Interplay between Cellular Metabolism and the DNA Damage Response in Cancer


Abstract
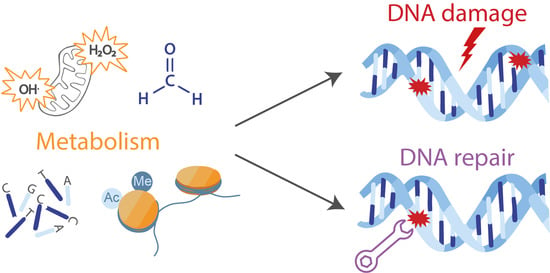
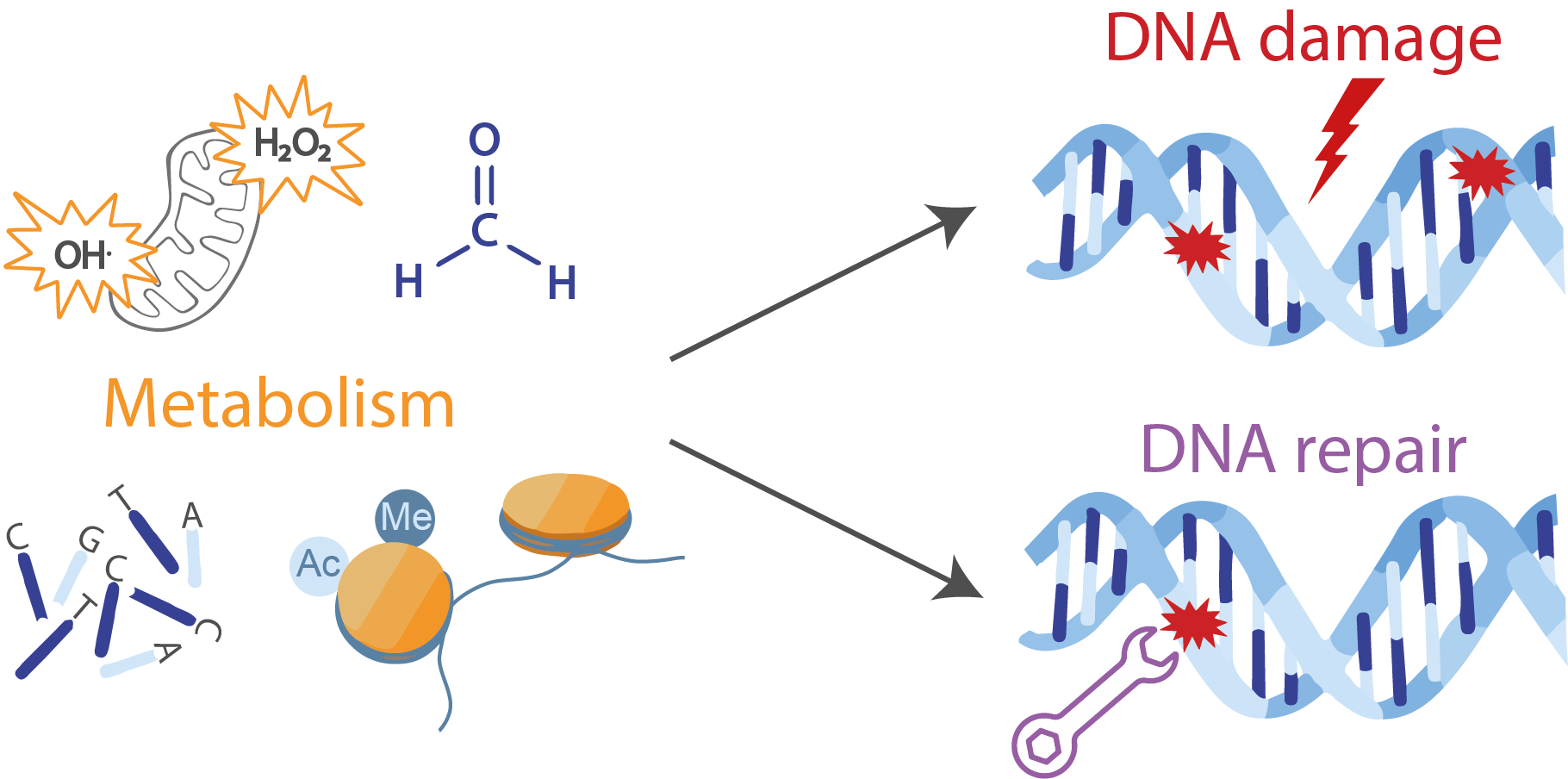
:
1. Introduction
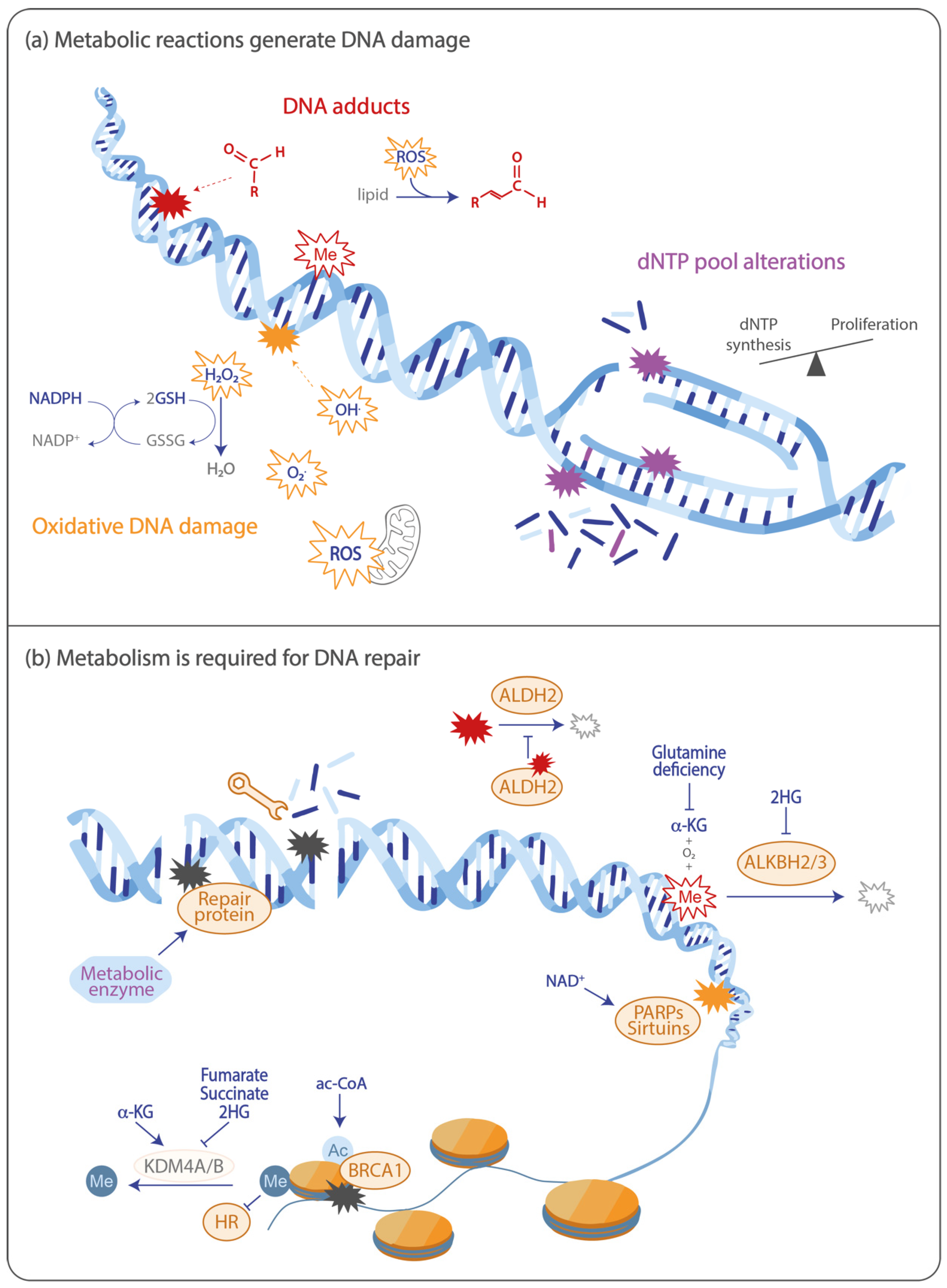
2. Oxidative Stress and the DNA Damage Response
2.1. Oxidative DNA Damage is Induced by Metabolic Reactions
2.2. Metabolism Functions in the Antioxidant Response
2.3. DNA Repair Following Oxidative Stress Depends on the Metabolite NAD+
3. DNA Adducts Are Produced through Metabolic Reactions
3.1. Aldehydes
3.2. Alkylating Agents
4. Alterations in dNTP Pools Generate DNA Damage
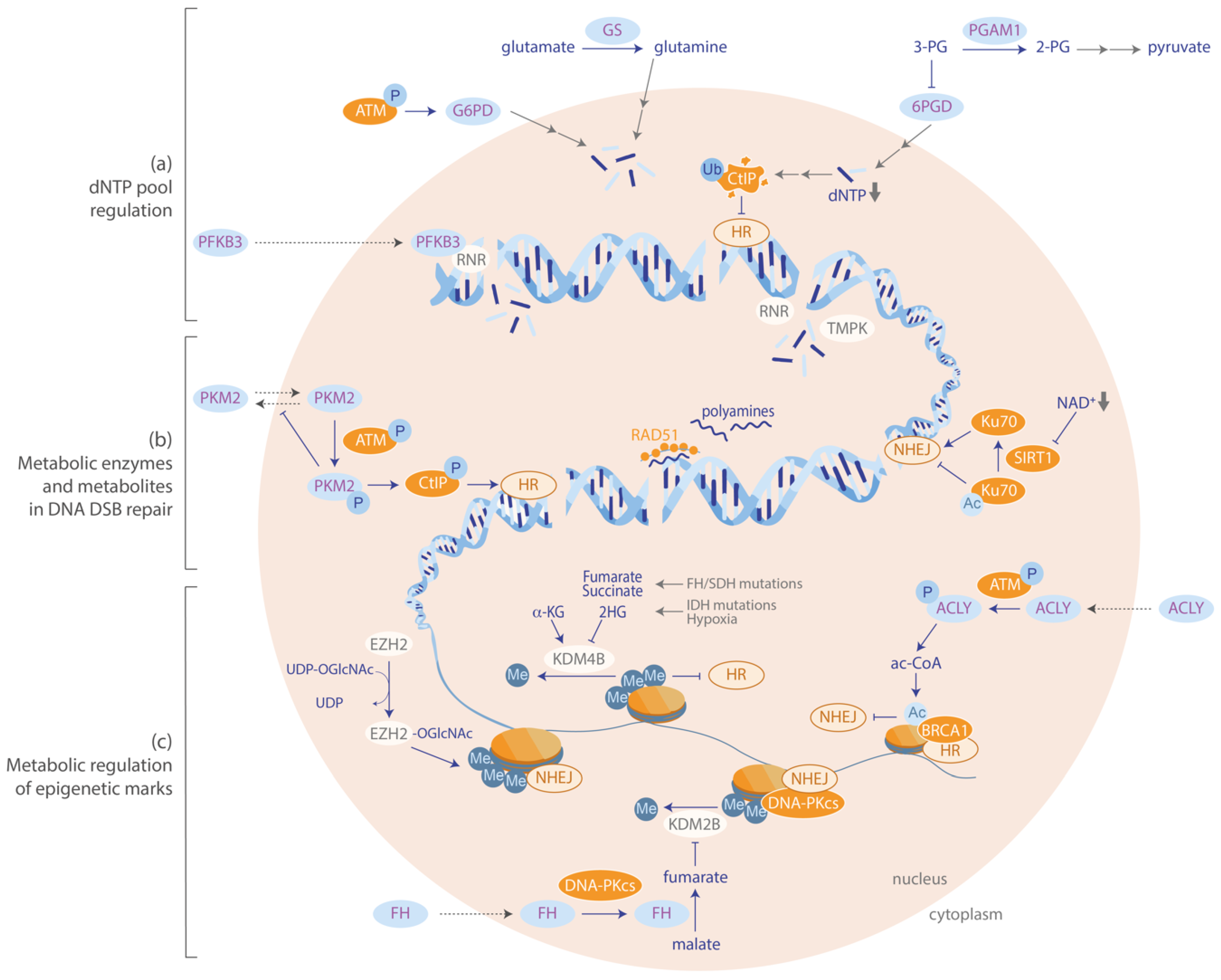
5. Metabolism is Involved in the Repair of DNA Double-Strand Breaks
5.1. Regulation of dNTP Pools is Critical for Efficient Repair of DSBs
5.2. Metabolic Regulation of Epigenetic Marks Influences DSB Repair
5.3. Metabolic Enzymes and Metabolites are Directly Involved in DSB Repair
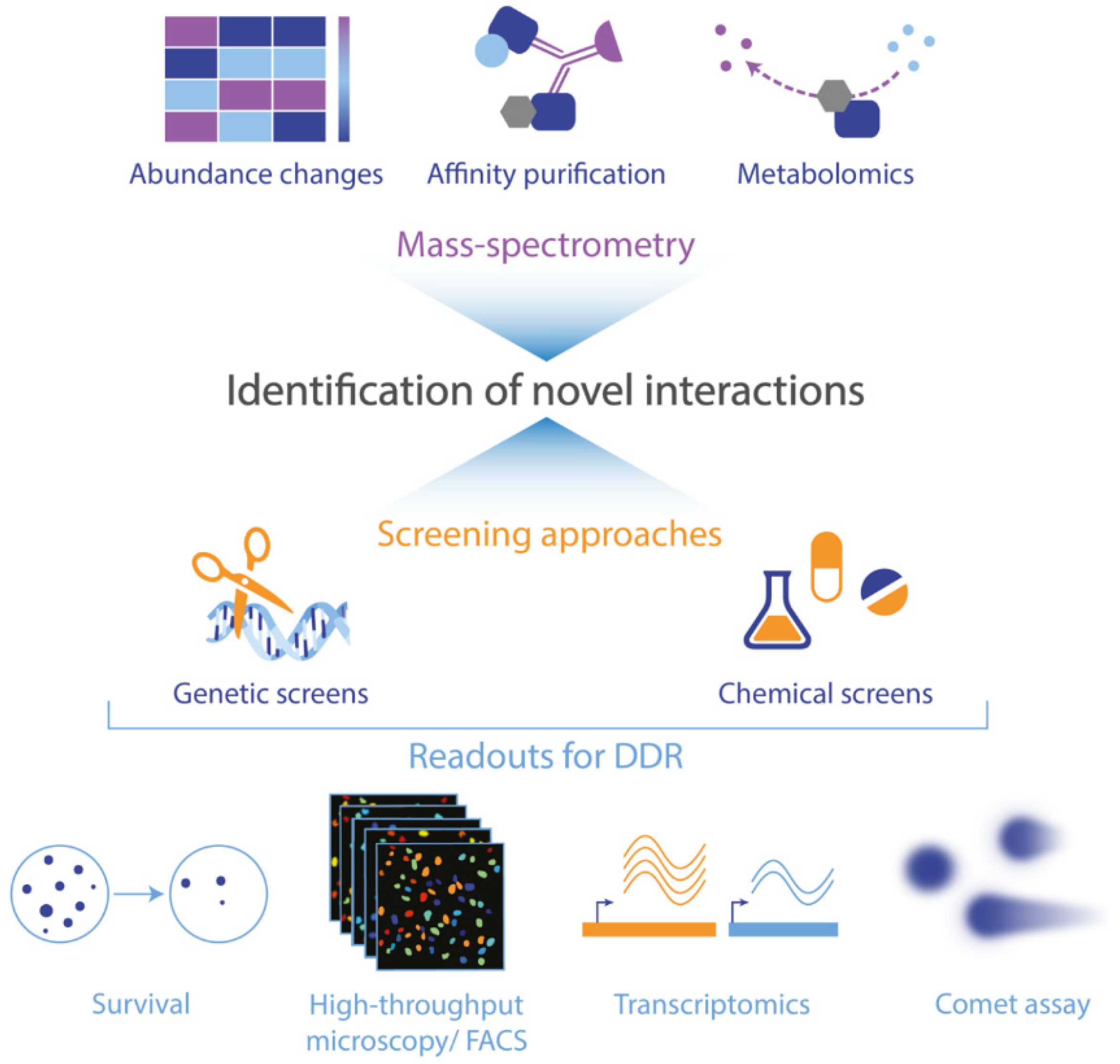
6. Outlook: Approaches to Identify Interactions between Metabolism and the DNA Damage Response
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Turgeon, M.O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Mosley, W.G.; Georgakila, S.; Ziech, D.; Panayiotidis, M.I. Viral-induced human carcinogenesis: An oxidative stress perspective. Mol. Biosyst. 2010, 6, 1162–1172. [Google Scholar] [CrossRef]
- Dizdaroglu, M. Oxidative damage to DNA in mammalian chromatin. Mutat. Res. 1992, 275, 331–342. [Google Scholar] [CrossRef]
- Georgakilas, A.G. Processing of DNA damage clusters in human cells: Current status of knowledge. Mol. Biosyst. 2008, 4, 30–35. [Google Scholar] [CrossRef]
- Cannan, W.J.; Tsang, B.P.; Wallace, S.S.; Pederson, D.S. Nucleosomes suppress the formation of double-strand DNA breaks during attempted base excision repair of clustered oxidative damages. J. Biol. Chem. 2014, 289, 19881–19893. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Adelman, R.; Saul, R.L.; Ames, B.N. Oxidative damage to DNA: Relation to species metabolic rate and life span. Proc. Natl. Acad. Sci. USA 1988, 85, 2706–2708. [Google Scholar] [CrossRef] [Green Version]
- Peto, R.; Roe, F.J.; Lee, P.N.; Levy, L.; Clack, J. Cancer and ageing in mice and men. Br. J. Cancer 1975, 32, 411–426. [Google Scholar] [CrossRef]
- Sulak, M.; Fong, L.; Mika, K.; Chigurupati, S.; Yon, L.; Mongan, N.P.; Emes, R.D.; Lynch, V.J. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. Elife 2016, 5. [Google Scholar] [CrossRef]
- Abegglen, L.M.; Caulin, A.F.; Chan, A.; Lee, K.; Robinson, R.; Campbell, M.S.; Kiso, W.K.; Schmitt, D.L.; Waddell, P.J.; Bhaskara, S.; et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA 2015, 314, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Maciak, S.; Michalak, P. Cell size and cancer: A new solution to Peto’s paradox? Evol. Appl. 2015, 8, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Francesco, A.; Di Germanio, C.; Bernier, M.; de Cabo, R. A time to fast. Science 2018, 362, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’yasova, D.; Ravussin, E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Rao, S.; Enot, D.P.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Jacquelot, N.; Yamazaki, T.; et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 2016, 30, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Maegawa, S.; Lu, Y.; Tahara, T.; Lee, J.T.; Madzo, J.; Liang, S.; Jelinek, J.; Colman, R.J.; Issa, J.J. Caloric restriction delays age-related methylation drift. Nat. Commun. 2017, 8, 539. [Google Scholar] [CrossRef]
- Algire, C.; Moiseeva, O.; Deschenes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev. Res. 2012, 5, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Ligibel, J.A.; Alfano, C.M.; Courneya, K.S.; Demark-Wahnefried, W.; Burger, R.A.; Chlebowski, R.T.; Fabian, C.J.; Gucalp, A.; Hershman, D.L.; Hudson, M.M.; et al. American Society of Clinical Oncology position statement on obesity and cancer. J. Clin. Oncol. 2014, 32, 3568–3574. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, M.; Nowicka, G. Obesity, DNA Damage, and Development of Obesity-Related Diseases. Int. J. Mol. Sci. 2019, 20, 1146. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Ikeda, T.; Enomoto, K.; Hosoda, K.; Nakashima, H.; Omae, K.; Watanabe, M.; Hibi, T.; Kitajima, M. Increased formation of oxidative DNA damage, 8-hydroxy-2’-deoxyguanosine, in human breast cancer tissue and its relationship to GSTP1 and COMT genotypes. Cancer Lett. 2000, 151, 87–95. [Google Scholar] [CrossRef]
- Roszkowski, K.; Jozwicki, W.; Blaszczyk, P.; Mucha-Malecka, A.; Siomek, A. Oxidative damage DNA: 8-oxoGua and 8-oxodG as molecular markers of cancer. Med. Sci. Monit. 2011, 17, CR329–CR333. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, R.; Brown, K.R.; Suarez, F.; Sayad, A.; Karamboulas, K.; Krzyzanowski, P.M.; Sircoulomb, F.; Medrano, M.; Fedyshyn, Y.; Koh, J.L.Y.; et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012, 2, 172–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Nobrega-Pereira, S.; Fernandez-Marcos, P.J.; Brioche, T.; Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Vina, J.; Serrano, M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016, 7, 10894. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubreuil, M.M.; Morgens, D.W.; Okumoto, K.; Honsho, M.; Contrepois, K.; Lee-McMullen, B.; Traber, G.M.; Sood, R.S.; Dixon, S.J.; Snyder, M.P.; et al. Systematic Identification of Regulators of Oxidative Stress Reveals Non-canonical Roles for Peroxisomal Import and the Pentose Phosphate Pathway. Cell Rep. 2020, 30, 1417–1433. [Google Scholar] [CrossRef] [PubMed]
- Heske, C.M. Beyond Energy Metabolism: Exploiting the Additional Roles of NAMPT for Cancer Therapy. Front. Oncol. 2019, 9, 1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 2019, 569, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.M.; Hwang, S.W.; Wang, T.; Park, C.W.; Ryu, Y.M.; Jung, J.H.; Shin, J.H.; Kim, S.Y.; Lee, J.L.; Kim, C.W.; et al. Increased nicotinamide adenine dinucleotide pool promotes colon cancer progression by suppressing reactive oxygen species level. Cancer Sci. 2019, 110, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, S.; Sasaki, T.; Minami, Y.; Ohsaki, Y. Nicotinamide phosphoribosyltransferase: A potent therapeutic target in non-small cell lung cancer with epidermal growth factor receptor-gene mutation. J. Thorac. Oncol. 2012, 7, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.M.; Park, C.W.; Kim, S.W.; Nam, Y.J.; Yu, J.H.; Shin, J.H.; Yun, C.H.; Im, S.H.; Kim, K.T.; Sung, Y.C.; et al. NAMPT suppresses glucose deprivation-induced oxidative stress by increasing NADPH levels in breast cancer. Oncogene 2016, 35, 3544–3554. [Google Scholar] [CrossRef]
- Cerna, D.; Li, H.; Flaherty, S.; Takebe, N.; Coleman, C.N.; Yoo, S.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J. Biol. Chem. 2012, 287, 22408–22417. [Google Scholar] [CrossRef] [Green Version]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent Advances in NAMPT Inhibitors: A Novel Immunotherapic Strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Kutzbach, C.; Stokstad, E.L. Feedback inhibition of methylene-tetrahydrofolate reductase in rat liver by S-adenosylmethionine. Biochim. Biophys. Acta 1967, 139, 217–220. [Google Scholar] [CrossRef]
- Pascale, R.M.; Peitta, G.; Simile, M.M.; Feo, F. Alterations of Methionine Metabolism as Potential Targets for the Prevention and Therapy of Hepatocellular Carcinoma. Medicina 2019, 55, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallaro, R.A.; Fuso, A.; Nicolia, V.; Scarpa, S. S-adenosylmethionine prevents oxidative stress and modulates glutathione metabolism in TgCRND8 mice fed a B-vitamin deficient diet. J. Alzheimers Dis. 2010, 20, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O. The molecular “Jekyll and Hyde” duality of PARP1 in cell death and cell survival. Front. Biosci. (Landmark Ed) 2009, 14, 72–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, M.; Hanashiro, K.; Kim, S.H.; Hanai, S.; Boulares, A.H.; Miwa, M.; Fukasawa, K. Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat. Cell Biol. 2007, 9, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Catalgol, B.; Wendt, B.; Grimm, S.; Breusing, N.; Ozer, N.K.; Grune, T. Chromatin repair after oxidative stress: Role of PARP-mediated proteasome activation. Free Radic. Biol. Med. 2010, 48, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Heske, C.M.; Davis, M.I.; Baumgart, J.T.; Wilson, K.; Gormally, M.V.; Chen, L.; Zhang, X.; Ceribelli, M.; Duveau, D.Y.; Guha, R.; et al. Matrix Screen Identifies Synergistic Combination of PARP Inhibitors and Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors in Ewing Sarcoma. Clin. Cancer Res. 2017, 23, 7301–7311. [Google Scholar] [CrossRef] [Green Version]
- Bajrami, I.; Kigozi, A.; Van Weverwijk, A.; Brough, R.; Frankum, J.; Lord, C.J.; Ashworth, A. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol. Med. 2012, 4, 1087–1096. [Google Scholar] [CrossRef]
- Bian, C.; Zhang, C.; Luo, T.; Vyas, A.; Chen, S.H.; Liu, C.; Kassab, M.A.; Yang, Y.; Kong, M.; Yu, X. NADP(+) is an endogenous PARP inhibitor in DNA damage response and tumor suppression. Nat. Commun. 2019, 10, 693. [Google Scholar] [CrossRef]
- Zhao, H.; Tang, W.; Chen, X.; Wang, S.; Wang, X.; Xu, H.; Li, L. The NAMPT/E2F2/SIRT1 axis promotes proliferation and inhibits p53-dependent apoptosis in human melanoma cells. Biochem. Biophys. Res. Commun. 2017, 493, 77–84. [Google Scholar] [CrossRef]
- Pan, J.H.; Zhou, H.; Zhu, S.B.; Huang, J.L.; Zhao, X.X.; Ding, H.; Qin, L.; Pan, Y.L. Nicotinamide phosphoribosyl transferase regulates cell growth via the Sirt1/P53 signaling pathway and is a prognosis marker in colorectal cancer. J. Cell. Physiol. 2019, 234, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hasan, M.K.; Alvarado, E.; Yuan, H.; Wu, H.; Chen, W.Y. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 2011, 30, 907–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.; Lee, S.; Kim, Y.N.; Lee, I.H. Deacetylation of CHK2 by SIRT1 protects cells from oxidative stress-dependent DNA damage response. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Asp. Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Agency for Research on Cancer (IARC Working Group). Review of Human Carcinogens: Volumes A-F.; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Bouvard, V.; Loomis, D.; Guyton, K.Z.; Grosse, Y.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K.; International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef] [Green Version]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, J.; Mutlu, E.; Sharma, V.; Collins, L.; Bodnar, W.; Yu, R.; Lai, Y.; Moeller, B.; Lu, K.; Swenberg, J. The endogenous exposome. DNA Repair 2014, 19, 3–13. [Google Scholar] [CrossRef]
- Tudek, B.; Zdzalik-Bielecka, D.; Tudek, A.; Kosicki, K.; Fabisiewicz, A.; Speina, E. Lipid peroxidation in face of DNA damage, DNA repair and other cellular processes. Free Radic. Biol. Med. 2017, 107, 77–89. [Google Scholar] [CrossRef]
- Beland, F. Rapid isolation, hydrolysis and chromatography of formaldehyde-modified DNA. J. Chromatogr. 1998, 308, 121–131. [Google Scholar] [CrossRef]
- Quievryn, G.; Zhitkovich, A. Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis 2000, 21, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Chaw, Y.F.; Crane, L.E.; Lange, P.; Shapiro, R. Isolation and identification of cross-links from formaldehyde-treated nucleic acids. Biochemistry 1980, 19, 5525–5531. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boor, P.J.; Trent, M.B.; Lyles, G.A.; Tao, M.; Ansari, G.A. Methylamine metabolism to formaldehyde by vascular semicarbazide-sensitive amine oxidase. Toxicology 1992, 73, 251–258. [Google Scholar] [CrossRef]
- Hazen, S.L.; Hsu, F.F.; d’Avignon, A.; Heinecke, J.W. Human neutrophils employ myeloperoxidase to convert alpha-amino acids to a battery of reactive aldehydes: A pathway for aldehyde generation at sites of inflammation. Biochemistry 1998, 37, 6864–6873. [Google Scholar] [CrossRef] [PubMed]
- Zhai, R.; Zheng, N.; Rizak, J.; Hu, X. Evidence for Conversion of Methanol to Formaldehyde in Nonhuman Primate Brain. Anal. Cell. Pathol. 2016, 2016, 4598454. [Google Scholar] [CrossRef] [Green Version]
- Speit, G.; Merk, O. Evaluation of mutagenic effects of formaldehyde in vitro: Detection of crosslinks and mutations in mouse lymphoma cells. Mutagenesis 2002, 17, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Hemminki, K.; Suni, R. Sites of reaction of glutaraldehyde and acetaldehyde with nucleosides. Arch. Toxicol. 1984, 55, 186–190. [Google Scholar] [CrossRef]
- Wang, M.; McIntee, E.J.; Cheng, G.; Shi, Y.; Villalta, P.W.; Hecht, S.S. Identification of DNA adducts of acetaldehyde. Chem. Res. Toxicol. 2000, 13, 1149–1157. [Google Scholar] [CrossRef]
- Lorenti Garcia, C.; Mechilli, M.; Proietti De Santis, L.; Schinoppi, A.; Kobos, K.; Palitti, F. Relationship between DNA lesions, DNA repair and chromosomal damage induced by acetaldehyde. Mutat. Res. 2009, 662, 3–9. [Google Scholar] [CrossRef]
- Sonohara, Y.; Yamamoto, J.; Tohashi, K.; Takatsuka, R.; Matsuda, T.; Iwai, S.; Kuraoka, I. Acetaldehyde forms covalent GG intrastrand crosslinks in DNA. Sci. Rep. 2019, 9, 660. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Gomi, T.; Fujioka, M. Serine hydroxymethyltransferase and threonine aldolase: Are they identical? Int. J. Biochem. Cell Biol. 2000, 32, 289–301. [Google Scholar] [CrossRef]
- Vaca, C.E.; Fang, J.L.; Conradi, M.; Hou, S.M. Development of a 32P-postlabelling method for the analysis of 2’-deoxyguanosine-3’-monophosphate and DNA adducts of methylglyoxal. Carcinogenesis 1994, 15, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Frischmann, M.; Bidmon, C.; Angerer, J.; Pischetsrieder, M. Identification of DNA adducts of methylglyoxal. Chem. Res. Toxicol. 2005, 18, 1586–1592. [Google Scholar] [CrossRef]
- Zheng, Q.; Omans, N.D.; Leicher, R.; Osunsade, A.; Agustinus, A.S.; Finkin-Groner, E.; D’Ambrosio, H.; Liu, B.; Chandarlapaty, S.; Liu, S.; et al. Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat. Commun. 2019, 10, 1289. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Shahabuddin; Hadi, S.M. Formation of strand breaks and interstrand cross-links in DNA by methylglyoxal. J. Biochem. Toxicol. 1990, 5, 161–166. [Google Scholar] [CrossRef]
- Murata-Kamiya, N.; Kamiya, H. Methylglyoxal, an endogenous aldehyde, crosslinks DNA polymerase and the substrate DNA. Nucleic Acids Res. 2001, 29, 3433–3438. [Google Scholar] [CrossRef] [Green Version]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Lyles, G.A.; Chalmers, J. The metabolism of aminoacetone to methylglyoxal by semicarbazide-sensitive amine oxidase in human umbilical artery. Biochem. Pharmacol. 1992, 43, 1409–1414. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, K.; Saito, S.; Sano, M.; Matsuura, T.; Hagiwara, M.; Tomita, I. Formation of methylglyoxal in reduced nicotinamide adenine dinucleotide phosphate-dependent lipid peroxidation of rat liver microsomes. Jpn. J. Toxicol. Environ. Health 1996, 42, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.K.; O’Hara, S.M.; Valladier, P.; Stone, K.; Mols, O.; Marnett, L.J. Identification of adducts formed by reaction of guanine nucleosides with malondialdehyde and structurally related aldehydes. Chem. Res. Toxicol. 1988, 1, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.K.; Reddy, G.R.; Blair, I.A.; Marnett, L.J. Characterization of an N6-oxopropenyl-2’-deoxyadenosine adduct in malondialdehyde-modified DNA using liquid chromatography/electrospray ionization tandem mass spectrometry. Carcinogenesis 1996, 17, 1167–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, K.; Uzieblo, A.; Marnett, L.J. Studies of the reaction of malondialdehyde with cytosine nucleosides. Chem. Res. Toxicol. 1990, 3, 467–472. [Google Scholar] [CrossRef]
- Nair, V.; Cooper, C.S.; Vietti, D.E.; Turner, G.A. The chemistry of lipid peroxidation metabolites: Crosslinking reactions of malondialdehyde. Lipids 1986, 21, 6–10. [Google Scholar] [CrossRef]
- Wang, M.; Dhingra, K.; Hittelman, W.N.; Liehr, J.G.; de Andrade, M.; Li, D. Lipid peroxidation-induced putative malondialdehyde-DNA adducts in human breast tissues. Cancer Epidemiol. Biomark. Prev. 1996, 5, 705–710. [Google Scholar]
- Pryor, W.A.; Stanley, J.P. Letter: A suggested mechanism for the production of malonaldehyde during the autoxidation of polyunsaturated fatty acids. Nonenzymatic production of prostaglandin endoperoxides during autoxidation. J. Org. Chem. 1975, 40, 3615–3617. [Google Scholar] [CrossRef]
- Diczfalusy, U.; Falardeau, P.; Hammarstrom, S. Conversion of prostaglandin endoperoxides to C17-hydroxy acids catalyzed by human platelet thromboxane synthase. FEBS Lett. 1977, 84, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Hashim, M.F.; Riggins, J.N.; Schnetz-Boutaud, N.; Voehler, M.; Stone, M.P.; Marnett, L.J. In vitro bypass of malondialdehyde-deoxyguanosine adducts: Differential base selection during extension by the Klenow fragment of DNA polymerase I is the critical determinant of replication outcome. Biochemistry 2004, 43, 11828–11835. [Google Scholar] [CrossRef]
- Winter, C.K.; Segall, H.J.; Haddon, W.F. Formation of cyclic adducts of deoxyguanosine with the aldehydes trans-4-hydroxy-2-hexenal and trans-4-hydroxy-2-nonenal in vitro. Cancer Res. 1986, 46, 5682–5686. [Google Scholar] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Benedetti, A.; Comporti, M.; Esterbauer, H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim. Biophys. Acta 1980, 620, 281–296. [Google Scholar] [CrossRef]
- Sodum, R.S.; Chung, F.L. Structural characterization of adducts formed in the reaction of 2,3-epoxy-4-hydroxynonanal with deoxyguanosine. Chem. Res. Toxicol. 1989, 2, 23–28. [Google Scholar] [CrossRef] [PubMed]
- el Ghissassi, F.; Barbin, A.; Nair, J.; Bartsch, H. Formation of 1,N6-ethenoadenine and 3,N4-ethenocytosine by lipid peroxidation products and nucleic acid bases. Chem. Res. Toxicol. 1995, 8, 278–283. [Google Scholar] [CrossRef]
- Chen, H.J.; Chung, F.L. Epoxidation of trans-4-hydroxy-2-nonenal by fatty acid hydroperoxides and hydrogen peroxide. Chem. Res. Toxicol. 1996, 9, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Pence, M.G.; Christov, P.P.; Wawrzak, Z.; Choi, J.Y.; Rizzo, C.J.; Egli, M.; Guengerich, F.P. Basis of miscoding of the DNA adduct N2,3-ethenoguanine by human Y-family DNA polymerases. J. Biol. Chem. 2012, 287, 35516–35526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, F.L.; Young, R.; Hecht, S.S. Detection of cyclic 1,N2-propanodeoxyguanosine adducts in DNA of rats treated with N-nitrosopyrrolidine and mice treated with crotonaldehyde. Carcinogenesis 1989, 10, 1291–1297. [Google Scholar] [CrossRef]
- Zhang, S.; Villalta, P.W.; Wang, M.; Hecht, S.S. Analysis of crotonaldehyde- and acetaldehyde-derived 1,n(2)-propanodeoxyguanosine adducts in DNA from human tissues using liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2006, 19, 1386–1392. [Google Scholar] [CrossRef] [Green Version]
- Stone, M.P.; Cho, Y.J.; Huang, H.; Kim, H.Y.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Minko, I.G.; Lloyd, R.S.; Harris, T.M.; et al. Interstrand DNA cross-links induced by alpha,beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 2008, 41, 793–804. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Y.; Chung, F.L.; Hecht, S.S. Identification of crotonaldehyde as a hepatic microsomal metabolite formed by alpha-hydroxylation of the carcinogen N-nitrosopyrrolidine. Chem. Res. Toxicol. 1988, 1, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, M.; Matsuda, T.; Sasaki, G.; Yagi, T.; Matsui, S.; Takebe, H. A spectrum of mutations induced by crotonaldehyde in shuttle vector plasmids propagated in human cells. Carcinogenesis 1998, 19, 69–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, F.L.; Young, R.; Hecht, S.S. A Study of Chemical Carcinogenesis.61. Formation of Cyclic 1,N2-Propanodeoxyguanosine Adducts in DNA Upon Reaction with Acrolein or Crotonaldehyde. Cancer Res. 1984, 44, 990–995. [Google Scholar] [PubMed]
- Nath, R.G.; Chung, F.L. Detection of exocyclic 1,N2-propanodeoxyguanosine adducts as common DNA lesions in rodents and humans. Proc. Natl. Acad. Sci. USA 1994, 91, 7491–7495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanishi, M.; Matsuda, T.; Nakayama, A.; Takebe, H.; Matsui, S.; Yagi, T. Molecular analysis of mutations induced by acrolein in human fibroblast cells using supF shuttle vector plasmids. Mutat. Res. /Genet. Toxicol. Environ. Mutagen. 1998, 417, 65–73. [Google Scholar] [CrossRef]
- Uchida, K.; Kanematsu, M.; Morimitsu, Y.; Osawa, T.; Noguchi, N.; Niki, E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J. Biol. Chem. 1998, 273, 16058–16066. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.M.; Hazen, S.L.; Hsu, F.F.; Heinecke, J.W. Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acrolein. A mechanism for the generation of highly reactive alpha-hydroxy and alpha,beta-unsaturated aldehydes by phagocytes at sites of inflammation. J. Clin. Invest. 1997, 99, 424–432. [Google Scholar] [CrossRef]
- Yang, I.Y.; Chan, G.; Miller, H.; Huang, Y.; Torres, M.C.; Johnson, F.; Moriya, M. Mutagenesis by acrolein-derived propanodeoxyguanosine adducts in human cells. Biochemistry 2002, 41, 13826–13832. [Google Scholar] [CrossRef]
- Barrows, L.R.; Magee, P.N. Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis 1982, 3, 349–351. [Google Scholar] [CrossRef]
- Rydberg, B.; Lindahl, T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982, 1, 211–216. [Google Scholar] [CrossRef]
- Kang, H.; Konishi, C.; Kuroki, T.; Huh, N. Detection of O6-methylguanine, O4-methylthymine and O4-ethylthymine in human liver and peripheral blood leukocyte DNA. Carcinogenesis 1995, 16, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, G.L. The Nature of the Active Methyl Donor Formed Enzymatically from L-Methionine and Adenosinetriphosphate1,2. J. Am. Chem. Soc. 1952, 74, 2942–2943. [Google Scholar] [CrossRef]
- Soll, J.M.; Sobol, R.W.; Mosammaparast, N. Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends Biochem. Sci. 2017, 42, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsden, D.A.; Jones, D.J.; Lamb, J.H.; Tompkins, E.M.; Farmer, P.B.; Brown, K. Determination of endogenous and exogenously derived N7-(2-hydroxyethyl)guanine adducts in ethylene oxide-treated rats. Chem. Res. Toxicol. 2007, 20, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Segerback, D. Reaction products in hemoglobin and DNA after in vitro treatment with ethylene oxide and N-(2-hydroxyethyl)-N-nitrosourea. Carcinogenesis 1990, 11, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, M.; Hochstein, P. Ethylene formation in rat liver microsomes. Science 1966, 152, 213–214. [Google Scholar] [CrossRef]
- Tornqvist, M.; Gustafsson, B.; Kautiainen, A.; Harms-Ringdahl, M.; Granath, F.; Ehrenberg, L. Unsaturated lipids and intestinal bacteria as sources of endogenous production of ethene and ethylene oxide. Carcinogenesis 1989, 10, 39–41. [Google Scholar] [CrossRef]
- Thier, R.; Bolt, H.M. Carcinogenicity and genotoxicity of ethylene oxide: New aspects and recent advances. Crit. Rev. Toxicol. 2000, 30, 595–608. [Google Scholar] [CrossRef]
- Manjanatha, M.G.; Shelton, S.D.; Chen, Y.; Parsons, B.L.; Myers, M.B.; McKim, K.L.; Gollapudi, B.B.; Moore, N.P.; Haber, L.T.; Allen, B.; et al. Dose and temporal evaluation of ethylene oxide-induced mutagenicity in the lungs of male big blue mice following inhalation exposure to carcinogenic concentrations. Environ. Mol. Mutagen. 2017, 58, 122–134. [Google Scholar] [CrossRef]
- Kumar, N.; Moreno, N.C.; Feltes, B.C.; Menck, C.F.; Houten, B.V. Cooperation and interplay between base and nucleotide excision repair pathways: From DNA lesions to proteins. Genet. Mol. Biol. 2020, 43, e20190104. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun Signal 2017, 15, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Katafuchi, A.; Matsubara, M.; Terato, H.; Tsuboi, T.; Masuda, T.; Tatsumoto, T.; Pack, S.P.; Makino, K.; Croteau, D.L.; et al. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J. Biol. Chem. 2009, 284, 27065–27076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanishi, M.; Matsuda, T.; Yagi, T. Genotoxicity of formaldehyde: Molecular basis of DNA damage and mutation. Front. Environ. Sci. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Cooper, D.E.; Cluntun, A.A.; Warmoes, M.O.; Zhao, S.; Reid, M.A.; Liu, J.; Lund, P.J.; Lopes, M.; Garcia, B.A.; et al. Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell 2018, 175, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Guengerich, F.P. Adduct size limits efficient and error-free bypass across bulky N2-guanine DNA lesions by human DNA polymerase eta. J. Mol. Biol. 2005, 352, 72–90. [Google Scholar] [CrossRef]
- Perrino, F.W.; Blans, P.; Harvey, S.; Gelhaus, S.L.; McGrath, C.; Akman, S.A.; Jenkins, G.S.; LaCourse, W.R.; Fishbein, J.C. The N2-ethylguanine and the O6-ethyl- and O6-methylguanine lesions in DNA: Contrasting responses from the “bypass” DNA polymerase eta and the replicative DNA polymerase alpha. Chem. Res. Toxicol. 2003, 16, 1616–1623. [Google Scholar] [CrossRef]
- Balbo, S.; Brooks, P.J. Implications of Acetaldehyde-Derived DNA Adducts for Understanding Alcohol-Related Carcinogenesis. In Biological Basis of Alcohol-Induced Cancer; Springer International Publishing: Cham, Switzerland, 2014; pp. 71–88. [Google Scholar] [CrossRef]
- Albertini, R.J. Vinyl acetate monomer (VAM) genotoxicity profile: Relevance for carcinogenicity. Crit. Rev. Toxicol. 2013, 43, 671–706. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxy radicals, lipid peroxidation and DNA damage. Toxicology 2002, 181–182, 219–222. [Google Scholar] [CrossRef]
- Winczura, A.; Czubaty, A.; Winczura, K.; Maslowska, K.; Nalecz, M.; Dudzinska, D.A.; Saparbaev, M.; Staron, K.; Tudek, B. Lipid peroxidation product 4-hydroxy-2-nonenal modulates base excision repair in human cells. DNA Repair 2014, 22, 1–11. [Google Scholar] [CrossRef]
- Tang, J.B.; Goellner, E.M.; Wang, X.H.; Trivedi, R.N.; St Croix, C.M.; Jelezcova, E.; Svilar, D.; Brown, A.R.; Sobol, R.W. Bioenergetic metabolites regulate base excision repair-dependent cell death in response to DNA damage. Mol. Cancer Res. 2010, 8, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Hu, W.; Tang, M.S. Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: A possible mechanism for lipid peroxidation-induced carcinogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 8598–8602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Hu, W.; Hu, Y.; Tang, M.S. Acrolein is a major cigarette-related lung cancer agent: Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc. Natl. Acad. Sci. USA 2006, 103, 15404–15409. [Google Scholar] [CrossRef] [Green Version]
- Doorn, J.A.; Hurley, T.D.; Petersen, D.R. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem. Res. Toxicol. 2006, 19, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.L.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell. Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.L.; Ye, D.; et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.Q.; Ishak Gabra, M.B.; Lowman, X.H.; Yang, Y.; Reid, M.A.; Pan, M.; O’Connor, T.R.; Kong, M. Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol. 2017, 15, e2002810. [Google Scholar] [CrossRef]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef] [Green Version]
- Franzolin, E.; Pontarin, G.; Rampazzo, C.; Miazzi, C.; Ferraro, P.; Palumbo, E.; Reichard, P.; Bianchi, V. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 14272–14277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashlan, O.B.; Scott, C.P.; Lear, J.D.; Cooperman, B.S. A comprehensive model for the allosteric regulation of mammalian ribonucleotide reductase. Functional consequences of ATP- and dATP-induced oligomerization of the large subunit. Biochemistry 2002, 41, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Tang, C.; Zhao, Q.; Wang, W.; Xiong, Y. Structural basis of cellular dNTP regulation by SAMHD1. Proc. Natl. Acad. Sci. USA 2014, 111, E4305–E4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, C.K. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 2015, 15, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannava, S.; Moparthy, K.C.; Wheeler, L.J.; Natarajan, V.; Zucker, S.N.; Fink, E.E.; Im, M.; Flanagan, S.; Burhans, W.C.; Zeitouni, N.C.; et al. Depletion of deoxyribonucleotide pools is an endogenous source of DNA damage in cells undergoing oncogene-induced senescence. Am. J. Pathol. 2013, 182, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef] [Green Version]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef]
- Gandhi, V.V.; Samuels, D.C. A review comparing deoxyribonucleoside triphosphate (dNTP) concentrations in the mitochondrial and cytoplasmic compartments of normal and transformed cells. Nucleosides Nucleotides Nucleic Acids 2011, 30, 317–339. [Google Scholar] [CrossRef] [Green Version]
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383. [Google Scholar] [CrossRef]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.F.; et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 2017, 546, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, K.M.; Worth, A.J.; Snyder, N.W.; Lee, J.V.; Sivanand, S.; Liu, Q.; Blair, I.A.; Wellen, K.E.; Zhang, R. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 2015, 11, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, X.; Duan, X.; Mao, W.; Li, X.; Li, Z.; Li, Q.; Zheng, Z.; Xu, H.; Chen, M.; Wang, P.G.; et al. O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nat. Commun. 2015, 6, 8468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.L.; Lu, F.Z.; Shen, X.Y.; Wu, Y.; Zhao, L.T. SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem. Biophys. Res. Commun. 2014, 455, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Buckland, R.J.; Watt, D.L.; Chittoor, B.; Nilsson, A.K.; Kunkel, T.A.; Chabes, A. Increased and imbalanced dNTP pools symmetrically promote both leading and lagging strand replication infidelity. PLoS Genet. 2014, 10, e1004846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, T.M.; Sharma, S.; Chabes, A.; Shcherbakova, P.V. Colon cancer-associated mutator DNA polymerase delta variant causes expansion of dNTP pools increasing its own infidelity. Proc. Natl. Acad. Sci. USA 2015, 112, E2467. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.N.; Marjavaara, L.; Knowels, G.M.; Schultz, E.M.; Fox, E.J.; Chabes, A.; Herr, A.J. dNTP pool levels modulate mutator phenotypes of error-prone DNA polymerase epsilon variants. Proc. Natl. Acad. Sci. USA 2015, 112, E2457–E2466. [Google Scholar] [CrossRef] [Green Version]
- James, S.J.; Cross, D.R.; Miller, B.J. Alterations in nucleotide pools in rats fed diets deficient in choline, methionine and/or folic acid. Carcinogenesis 1992, 13, 2471–2474. [Google Scholar] [CrossRef]
- James, S.J.; Basnakian, A.G.; Miller, B.J. In vitro folate deficiency induces deoxynucleotide pool imbalance, apoptosis, and mutagenesis in Chinese hamster ovary cells. Cancer Res. 1994, 54, 5075–5080. [Google Scholar]
- Blount, B.C.; Mack, M.M.; Wehr, C.M.; MacGregor, J.T.; Hiatt, R.A.; Wang, G.; Wickramasinghe, S.N.; Everson, R.B.; Ames, B.N. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 3290–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.W.; Tsao, N.; Huang, L.Y.; Yen, Y.; Liu, X.; Lehman, C.; Wang, Y.H.; Tseng, M.C.; Chen, Y.J.; Ho, Y.C.; et al. The Impact of dUTPase on Ribonucleotide Reductase-Induced Genome Instability in Cancer Cells. Cell Rep. 2016, 16, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kuilenburg, A.B.; van Lenthe, H.; van Gennip, A.H. Activity of pyrimidine degradation enzymes in normal tissues. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Basbous, J.; Aze, A.; Chaloin, L.; Lebdy, R.; Hodroj, D.; Ribeyre, C.; Larroque, M.; Shepard, C.; Kim, B.; Pruvost, A.; et al. Dihydropyrimidinase protects from DNA replication stress caused by cytotoxic metabolites. Nucleic Acids Res. 2020, 48, 1886–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gad, H.; Koolmeister, T.; Jemth, A.S.; Eshtad, S.; Jacques, S.A.; Strom, C.E.; Svensson, L.M.; Schultz, N.; Lundback, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Warpman Berglund, U.; Sanjiv, K.; Gad, H.; Kalderen, C.; Koolmeister, T.; Pham, T.; Gokturk, C.; Jafari, R.; Maddalo, G.; Seashore-Ludlow, B.; et al. Validation and development of MTH1 inhibitors for treatment of cancer. Ann. Oncol. 2016, 27, 2275–2283. [Google Scholar] [CrossRef]
- Niida, H.; Katsuno, Y.; Sengoku, M.; Shimada, M.; Yukawa, M.; Ikura, M.; Ikura, T.; Kohno, K.; Shima, H.; Suzuki, H.; et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev. 2010, 24, 333–338. [Google Scholar] [CrossRef] [Green Version]
- D’Angiolella, V.; Donato, V.; Forrester, F.M.; Jeong, Y.T.; Pellacani, C.; Kudo, Y.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012, 149, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.M.; Yeh, M.T.; Tsao, N.; Chen, C.W.; Gao, Q.Z.; Chang, C.Y.; Lee, M.H.; Fang, J.M.; Sheu, S.Y.; Lin, C.J.; et al. Tumor cells require thymidylate kinase to prevent dUTP incorporation during DNA repair. Cancer Cell 2012, 22, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Burkhalter, M.D.; Roberts, S.A.; Havener, J.M.; Ramsden, D.A. Activity of ribonucleotide reductase helps determine how cells repair DNA double strand breaks. DNA Repair 2009, 8, 1258–1263. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Li, Z.; Xiao, L.; Hu, W.; Zhang, L.; Xie, B.; Zhou, Q.; He, J.; Qiu, Y.; Wen, M.; et al. Glutamine Synthetase Promotes Radiation Resistance via Facilitating Nucleotide Metabolism and Subsequent DNA Damage Repair. Cell Rep. 2019, 28, 1136–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Sun, W.; Zhong, J.; Lv, H.; Zhu, M.; Xu, J.; Jin, N.; Xie, Z.; Tan, M.; Lin, S.H.; et al. Phosphoglycerate mutase 1 regulates dNTP pool and promotes homologous recombination repair in cancer cells. J. Cell Biol. 2017, 216, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, N.M.S.; Farnegardh, K.; Bonagas, N.; Ninou, A.H.; Groth, P.; Wiita, E.; Jonsson, M.; Hallberg, K.; Lehto, J.; Pennisi, R.; et al. Targeting PFKFB3 radiosensitizes cancer cells and suppresses homologous recombination. Nat. Commun. 2018, 9, 3872. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017, 67, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef]
- Chen, L.L.; Xiong, Y. Tumour metabolites hinder DNA repair. Nature 2020, 582, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.I.; Costa, A.S.H.; Ferguson, A.N.; Frezza, C. Fumarate hydratase loss promotes mitotic entry in the presence of DNA damage after ionising radiation. Cell Death Dis. 2018, 9, 913. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, X.; Shen, J.; Wang, Y.; Li, X.; Liu, R.; Xia, Y.; Chen, Q.; Peng, G.; Lin, S.Y.; et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat. Cell Biol. 2015, 17, 1158–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leshets, M.; Silas, Y.B.H.; Lehming, N.; Pines, O. Fumarase: From the TCA Cycle to DNA Damage Response and Tumor Suppression. Front. Mol. Biosci. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Efimova, E.V.; Appelbe, O.K.; Ricco, N.; Lee, S.S.; Liu, Y.; Wolfgeher, D.J.; Collins, T.N.; Flor, A.C.; Ramamurthy, A.; Warrington, S.; et al. O-GlcNAcylation Enhances Double-Strand Break Repair, Promotes Cancer Cell Proliferation, and Prevents Therapy-Induced Senescence in Irradiated Tumors. Mol. Cancer Res. 2019, 17, 1338–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sizemore, S.T.; Zhang, M.; Cho, J.H.; Sizemore, G.M.; Hurwitz, B.; Kaur, B.; Lehman, N.L.; Ostrowski, M.C.; Robe, P.A.; Miao, W.; et al. Pyruvate kinase M2 regulates homologous recombination-mediated DNA double-strand break repair. Cell Res. 2018, 28, 1090–1102. [Google Scholar] [CrossRef]
- Xia, L.; Qin, K.; Wang, X.R.; Wang, X.L.; Zhou, A.W.; Chen, G.Q.; Lu, Y. Pyruvate kinase M2 phosphorylates H2AX and promotes genomic instability in human tumor cells. Oncotarget 2017, 8, 109120–109134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.Y.; Su, G.C.; Huang, W.Y.; Ko, M.Y.; Yeh, H.Y.; Chang, G.D.; Lin, S.J.; Chi, P. Promotion of homology-directed DNA repair by polyamines. Nat. Commun. 2019, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Geck, R.C.; Foley, J.R.; Murray Stewart, T.; Asara, J.M.; Casero, R.A., Jr.; Toker, A. Inhibition of the polyamine synthesis enzyme ornithine decarboxylase sensitizes triple-negative breast cancer cells to cytotoxic chemotherapy. J. Biol. Chem. 2020, 295, 6263–6277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piacente, F.; Caffa, I.; Ravera, S.; Sociali, G.; Passalacqua, M.; Vellone, V.G.; Becherini, P.; Reverberi, D.; Monacelli, F.; Ballestrero, A.; et al. Nicotinic Acid Phosphoribosyltransferase Regulates Cancer Cell Metabolism, Susceptibility to NAMPT Inhibitors, and DNA Repair. Cancer Res. 2017, 77, 3857–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.H.; Min, B.H.; Lee, K.M.; Cho, M.H.; Park, G.H.; Lee, K.H. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Deng, X.; Sun, Y.; Bai, L.; Xiahou, Z.; Cong, Y.; Xu, X. Nampt is involved in DNA double-strand break repair. Chin. J. Cancer 2012, 31, 392–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannidou, A.; Goulielmaki, E.; Garinis, G.A. DNA Damage: From Chronic Inflammation to Age-Related Deterioration. Front. Genet. 2016, 7, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, J.H.; Brown, A.L.; Singh, S.K.; Chen, Y.; Gucek, M.; Lee, B.S.; Luckey, M.A.; Kim, M.K.; Park, J.H.; Sleckman, B.P.; et al. Metabolic sensor AMPK directly phosphorylates RAG1 protein and regulates V(D)J recombination. Proc. Natl. Acad. Sci. USA 2013, 110, 9873–9878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazouzi, A.; Battistini, F.; Moser, S.C.; Ferreira da Silva, J.; Wiedner, M.; Owusu, M.; Lardeau, C.H.; Ringler, A.; Weil, B.; Neesen, J.; et al. Repair of UV-Induced DNA Damage Independent of Nucleotide Excision Repair Is Masked by MUTYH. Mol. Cell 2017, 68, 797–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahar, O.D.; Kalousi, A.; Eini, L.; Fisher, B.; Weiss, A.; Darr, J.; Mazina, O.; Bramson, S.; Kupiec, M.; Eden, A.; et al. A high-throughput chemical screen with FDA approved drugs reveals that the antihypertensive drug Spironolactone impairs cancer cell survival by inhibiting homology directed repair. Nucleic Acids Res. 2014, 42, 5689–5701. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, K.; Huang, R.; Zhao, J.; Shahane, S.A.; Witt, K.L.; Smith-Roe, S.L.; Tice, R.R.; Takeda, S.; Xia, M. Identification of genotoxic compounds using isogenic DNA repair deficient DT40 cell lines on a quantitative high throughput screening platform. Mutagenesis 2016, 31, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Sdelci, S.; Lardeau, C.H.; Tallant, C.; Klepsch, F.; Klaiber, B.; Bennett, J.; Rathert, P.; Schuster, M.; Penz, T.; Fedorov, O.; et al. Mapping the chemical chromatin reactivation landscape identifies BRD4-TAF1 cross-talk. Nat. Chem. Biol. 2016, 12, 504–510. [Google Scholar] [CrossRef]
- Wishart, D.; Arndt, D.; Pon, A.; Sajed, T.; Guo, A.C.; Djoumbou, Y.; Knox, C.; Wilson, M.; Liang, Y.; Grant, J.; et al. T3DB: The toxic exposome database. Nucleic Acids Res. 2015, 43, D928–D934. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Neher, T.M.; Shuck, S.C.; Liu, J.Y.; Zhang, J.T.; Turchi, J.J. Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem. Biol. 2010, 5, 953–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihmaid, S.; Ahmed, H.E.A.; Al-Sheikh Ali, A.; Sherif, Y.E.; Tarazi, H.M.; Riyadh, S.M.; Zayed, M.F.; Abulkhair, H.S.; Rateb, H.S. Rational design, synthesis, pharmacophore modeling, and docking studies for identification of novel potent DNA-PK inhibitors. Bioorganic Chem. 2017, 72, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Juarez, E.; Chambwe, N.; Tang, W.; Mitchell, A.D.; Owen, N.; Kumari, A.; Monnat, R.J., Jr.; McCullough, A.K. An RNAi screen in human cell lines reveals conserved DNA damage repair pathways that mitigate formaldehyde sensitivity. DNA Repair 2018, 72, 1–9. [Google Scholar] [CrossRef]
- Kizilors, A.; Pickard, M.R.; Schulte, C.E.; Yacqub-Usman, K.; McCarthy, N.J.; Gan, S.U.; Darling, D.; Gaken, J.; Williams, G.T.; Farzaneh, F. Retroviral insertional mutagenesis implicates E3 ubiquitin ligase RNF168 in the control of cell proliferation and survival. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A genetic map of the response to DNA damage in human cells. Cell 2020. [Google Scholar] [CrossRef]
- Velimezi, G.; Robinson-Garcia, L.; Munoz-Martinez, F.; Wiegant, W.W.; Ferreira da Silva, J.; Owusu, M.; Moder, M.; Wiedner, M.; Rosenthal, S.B.; Fisch, K.M.; et al. Map of synthetic rescue interactions for the Fanconi anemia DNA repair pathway identifies USP48. Nat. Commun. 2018, 9, 2280. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakke, J.; Wright, W.C.; Zamora, A.E.; Oladimeji, P.; Crawford, J.C.; Brewer, C.T.; Autry, R.J.; Evans, W.E.; Thomas, P.G.; Chen, T. Genome-wide CRISPR screen reveals PSMA6 to be an essential gene in pancreatic cancer cells. BMC Cancer 2019, 19, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, D.; Singh, A.; Schmid-Burgk, J.L.; Carlson, R.J.; Mezger, A.; Garrity, A.J.; Zhang, F.; Blainey, P.C. Optical Pooled Screens in Human Cells. Cell 2019, 179, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Ivashkevich, A.; Redon, C.E.; Nakamura, A.J.; Martin, R.F.; Martin, O.A. Use of the gamma-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012, 327, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, S.K.; Zou, T.; Cao, B.; Chen, T.; To, W.P.; Yang, C.; Lok, C.N.; Che, C.M. Luminescent platinum(II) complexes with functionalized N-heterocyclic carbene or diphosphine selectively probe mismatched and abasic DNA. Nat. Commun. 2016, 7, 10655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herr, P.; Lundin, C.; Evers, B.; Ebner, D.; Bauerschmidt, C.; Kingham, G.; Palmai-Pallag, T.; Mortusewicz, O.; Frings, O.; Sonnhammer, E.; et al. A genome-wide IR-induced RAD51 foci RNAi screen identifies CDC73 involved in chromatin remodeling for DNA repair. Cell Discov. 2015, 1, 15034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, Z.D.; Margulies, C.M.; Chaim, I.A.; McRee, S.K.; Mazzucato, P.; Ahmad, A.; Abo, R.P.; Butty, V.L.; Forget, A.L.; Samson, L.D. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc. Natl. Acad. Sci. USA 2014, 111, E1823. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shiraishi, M.; Tsuchihashi, K.; Takatsuka, R.; Yamamoto, J.; Kuraoka, I.; Iwai, S. Fluorescence detection of DNA mismatch repair in human cells. Sci. Rep. 2018, 8, 12181. [Google Scholar] [CrossRef]
- Weingeist, D.M.; Ge, J.; Wood, D.K.; Mutamba, J.T.; Huang, Q.; Rowland, E.A.; Yaffe, M.B.; Floyd, S.; Engelward, B.P. Single-cell microarray enables high-throughput evaluation of DNA double-strand breaks and DNA repair inhibitors. Cell Cycle 2013, 12, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Sykora, P.; Witt, K.L.; Revanna, P.; Smith-Roe, S.L.; Dismukes, J.; Lloyd, D.G.; Engelward, B.P.; Sobol, R.W. Next generation high throughput DNA damage detection platform for genotoxic compound screening. Sci. Rep. 2018, 8, 2771. [Google Scholar] [CrossRef]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R.; et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 2016, 167, 1853–1866. [Google Scholar] [CrossRef]
- Elia, A.E.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; Everley, R.A.; Dephoure, N.; Zhou, C.; Koren, I.; Gygi, S.P.; Elledge, S.J. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Stechow, L.; Olsen, J.V. Proteomics insights into DNA damage response and translating this knowledge to clinical strategies. Proteomics 2017, 17, 1600018. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Xu, D.; Wang, W. Identification and analysis of new proteins involved in the DNA damage response network of Fanconi anemia and Bloom syndrome. Methods 2009, 48, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raschle, M.; Smeenk, G.; Hansen, R.K.; Temu, T.; Oka, Y.; Hein, M.Y.; Nagaraj, N.; Long, D.T.; Walter, J.C.; Hofmann, K.; et al. DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science 2015, 348, 1253671. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Main DNA Adducts and/or Crosslinks | Pathways Producing the Metabolite | Predicted Impacts on Genome Integrity |
|---|---|---|---|
| Formaldehyde | N2-hydroxymethyl-deoxyguanosine (N2- HOMe-dG) N6-hydroxymethyl-deoxyadenosine (N6-HOMe-dA) N4-hydroxymethyl-deoxycytosine (N4-HOMe-dC) [61] DNA–protein crosslinks [62] DNA intra and interstrand crosslinks [63] | Byproduct of enzymatic oxidative demethylation reactions [64] Methylamine metabolism [65] Myeloperoxidation [66] Methanol metabolism [67] | Base substitutions Frameshift mutations DNA breaks and chromosomal aberrations [68] Tandem bases substitutions |
| Acetaldehyde | N2-ethylidene-deoxyguanosine (reduced form: N2-ethyl-2′-deoxyguanosine) [69,70] DNA–protein crosslinks [71] DNA intra and interstrand crosslinks [70,72] | Ethanol metabolism [73] Pyruvate, threonine and other metabolic processes [74] | Base substitutions Frameshift mutations DNA breaks and rearrangements [73] Tandem bases substitutions [72] |
| Methylglyoxal | N2-(1-carboxyethyl)-2′-deoxyguanosine (CEdG) [75,76] Glycation of histones [77] Interstrand crosslinks [78] DNA–protein crosslinks [79] | Side product of glycolysis (Triosephosphate degradation) [80] Product of the degradation of acetone, aminoacetone and threonine [81] Degradation of glycated proteins [82] Lipid peroxidation [83] | Depurination of DNA: promutagenic Replication block: strand breaks, deletions Frameshift mutations |
| Malondialdehyde (MDA) | Pyrimido [1,2-α]purine-10(3H)-one-2′-deoxyribose (M1dG: main product) [84] N6-(3-oxoprenyl)deoxyadenosine (OPdA) [85] N4-(3-oxoprenyl)deoxycytidine (OPdC) [86] DNA interstrand crosslinks [87] DNA–protein crosslinks | Lipid peroxidation [88] Biosynthesis of prostaglandins [89,90] | Base substitutions [91] Frameshift mutations |
| 4-hydroxy-2-nonenal (HNE) | Substituted 1,N2-propano-2′-deoxyguanosine (4 diastereomers) [92,93] | Lipid peroxidation [60,94] | Base substitutions |
| 2,3-epoxy-4-hydroxynonanal (HNE epoxyde) | Etheno adducts: 1,N2-ethenodeoxyguanosine [95] 3,N4-ethenodeoxycytidine [96] 1,N6-ethenodeoxyadenosine [96] | Oxidation of HNE [60,97] | Base substitutions DNA replication blocade: by-pass by error-prone TLS polymerases [98] |
| Crotonaledyde (or 2 acetaldehydes) | 8-hydroxy-6-methyl-1,N2-propano-2′-deoxyguanosine (2 diastereomers) [99,100] DNA interstrand crosslinks [101] Protein–DNA crosslinks | Lipid peroxidation [60] Metabolite of N-nitrosopyrrolidine [102] | Base substitutions [103] Frameshift mutations [103] |
| Acrolein | γ-hydroxy-1,N2-propano-2′-deoxyguanosine (γ-OH-PdG) α-hydroxy-1,N2-propano-2′-deoxyguanosine (α-OH-PdG) [104,105] DNA intra and interstrand crosslinks [101,106] Protein–DNA crosslinks | Lipid peroxidation [60,107] Myeloperoxidation in neutrophils and monocytes [108] | Base substitutions [109] Frameshift mutations Tandem bases substitutions [106] |
| S-Adenosyl methionine (SAM) | N7-methylguanine (7meG) N1- and N3- methyladenine (1meA and 3meA) O6-methylguanine (O6meG) O4-methylthymine (O4meT) O4-ethylthymine [110,111,112] | Synthesized from ATP and methionine [113] | 7meG: Harmless but can become an abasic site, promutagenic 3meA: DNA replication block, by-pass by error-prone TLS polymerase O-adducts: Bases mispairing [114] |
| Ethylene oxide (EO) | N7-(2-hydroxyethyl)dG (main product) N3-(2-hydroxyethyl)dA N3-(2-hydroxyethyl)dU O6-(2-hydroxyethyl)dG [115,116] | Lipid peroxidation [60,117] Gut microflora [118] | Mutagenic (unclear mode of action) [119,120] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moretton, A.; Loizou, J.I. Interplay between Cellular Metabolism and the DNA Damage Response in Cancer. Cancers 2020, 12, 2051. https://doi.org/10.3390/cancers12082051
Moretton A, Loizou JI. Interplay between Cellular Metabolism and the DNA Damage Response in Cancer. Cancers. 2020; 12(8):2051. https://doi.org/10.3390/cancers12082051
Chicago/Turabian StyleMoretton, Amandine, and Joanna I. Loizou. 2020. "Interplay between Cellular Metabolism and the DNA Damage Response in Cancer" Cancers 12, no. 8: 2051. https://doi.org/10.3390/cancers12082051
APA StyleMoretton, A., & Loizou, J. I. (2020). Interplay between Cellular Metabolism and the DNA Damage Response in Cancer. Cancers, 12(8), 2051. https://doi.org/10.3390/cancers12082051