optimal active drug transport | Inflammatory response, degradation | [105,109,110] |
| Synthetic polymers (PLA, PLGA) | 50–200 nm | EPR effect, organ-specific targeting | Improved drug solubility, reduced toxicity, prolonged circulation time, thermosensitive delivery | Inflammatory response, immune reaction, degradation, cytotoxicity | [106,111,112,113,114] |
| Dendrimers | 50–100 nm | Optimal distribution and kidney elimination | Branches carrying the drug, controlled release, self-assembly | Immunological reaction, hematological toxicity, cytotoxicity | [105,109,120] |
| Micelles | 20–150 nm | Optimal distribution | Good biocompatibility and passive targeting, good drug solubilization and reduced degradation, self-assembly | Possible cytotoxicity | [111,112,121] |
| Inorganic NP (iron oxides, gold NP, carbon nanotubes) | 40–100 nm | Good distribution and tissue localization | Theranostic use, enhanced tumor imaging and radiotherapy | Metal toxicity, storage | [123,124,125,126,127] |
| N-BPs Form | Size nm | Cellular Biodistribution | In Vivo Biodistribution | In Vitro Functional Effects | In Vivo Functional Effects | Ref. |
|---|---|---|---|---|---|---|
| Free alendronic acid or free zoledronic acid | NA | Passive penetration and inhibition of FPPS, Generation of IPP and DMAPP | High plasma and renal excretion (min) and high bone concentration and release along time half-life (years) | Anti-tumor toxic effects triggering Vδ2 T cells | Potent anti-osteoporotic Effects Use for bone metastasis treatment | reviewed in [200] |
| high phase-transition phospholipids, cholesterol and pegylated liposomes for alendronic acid | 85–100 nm Neg charged | High cellular penetration | High Plasma half-life (18 h) Low renal excretion High extraskeletal tissue | Anti-tumor toxic effects triggering Vδ2 T cells | Murine models Anti-tumor effects Synergistic effects with Vδ2 T cell administration | [200,209,210,211] |
| high phase-transition phospholipids, cholesterol and pegylated liposomes for zoledronic acid | 85–100 nm Neg charged | High cellular penetration | High Plasma half-life (18 h) Low renal excretion High extraskeletal tissue | Anti-tumor toxic effects triggering Vδ2 T cells | Murine models anti-tumor effects and sudden death | [200,209,210,211] |
| ZOL-containing self-assembly PEGylated nanoparticles (NPs) or ZOL-encapsulating PEGylated liposomes DOTAP/chol/DSPE-PEG | 147–245 nm +1.8–17.5 mV ζ pot | Not performed specific studies | Not performed specific studies | Not performed specific studies | Potent anticancer effect in murine model of prostate adenocarcinoma | [212] |
| PEGylated ZOL- charged NPs with CaPZ NPs and DOTAP/chol/DSPEG2000 | 147 nm +18 mv ζ pot | Strong effects on mesenchymal stromal cells and tumor cells | Not performed specific studies | Not performed specific studies | Not performed specific studies | [213] |
| Spheric polymeric nanoparticles DPPC:DSPE-PEG7.5:2.5 molar ratiocharged with zol | 171 nm –43 mV ζpot | Penetration and generation of IPP in tumor cells and Mo, no toxicity on Mo | Not assessed as Zol charged NPs | Potent boost of Vδ2 T cell proliferation and cytolysis of 3D CRC spheroids and patient-derived organoids | Not assessed as Zol charged NPs Potent antitumor effects on orthotopic glioblastoma multiforme in murine models as docetaxel- and diclofenac-charged NPs | [214,215] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zocchi, M.R.; Tosetti, F.; Benelli, R.; Poggi, A. Cancer Nanomedicine Special Issue Review Anticancer Drug Delivery with Nanoparticles: Extracellular Vesicles or Synthetic Nanobeads as Therapeutic Tools for Conventional Treatment or Immunotherapy. Cancers 2020, 12, 1886. https://doi.org/10.3390/cancers12071886
Zocchi MR, Tosetti F, Benelli R, Poggi A. Cancer Nanomedicine Special Issue Review Anticancer Drug Delivery with Nanoparticles: Extracellular Vesicles or Synthetic Nanobeads as Therapeutic Tools for Conventional Treatment or Immunotherapy. Cancers. 2020; 12(7):1886. https://doi.org/10.3390/cancers12071886
Chicago/Turabian StyleZocchi, Maria Raffaella, Francesca Tosetti, Roberto Benelli, and Alessandro Poggi. 2020. "Cancer Nanomedicine Special Issue Review Anticancer Drug Delivery with Nanoparticles: Extracellular Vesicles or Synthetic Nanobeads as Therapeutic Tools for Conventional Treatment or Immunotherapy" Cancers 12, no. 7: 1886. https://doi.org/10.3390/cancers12071886
APA StyleZocchi, M. R., Tosetti, F., Benelli, R., & Poggi, A. (2020). Cancer Nanomedicine Special Issue Review Anticancer Drug Delivery with Nanoparticles: Extracellular Vesicles or Synthetic Nanobeads as Therapeutic Tools for Conventional Treatment or Immunotherapy. Cancers, 12(7), 1886. https://doi.org/10.3390/cancers12071886